")
Back to Journals » ImmunoTargets and Therapy » Volume 10
Cellular and Humoral Immune Responses in Covid-19 and Immunotherapeutic Approaches
Authors Hasan A , Al-Ozairi E , Al-Baqsumi Z, Ahmad R , Al-Mulla F
Received 6 September 2020
Accepted for publication 22 December 2020
Published 9 March 2021 Volume 2021:10 Pages 63—85
DOI https://doi.org/10.2147/ITT.S280706
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Professor Michael Shurin
Amal Hasan,1 Ebaa Al-Ozairi,2,3 Zahraa Al-Baqsumi,1 Rasheed Ahmad,1 Fahd Al-Mulla4
1Department of Immunology and Microbiology, Research Division, Dasman Diabetes Institute, Dasman, Kuwait City, Kuwait; 2Clinical Research Unit, Medical Division, Dasman Diabetes Institute, Dasman, Kuwait City, Kuwait; 3Department of Medicine, Faculty of Medicine, Jabriya, Kuwait City, Kuwait; 4Department of Genetics and Bioinformatics, Functional Genomics, Research Division, Dasman Diabetes Institute, Dasman, Kuwait City, Kuwait
Correspondence: Amal Hasan P.O. Box 1180, Dasman 15462, Kuwait
Tel +965 2224 2999 Ext. 4312
Email [email protected]
Abstract: Coronavirus disease 2019 (Covid-19), caused by the novel coronavirus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), can range in severity from asymptomatic to severe/critical disease. SARS-CoV-2 uses angiotensin-converting enzyme 2 to infect cells leading to a strong inflammatory response, which is most profound in patients who progress to severe Covid-19. Recent studies have begun to unravel some of the differences in the innate and adaptive immune response to SARS-CoV-2 in patients with different degrees of disease severity. These studies have attributed the severe form of Covid-19 to a dysfunctional innate immune response, such as a delayed and/or deficient type I interferon response, coupled with an exaggerated and/or a dysfunctional adaptive immunity. Differences in T-cell (including CD4+ T-cells, CD8+ T-cells, T follicular helper cells, γδ-T-cells, and regulatory T-cells) and B-cell (transitional cells, double-negative 2 cells, antibody-secreting cells) responses have been identified in patients with severe disease compared to mild cases. Moreover, differences in the kinetic/titer of neutralizing antibody responses have been described in severe disease, which may be confounded by antibody-dependent enhancement. Importantly, the presence of preexisting autoantibodies against type I interferon has been described as a major cause of severe/critical disease. Additionally, priorVaccine and multiple vaccine exposure, trained innate immunity, cross-reactive immunity, and serological immune imprinting may all contribute towards disease severity and outcome. Several therapeutic and preventative approaches have been under intense investigations; these include vaccines (three of which have passed Phase 3 clinical trials), therapeutic antibodies, and immunosuppressants.
Keywords: SARS-CoV-2, inflammation, immune regulation, neutralizing antibodies, cytokines, autoantibodies, type I interferon
Introduction
Coronavirus disease 2019 (Covid-19), a disease caused by the novel coronavirus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), emerged in China and rapidly spread across the world.1 The World Health Organization declared Covid-19 as a pandemic on 11 March 2020, and by 09 December 2020, there have been 67,780,361 confirmed cases worldwide, including 1,551,214 deaths. Daily new cases continue to emerge worldwide, with second spikes/waves in some areas. There have been considerable efforts to understand the pathogenicity of SARS-CoV-2, which is a respiratory coronavirus that belongs to the betacoronavirus genus and is closely related to other coronaviruses, namely SARS-CoV (79% genetic similarity) and middle east respiratory syndrome coronavirus (MERS-CoV).2,3 The main mode of transmission of SARS-CoV-2 is via respiratory droplets. The incubation period of SARS-CoV-2 ranges between 2 and 14 days4,5 with a median of 4–5 days;6–9 however, most (97.5%) infected patients develop symptoms within 11.5 days.8 Infected individuals can present with no symptoms, but most develop mild/moderate disease, while others succumb to severe/critical disease.4,5 Most patients with Covid-19 exhibit a fever and dry cough, but some can also experience loss of taste and/or smell,10–13 difficulty breathing, headache, dizziness, muscle/joint pain, nausea, diarrhoea, and coughing up of blood.6,14–19 In patients with severe disease, the clinical sequelae include immune dysfunction, lymphopenia, sustained inflammation, secondary bacterial infection, acute respiratory distress syndrome (ARDS), coagulation activation, thrombosis, myocardial injury, and hepatic and kidney injury.15,18,20,21–23 Limited knowledge is currently available to fully explain the disparity in symptom severity and fatality rates around the world especially pertaining to the host immune response to SARS-CoV-2. A better understanding of the immune response in patients with Covid-19 will pave the way for new treatments and prevention strategies. In this review, we discuss what is currently known (as per literature search conducted using PubMed) about the innate and adaptive immune responses to SARS-CoV-2 infection, and how these responses may be related to disease severity. In addition, novel immunotherapeutic approaches are presented.
SARS-CoV-2 Infection and the Inflammatory Response
Cell Entry, Cell Pyroptosis and NETosis
Analysis of the nucleic acid sequence within the spike protein receptor-binding domain (RBD) suggests that SAR-CoV-2 uses angiotensin-converting enzyme 2 (ACE2) as a cell receptor for cellular entry and infection,4,24,25 which is the same receptor used by SARS-CoV.4,26 SARS-CoV-2 infectivity may also be enhanced by ACE2 variant N720D,27 and by increased affinity of Furin to the mutated D614G spike protein of the virus.28 In addition, SARS-CoV-2 may gain entry via other cell receptors or utilize other cell entry modes such as antibody-dependent enhancement (ADE); however, currently, there is no evidence of this. ACE2 is mainly expressed by type 2 alveolar cells in the lung,5 and a minimal percentage of monocytes/macrophages also express ACE2.29 Cell pyroptosis, a form of programmed cell death which is extremely proinflammatory,30 is a likely pathogenic mechanism of SARS-CoV-2. After cellular entry, rapid viral replication may cause substantial epithelial and endothelial cell apoptosis and vascular leakage, which triggers the release of profuse proinflammatory cytokines and chemokines.6 Indeed, interleukin (IL)-1β, which is released during pyroptosis, is elevated in patients with SARS-CoV-2 infection.15
Moreover, pathogen-associated molecular patterns (PAMPs; such as ribonucleic acid (RNA)), and damage-associated molecular patterns (DAMPs; such as adenosine triphosphate, deoxyribonucleic acid (DNA), and antibody-secreting cell (ASC) oligomers) are detected by alveolar epithelial cells and macrophages, which results in the production of proinflammatory chemokines (monocyte chemoattractant protein-1 (MCP-1) and interferon (IFN)-γ-induced protein-10 (IP-10)), and cytokines (IL-6, IFN-γ,15,31 and type I IFN).32 These chemokines and cytokines attract immune cells, mainly T-cells and monocytes, from the circulation into the infected lungs.33,34 This may partly explain the lymphopenia observed in 80% of patients with SARS-CoV-2 infection.1,6 In addition, SARS-CoV-2 induces pyroptosis in lymphocytes and macrophages,6 which adds to the heightened inflammatory milieu,30,35 and exacerbates the lymphopenia in the vast majority of patients.6 This was also the case with SARS-CoV whereby infection of immune cells leads to aberrant cytokine production.36–39
Furthermore, SARS-CoV-2 infection, cell pyroptosis, and the accompanying hyperinflammation may trigger NETosis, which is a regulated cell death process by neutrophils that releases neutrophil extracellular traps (NETs).40 NETs are extracellular lattices of chromatin with histones and antimicrobial proteins, which are released upon stimulation. Pathogens such as respiratory viruses induce NETosis and the release of NETs that physically trap and kill microbes as part of the innate immune response.41 However, NETosis can become dysregulated in acute and chronic inflammatory disease.42 For example, platelets can trigger NET-mediated tissue damage, hypercoagulability, and thrombosis.43 Moreover, NETosis contributes to sepsis and ARDS pathogenesis, with NETs causing vascular tissue damage, microthrombi, and multiorgan failure.44–48 Pathogenic immunothrombosis described in severe cases of Covid-1949,23 may result from a dysregulated NET formation.50 Indeed, increased NET formation has been found to correlate with Covid-19-related ARDS and is therefore a potential marker of disease severity.51 Moreover, patients with severe disease have increased plasma platelet factor 4 (PF4) and regulated on activation, normal T-cell expressed and secreted (RANTES),51 which are known triggers of NETosis52 (Figure 1). Interestingly, neonatal NET-inhibitory factor, which is a NET-inhibitory peptide present in umbilical cord blood,53,54 inhibits NET formation induced by plasma from patients with Covid-19.51 Therefore, this knowledge may be used to discover novel treatments, for example, by the use of such peptides. Of note, since neutrophils can undergo pyroptosis,55,56 it is possible that this process also occurs in the inflamed lung tissue of patients with Covid-19.
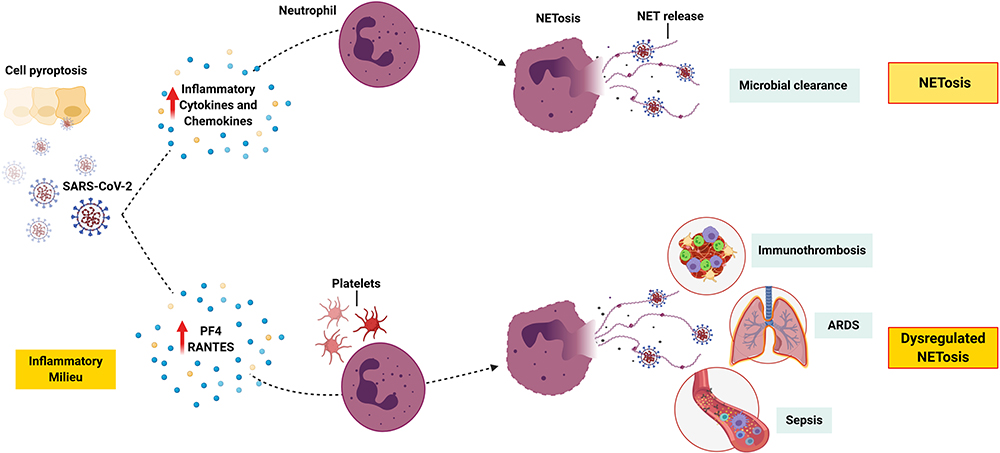 |
Figure 1 NETosis in severe Covid-19. SARS-CoV-2 infection and pyroptosis triggers the release of inflammatory cytokines and chemokines, which leads to the recruitment of neutrophils and other immune cells. Activation of neutrophils triggers NETosis, and the release of NETs to trap and kill the virus. However, a dysregulation in this process might lead to an increase in PF4 and RANTES, both of which are known to trigger NETosis. Activated platelets play a major role in NET-mediated tissue damage, and pathogenic immunothrombosis. In addition, NETosis contributes towards sepsis and ARDS.Note: This figure was created with BioRender.com. Abbreviations: ARDS, acute respiratory distress syndrome; NET, neutrophil extracellular trap; PF4, plasma platelet factor 4; RANTES, regulated on activation, normal T-cell expressed and secreted; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2. |
In addition, studies on SARS-CoV infection have shown that the spike protein can downregulate ACE2,57,58 and induce its shedding leading to the release of enzymatically active soluble ACE2 (sACE2).59 This loss of pulmonary ACE2 function enhances inflammation and vascular permeability leading to acute lung injury.60,61 Moreover, damage caused by the virus, coupled with inflammatory cytokines such as tumor necrosis factor (TNF) and IL-1β, enhances the shedding of ACE2 and the release of sACE2.59,62,63 It is thought that sACE2 may be involved in the inflammatory response of both SARS-CoV and SARS-CoV-2.62,64
Studies have shown that the viral load of SARS-CoV-2 reaches its peak within 5–6 days of symptom onset,65–67 and does not differ between patients with moderate and severe disease; however, a slower decline of the viral load has been observed in patients with critical disease. The viral load is strongly correlated with IFN-α, IFN-γ, and TNF, which suggests insufficient control of viral replication.68 Interestingly, SARS-CoV-2 RNA has been detected in patients’ samples up until death, which suggests a correlation between viral load persistence and poor disease outcome.69 Viral RNA has also been detected post active infection, and thus, its relationship with disease outcome is unclear.35
PAMPS/TLR Interaction and the IFN Response
The toll-like receptor (TLR) family is comprised of 10 members (TLR-1–TLR-10), which are expressed by innate immune cells such as macrophages, and also by epithelial and fibroblast cells. TLRs can be located on the cell surface (such as TLR-1, TLR-2, TLR-4, TLR-5, TLR-6, and TLR-10), or in the endosome compartment (such as TLR-3, TLR-7, TLR-8, and TLR-9),70 and can be activated by a multitude of PAMPs present in pathogens. TLR-7 recognizes PAMPS in the form of viral single-stranded RNA and is therefore implicated in the clearance of SARS-CoV-2.71 TLR activation via the myeloid differentiation primary response 88 and TIR-domain-containing adapter-inducing IFN-β-dependent pathways causes nuclear translocation of the transcription factors nuclear factor kappa B (NF-κB), IFN regulatory factor (IRF)-3 and IRF-7 resulting in the production of proinflammatory cytokines (such as IL-6 and TNF) and type I IFNs, which are critical in anti-viral immunity.71 In this regard, the type I IFN response is the first line of defence against viralVaccine leading to suppression of viral replication at an early stage.32 The binding of type I IFN with IFN-α/β receptor (IFNAR) on immune cells activates downstream signalling pathways and initiates the transcription of IFN-stimulated genes.32
In SARS-CoV and MERS-CoVVaccine, type I IFN responses were suppressed and associated with disease severity.72 Similarly, patients with SARS-CoV-2 infection were found to have suboptimal type-I IFN and type-III IFN responses.50,73–77 These findings suggest that such coronaviruses might interfere with key signalling pathways that are involved in type I IFN production. Accordingly, TLRs and/or down-stream signalling molecules might be implicated in the failure to induce an effective anti-SARS-CoV-2 immune response. In this regard, SARS-CoV-2 may prevent a type I IFN response, in patients who progress to severe Covid-19, by inhibiting TNF-receptor-associated factors (TRAF)-3 and −6 which are critical in inducing IRF-3/7 in response to TLR-7 activation.78 A similar mechanism has been described with SARS-CoV whereby the virus prevented pattern-recognition receptor (PRR) signalling via TANK-binding kinase 1/inhibitor of NF-κB kinase subunit-ε, TRAF-3, and IRF-3.79,80
Interestingly, experimental mice models of ARDS have shown that genetic inactivation of the TLR-4 gene, but not TLR-3 or 9 genes, was associated with reduced acute lung injury.81 This kind of improvement was also observed in IL-6-/- mice,82 however, no benefit has been confirmed with the use of a humanized monoclonal antibody (tocilizumab) against IL-6R in humans.83 In patients with severe Covid-19, macrophages play a key role in the production of cytokines such as IL-6, TNF, IL-1β, IL-10, and IL-12 via TLRs activation.82 Importantly, stimulation of lung macrophages with TLR agonists showed that TLR-4 activation had the most profound effect on cytokine production. Although SARS-CoV-2 is unlikely to activate TLR-4, it has been hypothesized that oxidized phospholipids may be responsible for TLR-4 activation and onset of ARDS.81 Since neutrophil myeloperoxidase, reported to be elevated in patients with severe/critical Covid-19,84 is capable of oxidising phospholipids85 abundant in alveolar surfactant,86 TLR-4 may represent a novel target against Covid-19 via the use of TLR-4 antagonists.78
Alternatively, pathogenic coronaviruses may employ other mechanisms that hamper the immune response, for example, by interfering with signalling molecules downstream of IFNAR.72 In SARS-CoV infection, the virus prevents PRR recognition of viral RNA,79,87,88 prevents downstream IFN signalling via signal transducer and activator of transcription-1,89 and promotes host messenger RNA degradation and thus hindering protein translation.90 Such hampering with the IFN response promotes viral replication and increases pyroptosis leading to a profound inflammatory response.35 In SARS-CoV and MERS-CoVVaccine, a dysregulated type I IFN response and increased neutrophil and monocyte/macrophage influx into the lung72,91,92 are the main cause of lethal pneumonia. In this regard, a delayed type I IFN response may lead to influx of hyperinflammatory neutrophils and monocytes/macrophages resulting in lung immunopathology including pneumonia and ARDS.72,91,92 Moreover, active viral replication at a later stage results in hyperproduction of type I IFN and influx of neutrophils and macrophages, which are the main sources of proinflammatory cytokines. In SARS-CoV-2 infection, similar changes in total neutrophils and lymphocytes are described, and therefore, it is likely that SARS-CoV-2 induces a delayed type I IFN response, and thus, loss of viral control in the early stages of infection. Importantly, SARS-CoV-2 infection appears to be associated with an inappropriate inflammatory response defined by low levels of type I and III IFNs but high levels of IL-6. Therefore, a reduced innate antiviral immunity simultaneously with profound inflammatory cytokine production are the defining features of Covid-19.76 Given that young children do not succumb to severe disease, when the innate immune response is highly effective suggests that innate immunity is critical for disease outcome.72
Cytokine Release Syndrome and Immunopathology
Infection with SARS-CoV-2 causes lung tissue damage which triggers a local immune response. This constitutes the recruitment of monocytes and macrophages, which release inflammatory cytokines and prime adaptive T-cell and B-cell responses. In the majority of cases, the immune response resolves the infection without any further sequelae; however, in some patients, the immune system goes into overdrive leading to a “cytokine storm” and widespread lung inflammation. In pathogenic coronavirusVaccine, including SARS-CoV, MERS-CoV,72 and SARS-CoV-2,15,18,21 the influx of hyperinflammatory monocytes/macrophages into the lung can lead to severe immunopathology, pneumonia, and ARDS in susceptible individuals. Indeed, the bronchoalveolar lavage fluid of patients with severe disease contains a highly inflammatory monocyte-derived FCN1+ macrophage population, which is not present in patients with mild disease.93 Increased inflammatory cell infiltration and excessive secretion of proteases and reactive oxygen species can lead to substantial alveolar damage, breathing difficulties, and increase the risk of secondary infections.33,34 Elevated levels of inflammatory cytokines can cause septic shock and multi-organ failure, which might be the reason why some patients develop myocardial damage and circulatory failure.94
It has been shown that patients with severe Covid-19 have higher levels of circulating CD14+CD16+ inflammatory monocytes compared to persons with mild disease. It is thought that these inflammatory monocytes contribute to the “cytokine storm” by secreting high levels of MCP-1, IP-10, and macrophage inflammatory protein 1α (MIP1α).95 This was confirmed by another study which showed that patients with severe/critical Covid-19 exhibit higher plasma levels of TNF, IL-2, IL-7, IL-10, granulocyte macrophage-colony stimulating factor (GM-CSF), IP-10, MCP-1, and MIP1α compared to patients who do not require intensive care.15
Moreover, increased neutrophils, lymphopenia, increased IL-6, and C-reactive protein (CRP) are correlated with disease severity and mortality.4,5 The level of IL-6 in patients with severe disease increases with time and is higher in non-survivors compared to survivors.69 Moreover, excessive activation of neutrophils infiltrating the lungs, dysregulation of myeloid cells, and T-cell and natural killer (NK) cell exhaustion have been described in patients hospitalized with Covid-19.50,73–77 As described previously, IP-10 is upregulated in severe disease74 in spite of depletions in plasmacytoid dendritic cells (pDC), T helper (Th)-1, and Th17.1 cells, which may have led to deficiencies in type I IFN and type III IFN (IFNs are the main inducers of IP-10). This suggests that elevated IP-10 may have been induced via other virus-related mechanisms.74 Similarly, IP-10 was elevated in SARS-CoV74 and MERS-CoV96,97 but not in other lower respiratory tractVaccine. Accordingly, IP-10 dysregulation, which interferes with chemotaxis of immune cells, is another signature of coronavirus infections.74
In addition, basophils are depleted in patients with Covid-19, which may reflect increased recruitment into damaged lungs. Since basophils play a role in tissue repair, and in producing anti-coagulants, their depletion may contribute to sustained pneumonitis and to thromboses which are often associated with Covid-19. Indeed, basophil depletion frequently preceded clinical signs of disease severity.74 Of note, most patients with Covid-19 display low numbers of eosinophils in the early stages of infection. This is thought to be a stress response to acute lung injury which inhibits eosinophil release from the bone marrow. However, eosinophil numbers improve in the later stages of infection, which coincides with improved viral load and radiographic images. Moreover, this improvement is earlier in patients with mild disease.98
T-Cell Responses to SARS-CoV-2
T-cells play an important role in viralVaccine. CD8+ T-cells directly kill virus-infected cells and produce cytokines to recruit immune cells to the site of infection. CD4+ T-cells provide help to B-cells for antibody production and prime the response of CD8+ T-cells and other immune cells.35 Currently, there is limited knowledge about the T-cell response to SARS-CoV-2 infection. In general, the immune response can be protective, pathogenic or both depending on the timing, components, and/or magnitude of the elicited immune response.99 Since most acute viralVaccine result in protective immunity,100 it is likely that substantial T-cell and B-cell responses develop to SARS-CoV-2 and result in clearance of the viral infection.101 It is expected that some T-cells and B-cells will be retained long term as immunological memory and provide protective immunity against future infection with SARS-CoV-2.101 However, adaptive immune responses to human coronaviruses may fail to occur,102–104 and thus protective immunity may not develop.105 Failure to develop long-term immunity might be due to an insufficient CD4+ T-cell response, and consequently an inadequate neutralizing antibody response.106,107 Accordingly, it is important to understand all aspects of T-cell and B-cell responses to SARS-CoV-2, including their link with mild and/or moderate versus severe disease. This knowledge would aid in the development of vaccines or other therapeutic modalities.
Epitope Specificity of T-Cells
In someVaccine, T-cell responses can be strongly biased towards specific viral proteins with substantial differences between CD4+ and CD8+ T-cells.108,109 Identification of such proteins and epitopes is important for monitoring immune responses to viral infections, and for the design of vaccine candidates.99 Currently, most Covid-19 vaccine candidates are focused on the spike protein.99 A recent study investigated the epitope specificity of SARS-CoV-2-specific CD4+ and CD8+ T-cells. This study used a prediction strategy to predict dominant epitopes independently of ethnicity and human leukocyte antigen (HLA) polymorphism, which makes findings feasible to be used worldwide. There was no significant difference in the CD4+ and CD8+ T-cell frequencies between patients with Covid-19 and those who were not exposed. Importantly, spike-specific CD4+OX40+CD137+T-cell responses were detected in 100% of patients, and with a similar proportion to the remainder of the virus orfeome.99 This suggests that the costimulatory molecules OX40 and CD137 are critical in mounting an efficient CD4+ T-cell response to SARS-CoV-2. It also raises the question as to whether patients with severe disease fail to upregulate these costimulatory molecules, and thus, fail to mount an efficient CD4+T-cell response and consequently neutralizing antibody response. Furthermore, T-cells were polarized towards a Th1 type immune response as evidenced by their production of high levels of IFN-γ. With regards to antigen specificity, the CD4+ T-cell response was not only restricted to the spike protein, which was ~50%, but also to other antigens. In addition, CD8+CD69+CD137+ T-cell responses were detected in 70% of patients with Covid-19, and the majority of these cells expressed IFN-γ, granzyme B and TNF. Although the spike protein was a target of SARS-CoV-2-specific CD8+ T-cells, it was not dominant as other antigens were just as strongly recognized.99
T-Cell Populations and Disease Severity
Patients with Covid-19 have marked reductions in the number and frequency of both CD4+ and CD8+ T-cells, and increased activation of T-cells.68 An autopsy report of a patient who died of severe Covid-19 showed an accumulation of mononuclear cells in the lungs and low levels of hyperactive T-cells in peripheral blood.33 These findings suggest that T-cells are recruited from the circulation into the infected lung tissues to control viral infection.35 It has been reported that increased T-cell exhaustion and decreased functional diversity are associated with severe disease in patients with Covid-19.110 Moreover, the immune response shows elements of protective anti-viral immunity along with subtle elements of sepsis. These include T-cell cytopenia, high cycling, activation and depletion of effector-memory CD4+ and CD8+ T-cells, overexpression of IL-8,74 and reduction of γδ-T-cells,111,112 mainly the Vγ9Vδ2+ T-cell subset.74 γδ-T-cells have been shown to have protective antiviral function in influenza pneumonia113,114 and thus are likely to play a protective role in Covid-19. Moreover, selective T-cell proliferation, exhaustion, and depletion are correlated with Covid-19 severity. Activated T-cells seem to have a protective potential as evidenced by the finding that cycling CD4+ T-cells are correlated with immunoglobin (Ig)-G antibodies against SARS-CoV-2, as well as cytolytic CD8+ T effector memory cells, in patients compared to healthy controls.74 Therefore, such immunological correlates along with other tests (such as CRP and D-dimer) might be useful to identify patients at risk of severe disease.74
The notion that activated T-cells are key determinants of protection may explain the increased susceptibility of older individuals to severe Covid-19. Aging is associated with thymic involution which depletes the potential to generate new T-cell repertoires. In contrast, T-cell repertoires are abundant in children which may explain their resistance to severe disease.74 In addition, the ageing lung microenvironment may hinder DC maturation and migration to the lymphoid organs,115 and consequently impair T-cell activation.35 Moreover, adults rely heavily on memory T-cells, which is undermined by the dysregulation of T effector memory cells in Covid-19. The activated/proliferative phenotype of high proportions of memory T-cells in most patients with Covid-19 may override SARS-CoV-2-specific T-cells. The underlying cause of such dysregulation of subset-specific T-cells needs to be investigated.75 Although there is no evidence that effector memory T-cells from patients with Covid-19 express a different homing profile to those from healthy controls, cells from patients show gene expressions related to apoptosis. Activation-induced cell death of memory T-cells is difficult to restore in the presence of profound depletions of CD4+ and CD8+ naïve T-cells. Moreover, the expression of Fas ligand, granzymes, and perforin by CD8+ effector memory T-cells reflects killing capabilities. Additionally, different metabolic profiles of distinct T-cell subsets might be differentially susceptible to inflammatory mediators, hypoxia, or dysoxia.75 On a different note, a recent study showed that the ratio of IL-2R to T-cells is substantially increased in patients with severe/critical Covid-19. Moreover, the IL-2R/T-cells and cytokine profiles were decreased in recovered patients but increased in those with severe disease.116
In SARS-CoV infection, patients who had severe disease but later recovered developed coronavirus-specific memory T-cells which lasted for up to 2 years.117,118 CD4+ T-cells from patients with SARS-CoV exhibit a Th1 cell response and produce IL-2, IFN-γ, and TNF, which indicates that cellular immunity is critical for the control of the infection.97,119 Although these proinflammatory cytokines contribute to immunopathogenesis, this response is critical for the clearance of the virus as depletion of these cells in mice results in severe lung inflammation.35,120 Moreover, immunization with DCs bearing SARS-CoV peptides increases the number of CD4+ and CD8+ T-cell accumulation in the lungs and enhances survival.121,122 Furthermore, transfer of these cells into immunodeficient mice leads to better protection against SARS-CoV infection,122 which further highlights the importance of T-cells in controlling infection.
CD8+ T-cells show limited expression of inhibitory receptors in patients with Covid-19 when compared to healthy controls.123 It has been reported that CD8+ T-cells from patients with severe disease produce less cytokines upon stimulation.77 However, other studies showed a hyperactive CD8+ T-cell response with increased expression of markers associated with NK cells and increased toxicity.77,124,125 In addition, increased numbers of CD38+HLA-DR+ activated CD8+ T-cells or proliferating CD8+ T-cells were found in most patients with Covid-19.73,124,126,127 Of note, this was not the case in some patients, which indicates the presence of diverse patterns of CD8+T-cell responses in Covid-19.73 Therefore, there seems to be a heterogeneity in the immune response to SARS-CoV-2 and there might be distinct immunotypes linked to different disease features.73,128 Unsurprisingly, analysis of the bronchoalveolar lavage fluid confirmed the importance of respiratory CD8+ T-cell responses,93,129 which involves interactions (particularly IFN-γ axis) between CD8+Tcells and epithelial cells in the upper respiratory tract.130 Indeed, a more robust clonal expansion of CD8+T-cells in bronchoalveolar lavage93 or peripheral blood131 might be associated with mild Covid-19. Moreover, SARS-CoV-2-specific CD8+ T-cells have been detected in patients who have recovered, which confirms the induction of virus-specific CD8+T-cell responses and the existence of CD8+T-cell memory.99,132–134 The exact role of SARS-CoV-2-specific CD8+T-cells in the control of the early acute infection and the degree of protection from future infections remains to be elucidated.135
CD4+T-cells show functional impairment and elevated expression of activation and/or exhaustion markers in patients with Covid-19.136,137 A higher proportion of IFN-γ-producing Th1-like cells have been reported in patients with moderate disease compared to those with severe disease.16 CD4+T-cells specific to the spike protein of SARS-CoV-2 have been described during acute infection and shown to have a Th1 cell cytokine profile.138 Moreover, patients with mild Covid-19 show a normal Th2 cell response; however, the role of these cells in severe disease is unclear.126 Other reports have described a profound response by CD4+ T-cells that co-express C-C chemokine receptor 6 in severe Covid-19,1,33 which supports a role for pathologic Th17 cell responses. Interestingly, there are reports of increased CD4+T-cell responses producing transforming growth factor-β,139 and a subset of CD4+T-cells producing GM-CSF or IL-6 in patients with Covid-19.140 The lymphopenia described in most patients with Covid-19 impacts CD4+ T-cells,73,141 however, it is still unclear whether this is related to CD4+ T-cell activation, dysfunction, or both. Importantly, after recovery from Covid-19, patients have virus-specific memory CD4+ T-cells,99,132–134 which indicates the development of protective immunity. Moreover, patients who recovered from mild Covid-19 developed memory CD4+ T-cells that expressed high levels of IL-7Rα.134 Lastly, cross-reactive memory CD4+ T-cells have been detected in individuals who have not been exposed to SARS-CoV-2;99 however, it is not known how these preexisting cells affect disease outcome.
In addition, ICOS+CD38+ circulating T follicular helper (TFH) cells may be altered in patients with Covid-19.73 Increased numbers of TFH cells might be related to an increase in the number of circulating plasmablasts.124 A recent study has demonstrated that the majority of patients convalescing from Covid-19 maintained SARS-CoV-2 spike S1- and S2-specific antibodies, which showed neutralizing activity against the SARS-CoV-2 pseudotyped virus. Some of these antibodies also cross-neutralized the other related coronaviruses including SARS-CoV and MERS-CoV or both pseudotyped viruses. Interestingly, individuals convalescing from severe Covid-19 showed higher titres of neutralizing antibody, a rapid increase in lymphocyte numbers, and a higher frequency of CXCR3+ TFH cells compared to individuals convalescing from mild disease. Moreover, TFH cells were specific to the spike protein and their frequency was directly correlated with neutralizing antibody titres in individuals convalescing from Covid-19.142 In this study, no autoantibodies (auto-Abs) were detected. These new insights are useful in the development of new treatments and vaccines for SARS-CoV-2 infection.142
Vigorous immune responses to pathogens such as SARS-CoV-2 can be detrimental to the host due to tissue damage. Therefore, regulatory mechanisms, particularly those related to regulatory T-(reg) cells, are in place to ensure that inflammation is under control. Treg cells are either thymus-derived or induced in the periphery to induce immune tolerance.143 The level of peripheral Treg cells is decreased in patients with severe Covid-19 compared to patients with mild disease.1,16,139,144 Although the reasons for this reduction in Treg cells remain to be elucidated, it is possible that the cells might have migrated to the site of inflammation in the lung to prevent tissue damage. Moreover, Treg cells have been shown to help resolve ARDS inflammation in animal models,145 which suggests that a loss of Treg cells might promote the development of lung immunopathology in Covid-19. Transcriptomic analysis of T-cells from the bronchoalveolar lavage showed reduced IL-2 transcripts in severe cases compared to mild cases.146 In this regard, reduced IL-2 may lead to Treg cell apoptosis as evidenced by reduced levels of forkhead box P3. It has also been shown that patients with severe Covid-19 have elevated levels of soluble IL-2R (CD25),1,16,144 which is thought to be due to enhanced proteolytic cleavage of cell surface CD25 as a result of inflammation. Elevated levels of soluble CD25 might interfere with IL-2 availability and thus promote apoptosis of Treg cells.147
B-Cell Responses to SARS-CoV-2
Epitope Specificity of B-Cells
Neutralization of the virus by antigen-specific antibodies is an important step in the eradication of the viral infection; however, the specificity/target of the neutralizing antibodies remains to be elucidated.35 In SARS-CoV infection, the main target of neutralizing antibodies is the RBD,148 which comprises a region of 193 amino acids in the spike protein that binds to the ACE2 receptor.149–151 Importantly, previously identified monoclonal antibodies to SARS-CoV can also bind to SARS-CoV-2,152 however, the epitope specificity of these antibodies may not overlap with ACE2 binding site. In fact, some of the most potent neutralizing antibodies to SARS-CoV do not bind to SARS-CoV-2;153 which is likely due to differences in the RBDs between the two viruses. Out of 33 amino acids in the spike protein of SARS-CoV, a region that contains the critical residues that contact ACE2,154 only 15 were conserved in SARS-CoV-2. Nonetheless, antibodies raised against SARS-CoV protein can cross-react and neutralize SARS-CoV-2, which suggests overlapping epitopes between the two viruses.155,156
B-Cell Populations and Disease Severity
B-cell activation and rapid production of antigen-specific antibodies by ASCs are critical for the control of viral infections. Various cohorts reported reduced CD5+ B-cell frequencies, elevated plasmablasts, and the production of SARS-CoV-2-specific antibodies.73–75,77,157,158 More recently, studies have focused on characterising B-cell responses in terms of being protective or non-protective. Despite reports of lymphopenia,6 patients with Covid-19 display higher numbers of peripheral blood mononuclear cells with elevated CD19+ B-cells compared to healthy controls.159 B-cell profiling revealed 5 primary populations including transitional, naïve, double-negative, memory, and ASCs, which can further be divided into 14 secondary populations. Importantly, distinct B-cell profiles were identified among patients with mild disease, patients in intensive care unit (ICU), and healthy persons. B-cells from ICU patients displayed expansions of ASC and double-negative 2 cells that lack the follicular homing marker C-X-C chemokine receptor type 5 and are thus extrafollicular, while those from persons with mild disease displayed transitional cells.159 Furthermore, activated-naïve and activated effector double-negative 2 B-cells, which also express high levels of CD11c and the IFN-γ-inducible T-bet, were highly enriched in ICU patients.159 These cell types are known to participate in extrafollicular responses,160 and interestingly, similar B-cells have been identified in autoimmunity and in viral clearance in animal models.161–163 Therefore, the induction of activated- naïve and effector double-negative 2 B-cells in the early stages of human acute viral infections159 may provide some insight into the link between viral infections and autoimmune disease such as new-onset type 1 diabetes. Accordingly, patients with a history of SARS-CoV-2 infection, in particular severe/critical disease, should be monitored for autoimmunity.
Moreover, patients in ICU displayed higher levels of ASCs compared to persons with mild disease, which suggests an immunopathologic role for circulating ASCs in severe Covid-19. A high number of ASCs expressed CD138 (a plasma cell maturation marker), which indicates a highly inflammatory milieu of ASCs in ICU patients.159 Similarly, 21lo transitional B-cells were elevated in ICU patients compared to those with mild disease - where the cells did not reach more than 25% of the B-cell compartment. Of note, although 21lo transitional cells express the same levels of developmental markers in both healthy controls and ICU patients, cells from the latter also expressed CD138. Correlation of this B-cell subset with milder disease suggests a protective role for these cells, probably through regulatory functions.159,164 In addition, higher expression of CD138 may indicate that these cells have converted into ASCs.159,165 Future studies should elucidate the mechanisms by which 21lo transitional cells act protective.159
Anti-SARS-CoV-2 Neutralizing Antibody Titre and Kinetics
Antigen-specific antibody response plays a protective role by limiting infection and preventing re-infection. Until recently, little was known about the kinetics/titer of the antibody response to SARS-CoV-2, and whether the antibody titer correlates with disease severity. An initial study showed that approximately 22.0% of patients who tested positive by quantitative polymerase chain reaction were negative for IgM antibody; however, most of the patients were in the early phase of infection and thus may not have produced antibodies in sufficient amounts. In fact, 2 patients with severe illness failed to generate an antibody response even at days 19 to 22 post symptom onset, which might have contributed to disease severity. Other patients who were sampled between 9 and 17 days still showed negative IgM results. This study highlights the importance of the test timing, and the contribution of host factors in the antibody response.166 These initial studies were followed by a more comprehensive study which investigated the titer/kinetics of total antibody, IgG and IgM responses. This study showed strong evidence that the antibody response to SARS-CoV-2 is similar to that of other viruses. The seroconversion rate and antibody levels increased rapidly during the first 2 weeks post symptom onset with the cumulative seropositive rate reaching 50% by day 11 and 100% by day 39. Moreover, the seroconversion time of total antibody, IgM, and IgG were observed at day 11, 12, and 14, respectively.167
More recent titer/kinetic studies show that after 8 days of symptom onset, patients develop antibodies with a neutralizing titer of 50% inhibitory dose, and a range of 98–32,000,168 which is similar to other cohorts.169,170 Importantly, the antibody titer was higher in patients with severe disease,168 and strongly correlated with disease severity.167 However, the average time to reach peak neutralization was the same regardless of disease severity. Therefore, whereas disease severity increases the magnitude of the neutralizing antibody response, it does not appear to have an impact on the kinetics.168 Accordingly, a high titer of antibodies against SARS-CoV-2 may be used as an indicator of critical illness, independently of old age, male gender, and preexisting conditions. The relation between high antibody titer and disease severity is still under investigation. It has been speculated that a higher viral load and increased viral antigens may lead to severe disease and thus induce a stronger antibody response.168 However, although a slower decline of viral load has been observed in patients admitted to ICU, the viral load does not differ between patients with moderate and severe disease.68 An alternative explanation is that antibodies may contribute towards disease severity by ADE,167 which was the case in SARS-CoV;171,172 however, there is currently no evidence that this is taking place in Covid-19.173 Nevertheless, a high antibody titer may be used to identify patients most at risk of worse clinical outcomes. On the other hand, a recent study found that 3 patients who died failed to produce SARS-CoV-2 antibodies,74 which confirms data from earlier studies.166 Therefore, any type of antibody response that is either too low/non-existent or too high appears to be associated with severe disease. Interestingly, asymptomatic patients were also able to generate neutralizing antibody titres >1000;168 in fact, potent neutralizing monoclonal antibodies have been isolated from these patients.174
Limited information is available with regards to the long-term persistence of anti-SARS-CoV-2 antibodies. It has been reported that previous exposure to SARS-CoV-2 might not confer total immunity against reinfection in all cases, which could be relevant for vaccine development.175 Additionally, both symptomatic and asymptomatic SARS-CoV-2 reinfection can occur in the presence of SARS-CoV-2 antibodies.176 In spite of these reports, antibodies are likely to be effective against SARS-CoV-2 as evidenced by the fact that convalescent serum therapy has provided good clinical efficacy in patients with Covid-19,177 as well as SARS-CoV.178–180 However, longitudinal studies are required to elucidate the long-term persistence of the antibodies.
Effector Functions of Antibodies and Risk of ADE
Antiviral neutralizing antibodies play an important role in viral clearance. However, studies using animals infected with SARS-CoV have shown that anti-spike protein neutralizing antibodies (anti-spike IgG) can alter inflammatory responses and cause severe acute lung injury. Moreover, animals vaccinated with SARS-CoV protein showed more pronounced acute lung injury compared to those that were not vaccinated. Although the adoptive transfer of anti-spike IgG neutralizing antibodies to the animals reduced viral load following subsequent challenge, it resulted in acute diffuse alveolar damage, whereas control groups (injected with non-specific IgG) had minor to moderate lung inflammation.172 Therefore, despite viral suppression, anti-spike IgG at the acute stage of SARS-CoV infection can cause persistent severe lung injury.64 This is because the presence of anti-spike IgG prior to viral clearance may alter the polarization of alveolar macrophages, and lead to the accumulation of proinflammatory monocytes/macrophages and the production of MCP-1 and IL-8. This inflammatory response is mediated by the binding of the anti-spike IgG-virus complex to the Fc receptors (FcR) on the surface of monocytes/macrophages. This mechanism is confirmed by the finding that FcR blockade reduces the production of inflammatory cytokines.172 In addition, anti-spike IgG-virus complexes may activate the classical pathway of the complement system leading to cellular damage. Another possible mechanism may be antibody-dependent cell-mediated cytotoxicity.64 In humans with SARS-CoV-2 infection, 2 weeks post symptom onset, a strong positive correlation is observed between antibody titer and clinical severity.167 Therefore, and as was the case with SARS-CoV, a possible underlying mechanism could be related to ADE of viral infection in patients who produce early, sub-optimal antibodies that fail to clear the virus but instead lead to persistent viral replication and inflammation.64 In other viral infections, ADE promotes cellular uptake of virus-antibody complexes after their interaction with FcR, which leads to enhanced infection of target cells.181–185 Therefore, interaction of anti-spike IgG-virus complexes with FcR may facilitate cell infection and viral replication along with lung inflammation of patients who succumb to severe Covid-19.64 In SARS-CoV, anti-spike neutralizing antibodies are responsible for the infection of immune cells;186,187 moreover, ADE exacerbates lung inflammation, thereby increasing the risk of ARDS and other clinical sequelae.188,189 Currently, studies need to elucidate whether ADE is a mechanism of pathogenesis of SARS-CoV-2. Moreover, this phenomenon needs to be taken into account when developing new treatments.190
Autoantibodies Against Type I Interferons
A recent study reported that at least 10% of patients with severe Covid-19 pneumonia have neutralizing auto-Abs against type I IFNs.191 Additionally, patients with inborn errors of type I IFNs develop severe Covid-19.192 Taken together, these findings highlight the crucial role that type I IFNs play in protective immunity against SARS-CoV-2. Auto-Abs against type I IFNs were clinically silent until patients were infected with SARS-CoV-2, which is a poor inducer of type I IFNs.76 Therefore, the little IFNs induced by the virus are sufficient for protection against severe disease. As was the case with inborn errors of type I IFN production, neutralizing auto-Abs against type I IFNs tip the balance in favor of the virus, which results in insufficient innate and adaptive immune responses and severe disease.191 Importantly, patients infected with SARS-CoV-2 can be screened for auto-Abs to identify those at increased risk of developing severe pneumonia. Once identified, these patients can be treated early with nebulized or injected IFN-β, which may prove to be beneficial given that auto-Abs against IFN-β is rare in patients with auto-Abs against type I IFNs.191 Other preventive or treatment modalities include the specific inhibition of type I IFN-reactive B-cells, plasmapheresis, and monoclonal antibodies aimed at depleting plasmablasts.193
Prior Infections/Vaccines and Cross-Reactive Immunity
It has been observed that severe Covid-19, as a result of a dysfunctional immune response and failure to eradicate the virus, occurs mostly in older people and in individuals with comorbidities such as diabetes, cardiovascular disease, and hypertension. Notably, males tend to suffer more severe disease than females.4,5 Ethnicity has also been associated with disparity of outcomes.218 Interestingly, a small increase within the normal range of fasting blood glucose was also associated with increased risk of ICU admission.219 Young healthy adults with no preexisting conditions have also been reported to develop severe disease. Intriguingly, children are less likely to develop severe disease despite having high viral titres.194 A study of 44,672 people with Covid-19 found that children (under the age of 10 years old) made up less than 1% of those cases and none of the 1023 deaths.195 Moreover, more than 50% of children under the age of 18 years are asymptomatic or experience mild symptoms, and less than 6% of children develop severe disease.196,197 However, the underlying mechanisms leading to a dysfunctional immune response to SARS-CoV-2 remains poorly understood. An important aspect to consider is whether previous exposure to infections and/or vaccines has an impact on susceptibility to severe Covid-19.
Trained Innate Immunity
Innate immunity is mediated by different cell types where pattern recognition molecules play a key role in resistance against pathogens and tissue repair.198 Exposure to certain vaccines such as the bacilli Calmette–Guérin (BCG) vaccine or microbial components can enhance the baseline tone of innate immunity and induce antimicrobial resistance, known as trained innate immunity.199 This training is relevant to resistance against infectious diseases such as Covid-19. Studies have described the existence of innate immune memory conferred by myeloid cells (monocytes, macrophages, and neutrophils), which are the central players of innate immunity.200 A fundamental property of myeloid cells (particularly macrophages) is diversity and plasticity, which are to some extent imprinted through ontogenetic origin but can also be influenced by environmental cues. In response to certain triggers such as microbial components and/or cytokines, macrophages increase their effector function, get primed for short-term responses, or become unresponsive. Importantly, microbial components can confer long-term imprinting or training of innate immunity and myeloid cell function.201 Therefore, the cellular basis of trained innate immunity and protection against secondary infections resides in the reprogramming of innate immune cells.199
Certain live attenuated vaccines such as the BCG can boost antimicrobial function in myeloid cells, which is associated with changes in chromatic conformation, and alteration in accessibility to the regulatory elements in DNA that govern gene transcription in these cells.199 Specifically, stimulation of innate immune cells can leave an “epigenetic scar” with heightened ability to influence gene expression. In this regard, the long-term induction of innate immune memory can be generated by transcriptomic, epigenetic, and functional imprinting of myeloid progenitors in the bone marrow. Therefore, vaccines with live attenuated microorganisms may cause a long-term effect on cells conferring innate immunity. Indeed, the link between epigenetic memory and innate immunity at the bone marrow progenitors’ level is consistent with a myeloid bias in bone marrow hematopoietic stem cells of healthy individuals 3 months after BCG vaccination.202 Since children undergo a vaccination program that involves adjuvants, this may explain their relative resistance to Covid-19.203 Likewise, elderly persons who received the influenza vaccine with an adjuvant may have a better outcome to Covid-19 compared to those who received the vaccine without an adjuvant.199 Interestingly, it has been suggested that the BCG vaccine has a pleiotropic effect that reduces the incidence of other infections.204 Accordingly, trials to ascertain the efficacy of BCG in preventing Covid-19 are underway. If proven to be effective, it will reinforce a role of trained immunity in illness prevention.199
Serological Immune Imprinting
Humans are exposed to different strains of viruses from an early age leading to the production of neutralizing antibodies to the encountered strains.205,206 This preexisting immunity from earlier infections/vaccinations exerts a major influence on the nature of the antibody response elicited by subsequent challenge,206,207 a phenomenon known as “immune imprinting”.209 Preexisting serological immunity may impede the elicitation of antibodies against new strains.210 Persistent antibody clonotypes dominate the response to viral infections such as influenza virus infection and repeated vaccinations over multiple years.211 Accordingly, multiple exposure to coronavirus infections and/or influenza vaccinations during a lifetime, as would be expected in older adults, may result in a weak or mismatched antibody response to SARS-CoV-2, and thus, lead to severe disease. This may partly explain why children are protected against severe disease; children may be benefiting from lack of multiple exposure to coronaviruses whereas adults, having lived longer, are more likely to have encountered other coronaviruses such as those that cause coughs and colds, and therefore, already have antibodies against these milder viruses.
It has been suggested that patients with severe Covid-19 may have been primed by prior coronavirus exposures, and due to antigenic heterogeneity, are suffering from ADE.190 This was certainly the case with SARS-CoV where ADE was identified,212 and the priming strains were thought to be coronaviruses that cause mild infections.213 Moreover, ADE may explain the geographic discrepancy in severity of Covid-19.190 The priming coronavirus, at least in Hubei Province, could have been SARS-CoV;214 however, it does not explain the severe cases in other parts of the world. It is more likely that SARS-CoV is not the predominant coronavirus. There may have been mild strains of similar coronaviruses in the circulation that were either asymptomatic or mistaken for a regular common cold virus.190,215 In addition, a SARS-CoV-specific antibody, namely CR3022, that was isolated from a convalescent patient was able to bind potently to the RBD of SARS-CoV-2.153 However, CR3022 recognizes an epitope that does not overlap with ACE2 binding site of SARS-CoV-2.153 This may help explain why some people who have previously been infected with SARS-CoV or other coronaviruses end up with worse outcomes. The binding of CR3022 antibody with SARS-CoV-2 may lead to virus-antibody complexes, which instead of neutralizing the virus, it enhances its pathogenicity by allowing the virus to bind to ACE2 and/or leading to ADE.190
Preexisting CD4+T-Cell Immunity
Previous infections with coronaviruses may offer some degree of cross-reactive immunity. A recent study showed that some unexposed individuals appear to have immunity to SARS-CoV-2, which indicates that cross-reactive immunity to coronaviruses may exist in a fraction of the population, and may influence susceptibility to Covid-19 and its severity.216 Indeed, CD4+ T-cell responses have been detected in 40–60% of unexposed individuals, which suggests the presence of cross-reactive preexisting immunity to SARS-CoV-2. It has been speculated that cross-reactive CD4+ T-cells may be protective and prevent severe disease.99 These findings are important because the presence of cross-reactive immunity may influence responsiveness to candidate vaccines.217 Further studies are required to determine how this affects clinical outcome. It is reasonable to speculate that previous infections with milder forms of coronaviruses and the resultant cross-reactive CD4+T-cells in the circulation may be protective and prevent severe disease. However, as aforementioned, one must keep in mind that preexisting antibodies may in fact hinder protective immunity by means of ADE leading to severe disease.
Immunotherapeutic Approaches for Covid-19
Vaccine Development
A good vaccine candidate against viral infections would be expected to induce a type of protective immune response similar to that elicited by natural infection. Therefore, a better understanding of both protective and pathogenic immune responses elicited by SARS-CoV-2 is essential. This knowledge can aid in the design of vaccines that are most likely to elicit protective immunity. Current understanding of immune responses during Covid-19, although very limited, can help in the selection of strategies most likely to elicit protective immunity against SARS-CoV-2. If natural infection elicits potent T-cell responses, which are commonly associated with protective antiviral immunity, then this would aid in rapid vaccine development.99 Importantly, when previous studies tested vaccine formulations against SARS-CoV in animal models, they showed increased immunopathology associated with Th2 cell-mediated eosinophil infiltration.220–222 These studies highlight the importance of elucidating the nature of protective versus pathogenic T-cell responses which is important for the design of vaccines that are both effective and safe.223 T-cell responses are critical in eliminating the virus and should be considered in vaccine strategies. However, whether T-cell responses alone are capable of preventing infection in human settings remains to be investigated.35 Although studies have reported concerns regarding the possibility of vaccine enhancement of disease via ADE,190 there is currently no evidence of this. In addition, this may not be a concern since there appears to be a predominant Th1 response to SARS-CoV-2 in convalescing Covid-19 patients.99 Currently, there are three candidate vaccines with excellent efficacy and little side effects, these include: BioNTech/Fosun Pharma/Pfizer (clinical phase 3), Moderna/NIAID (clinical phase 3), and University of Oxford/AstraZeneca (clinical phase 3).
Therapeutic Antibodies
In some parts of the world, convalescent plasma has been used as a source of therapeutic polyclonal antibodies for the treatment of Covid-19, and this was shown to reduce mortality.224 Currently, a clinical trial is assessing the efficacy of using convalescent plasma by the infusion of polyclonal antibodies derived from convalescent patients' plasma.225 However, one trial of convalescent plasma was stopped early because most patients had already developed high titers of neutralizing antibodies at the time of transfusion.225 Importantly, given the link between auto-Abs against type I IFN and severe Covid-19, patients recovering from severe disease should be excluded from donating convalescent plasma for ongoing clinical trials. Alternatively, such patients should be tested before their plasma donations are accepted.226
An alternative approach would be the use of therapeutic monoclonal antibodies to SARS-CoV-2. There are various approaches that can be employed to develop therapeutic neutralizing monoclonal antibodies; these include cloning of B-cell sequences from convalescent patients, phage library display, and mouse immunization.227 However, if the spike protein undergoes any alterations, monoclonal antibodies may not be effective, which is possible as the virus spreads and mutates. So far, the RBD has remained relatively conserved with only four alterations identified in the spike protein, these include V483A, L455I, F456V, and G476S.35 These alterations are similar to the ones found in MERS-CoV228 and SARS-CoV.229 The alteration in MERS-CoV (1529T) reduces binding of the viral protein to the receptor target and enhances resistance to antibody neutralization.228 Likewise, the alteration in SARS-CoV (L443R and D463G) allows the virus to escape from antibody neutralization.229
In SARS-CoV infection, patients were generally able to produce neutralizing antibodies. Moreover, the spike protein fragment that consisted of the RBD of SARS-CoV was more immunogenic than the other fragments of the spike protein; this suggests that the immune system was capable of targeting neutralizing epitopes effectively.35,229 Importantly, SARS-CoV was not capable of preventing antibody neutralization by means of glycan shielding of the RBD.231 In spite of this, therapeutic antibody candidates should be selected with caution. In SARS-CoV infection, preexisting antibodies to other coronaviruses may exacerbate the disease through ADE.190,212,213 Similarly, studies in animal models demonstrated that neutralizing antibodies to the spike protein may exacerbate inflammatory responses leading to severe lung injury.172 In addition, in SARS-CoV infected individuals, antiviral IgG seroconversion coincides with acute respiratory disease in 80% of patients.171 Moreover, patients who developed anti-spike neutralizing antibodies and reached peak levels more rapidly (within 2 weeks) had higher mortality rates compared to those who took longer to reach peak levels (20 days).232 Similarly, patients with severe MERS-CoV have higher antibody titers than those with mild disease;233,234 however, it is thought that this may have been due to a delay in the development of antibody responses.235 Crucially, virus-antibody immune complexes may bind to and activate Fc receptors on alveolar macrophages, which leads to the expression of proinflammatory MCP-1 and IL-8.236 Also, these complexes may activate the complement system, further adding to the inflammatory milieu.172 Therefore, therapeutic antibody candidates should be capable of neutralizing the virus without having any inflammatory activity as a side effect.237 One example by which this can be achieved is by altering the Fc region to change its binding affinity for activating Fc receptors.236,238
Cell-Based Therapies
Immunotherapy is an important method of treatment to combat viral infections. Most attempts at immunotherapy have been effective in combating related coronaviruses such as SARS-CoV and MERS-CoV. Acquired cellular immune surveillance is a precisely controlled series of processes and activities affecting particular T-cell populations and subpopulations. The main obligation for the cytotoxic removal of virally compromised and tumor cells rests with CD3+CD8+TCR+ T-cells.239 One part of the regulatory arm of cellular immunity results in “exhausted T-cells”, which mainly belong to the CD8+ population and occur in persistent high-grade viral infections, such as Covid-19.240 In patients with Covid-19, their data point to major T cytopenia in CD4+ and CD8+ T-cells in the circulation.241 Such patients’ serum had considerably elevated amounts of IL-6, IL-10, and TNF. Further analyses revealed a steady rise in the subpopulation PD-1+CD8+ and TIM-3+CD8+ as patients progressed from prodromal to symptomatic Covid-19 needing intensive treatment.136,242 Research demonstrates a combination of both immunotherapy and lifestyle-dependent treatments can reduce prevalence and improve obesity-related malignancy outcomes. New findings are also updated to exploit obesity for favorable benefits, although associated with several harmful cancer causes, provided the involved mechanisms are handled properly. In fact, a large body of literature relates to a “meta-inflammatory” state of obesity associated with a deficit in the immune response.243 It is important to understand how obesity shapes immune responses and how obesity can change both the responses to immunotherapy and the potential toxicity. Therefore, all this suggests a surprisingly positive association between obesity, Covid-19, and immunotherapy.243
Immunosuppressants and Other Therapeutic Modalities
Based on current understanding of the underlying mechanisms that may be involved in severe outcomes, several therapeutic approaches may be effective. One approach is to inhibit FcR activation which can be achieved by several methods. Firstly, the FcR can be blocked by specific antibodies to prevent binding of the virus-IgG antibody complex to the receptor;236,244 secondly, the FcR can be blocked by the use of small-molecule inhibitors to interact with the Ig-binding domains of the FcR; thirdly, targeting Fcc receptor IIb (FccRIIb), which is the inhibitory FcR, by the use of anti-FccRIIb antibodies;245,246 lastly, targeting the neonatal FcR which is critical for extending the half-life of IgG by using antibody or small molecule-mediated blockade of neonatal FcR. This prevents IgG interaction with neonatal FcR, and decreases circulating IgG levels.247 In addition, intravenous immunoglobulin may be used to saturate the IgG recycling capacity of neonatal FcR and thus reduce the levels of antiviral neutralizing antibodies.248
In addition, several clinical trials are underway aimed at elucidating the efficacy of cytokine antagonists, including the GM-CSF antagonists gimsilumab,249,250 lenzilumab,251 and namilumab.252 Moreover, tocilizumab, which is a humanized monoclonal antibody against IL-6R,253,254 was recently assessed and found to be ineffective for preventing intubation or death in hospitalized patients; however, some benefit or harm has not been ruled out.83 Another approach could be the use of agonists against TLR-7 which may prevent severe Covid-19.78 In addition, a novel adjunctive therapy, namely cytosorb, can absorb a broad spectrum of cytokines,255 DAMPs and PAMPs, which reduces their circulating levels. Also, the immunomodulatory agent thalidomide regulates immune function, and has a calming effect which reduces oxygen consumption and relieves digestive symptoms, in patients with Covid-19. Therefore, it is thought that thalidomide may be helpful as an adjuvant treatment for patients with critical Covid-19. Accordingly, a randomized controlled clinical trial to investigate the efficacy of thalidomide combined with glucocorticoid for the treatment of Covid-19 is warranted.256 Moreover, an open-label study showed preliminary data suggesting that a combination of hydroxychloroquine and azithromycin may help treat patients with severe Covid-19,257 however, this study was later withdrawn. Notably, there are some trials assessing the use of corticosteroids for the treatment of Covid-19.35,225 Given that the virus-neutralizing complex may initiate inflammatory responses through the classical pathway of the complement system, therapeutic approaches could also be aimed at targeting this pathway.258 Moreover, interventions such as type I IFNs, when administered at the correct timing, may be effective.72 Other recent findings support strategies to boost T-cell competence by the use of IL-7 and/or IP-10 antagonism, which is currently being trialed.74
Moreover, current clinical trials are assessing the efficacy of recombinant human (rh) ACE2 in regulating damage caused by its downregulation by the virus. Other trials are aimed at blocking SARS-CoV-2 infectivity by the use of Bromhexine Hydrochloride combined with umifenovir hydrochloride, rhIFN-α2b, and favipiravir in patients with suspected/mild pneumonia related to Covid-19. Furthermore, clinical trials are assessing medicinal or herbal compounds aimed at reducing inflammation. These include methylprednisolone, fingolimod (a sphingosine 1-phosphate receptor modulator), thalidomide/methylprednisolone, complement C5 inhibitor eculizumab, an extract of Trametes robiniophila Murr (Huaier), an extract from Salvia miltiorrhiza (Chinese red sage), and finally, the effect of honeysuckle oral liquid on boosting immunity to SARS-CoV-2.225
Conclusion
There is accumulating knowledge delineating the differences in the innate and adaptive immune response between patients with mild/moderate versus severe Covid-19. Severe disease has been attributed to a dysfunctional innate immune response coupled with an exaggerated and/or dysfunctional adaptive immunity. Most patients, including those with severe disease, appear to mount CD4+ and CD8+ T-cell responses as well as B-cell responses; however, there is mounting evidence that such responses might be dysfunctional in severe disease. Distinct populations of T-cell and B-cell responses may exist among patients, and thus, may necessitate different clinical approaches. Moreover, differences in the kinetic/titer of neutralizing antibodies have been described in severe disease, which might potentially be confounded by antibody-dependent enhancement. Importantly, the presence of preexisting autoantibodies against type I IFN has recently been identified as a major cause of severe/critical disease (an overall depiction of the innate and adaptive immune response in mild versus severe Covid-19 is shown in Figure 2). Overall, knowledge has been gained with regards to the immune response to SARS-CoV-2 in mild versus severe disease. Additionally, several therapeutic and preventative approaches have been under investigation, including vaccines (three of which have passed phase 3 clinical trials), therapeutic antibodies, and immunosuppressants. Future studies should continue to dissect the impact of SARS-CoV-2 infection on different aspects of the immune system, including the monitoring of recovered patients for early detection of potential complications such as autoimmunity. Additionally, studies need to elucidate the efficacy of the novel vaccines in terms of (1) protection against SARS-CoV-2 infection in the wider community, (2) induction of long-term immunological memory, (3) prevention of asymptomatic viral carriage/opportunistic infection, and potential spread, (4) long-term impact on health in general. In addition, studies should evaluate the efficacy and safety of other novel interventions that may prevent or reduce disease severity.
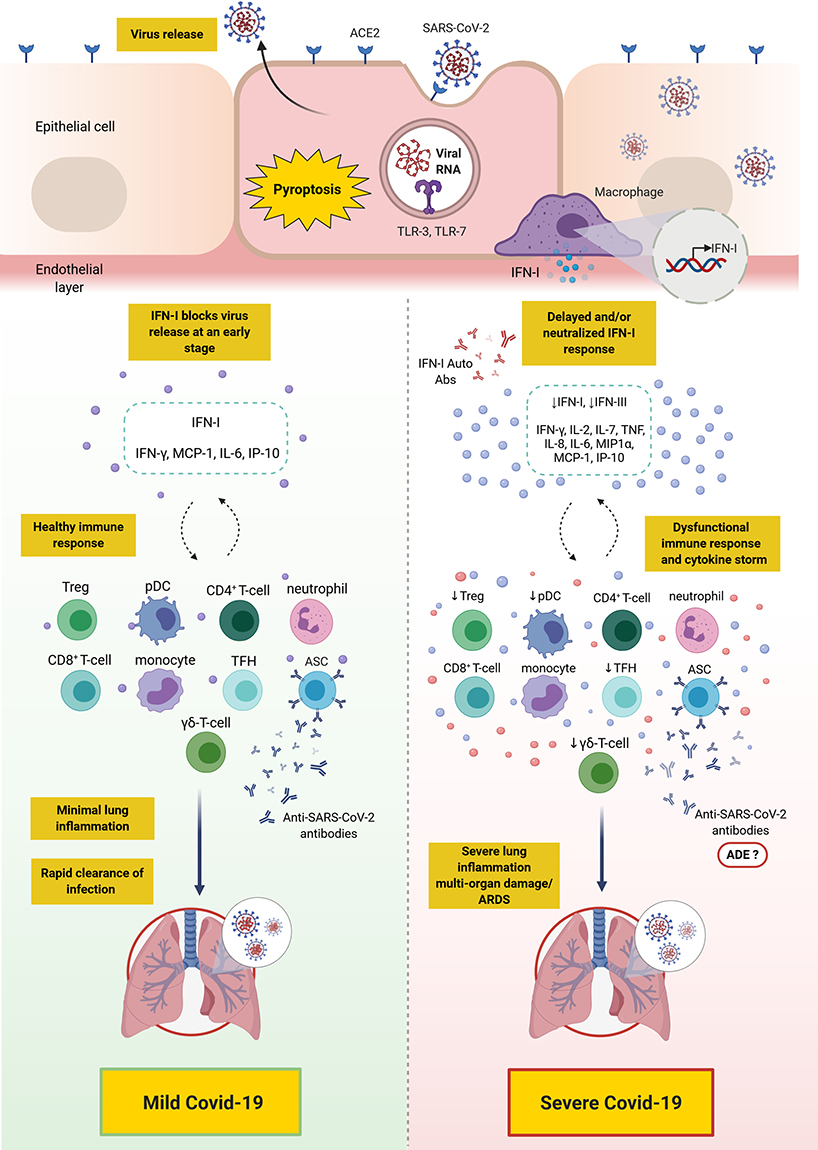 |
Figure 2 Overall depiction of the immune response in mild versus severe Covid-19. SARS-CoV-2 binds to ACE2 expressing cells. Upon entry, viral RNA is recognized by TLR-7 which results in the production of proinflammatory cytokines (such as IL-6 and TNF) and IFN-I. Alveolar macrophages trigger the release of proinflammatory cytokines and chemokines attracting T-cells, pDC and monocytes to the site of infection. In mild Covid-19, the release of IFN-I blocks viral replication and its release at an early stage, thus, controlling the infection. However, in severe Covid-19, due to a defective immune response, a delay in IFN-I production and/or its neutralization with anti-IFN-I autoantibodies may lead to continued viral replication and a profound inflammatory response, namely “cytokine storm”. This is also accompanied with the accumulation of neutrophils, monocytes and T-cells in the lungs leading to lung damage. Ineffective anti-SARS-CoV-2 antibodies produced by ASCs may play a role in the pathogenesis of SARS-CoV-2 and its clinical sequalae through ADE.Note: This figure was created with BioRender.com. Abbreviations: ACE2, angiotensin-converting enzyme 2; ADE, antibody-dependent enhancement; ARDS, acute respiratory distress syndrome; Auto-Abs, autoantibodies; ASC, antibody secreting cell; Covid-19, coronavirus disease 2019; IFN, interferon; IP-10, interferon γ-induced protein-10; IL, interleukin; MCP-1, monocyte chemoattractant protein-1; MIP1α, macrophage inflammatory protein 1α; pDC, plasmacytoid dendritic cell; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; TLR, toll-like receptor; TNF, tumor necrosis factor; Treg, regulatory T-cells; TFH, T follicular helper. |
Abbreviations
ACE2, angiotensin-converting enzyme 2; ADE, antibody-dependent enhancement; Auto-Abs, autoantibodies; ASC, antibody-secreting cell; BCG, bacilli Calmette–Guérin; CD, cluster of differentiation; Covid-19, coronavirus disease 2019; CRP, C-reactive protein; DAMPs, damage-associated molecular patterns; DNA, deoxyribonucleic acid; FcR, Fc receptor; FccRIIb, Fcc receptor IIb; GM-CSF, granulocyte macrophage-colony stimulating factor; HLA-DR, human leukocyte antigen DR isotype; TCR, T-cell receptor; Treg, regulatory T-cells; TRAF, TNF-receptor-associated factor; ICU, intensive care unit; IFN, interferon; IFNAR, interferon-α/β receptor; IFN-γ, interferon-γ; IL, interleukin; IgG, immunoglobulin G; IgM, immunoglobulin M; IP-10, interferon-γ-induced protein 10; IRF-3, interferon regulatory factor 3; MCP-1, monocyte chemoattractant protein-1; MERS-CoV, middle east respiratory syndrome coronavirus; MIP1α, macrophage inflammatory protein 1α; mRNA, messenger ribonucleic acid; NET, neutrophil extracellular trap; NF-κB, nuclear factor kappa B; NK, natural killer; PAMPs, pathogen-associated molecular patterns; PF4, plasma platelet factor 4; PD-1, programmed cell death protein 1; pDC, plasmacytoid dendritic cell; PRR, pattern-recognition receptor; RANTES, regulated on activation, normal T-cell expressed and secreted; rh, recombinant human; RBD, receptor-binding domain; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; STAT1, signal transducer and activator of transcription 1; TFH, T follicular helper; Th1, T-helper 1; TLR, Toll-like receptor; TNF, tumor necrosis factor; TIM-3, T-cell immunoglobulin and mucin domain-3.
Acknowledgments
This work was supported by a grant (CORONAPROP10) from the Kuwait Foundation for the Advancement of Sciences (KFAS).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Qin C, Zhou L, Hu Z, et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin Infect Dis. 2020.
2. Center for disease control and prevention link; 2020. Avaialable from: https://wwwcdcgov/coronavirus/2019-ncov/hcp/indexhtml
3. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species severe acute respiratory syndrome- related coronavirus: classifying 2019- nCoV and naming it SARS- CoV-2. Nat Microbiol. 2020.
4. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273.
5. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269.
6. Guan WJ, Ni ZY, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708–1720.
7. Pung R, Chiew CJ, Young BE, et al. Investigation of three clusters of COVID-19 in Singapore: implications for surveillance and response measures. Lancet. 2020;395(10229):1039–1046.
8. Lauer SA, Grantz KH, Bi Q, et al. The incubation period of coronavirus disease 2019 (covid-19) from publicly reported confirmed cases: estimation and application. Ann Intern Med. 2020;172(9):577–582.
9. Li Q, Guan X, Wu P, et al. Early transmission dynamics in Wuhan, China, of Novel Coronavirus-infected pneumonia. N Engl J Med. 2020;382(13):1199–1207.
10. Menni C, Valdes AM, Freidin MB, et al. Real-time tracking of self-reported symptoms to predict potential COVID-19. Nat Med. 2020;26(7):1037–1040.
11. Eliezer M, Hautefort C, Hamel AL, et al. Sudden and complete olfactory loss of function as a possible symptom of COVID-19. JAMA Otolaryngol Head Neck Surg. 2020;146(7):674–675.
12. Gautier JF, Ravussin Y, New A. Symptom of COVID-19: loss of Taste and Smell. Obesity. 2020;28(5):848.
13. Pellegrino R, Cooper KW, Di Pizio A, Joseph PV, Bhutani S. Corona viruses and the chemical senses: past, present, and future. Chem Senses. 2020.
14. Chan JF, Yuan S, Kok KH, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395(10223):514–523.
15. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506.
16. Chen G, Wu D, Guo W, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020;130(5):2620–2629.
17. Liu Y, Yang Y, Zhang C, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. 2020;63(3):364–374.
18. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395(10223):507–513.
19. Phan LT, Nguyen TV, Luong QC, et al. Importation and human-to-human transmission of a novel coronavirus in Vietnam. N Engl J Med. 2020;382(9):872–874.
20. Zhang B, Zhou X, Qiu Y, et al. Clinical characteristics of 82 cases of death from COVID-19. PLoS One. 2020;15(7):e0235458.
21. Wang D, Hu B, Hu C, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. 2020.
22. Alshukry A, Ali H, Ali Y, et al. Clinical characteristics of coronavirus disease 2019 (COVID-19) patients in Kuwait. PLoS One. 2020 Nov 20;15(11):e0242768.
23. Abu-Farha M, Al-Sabah S, Hammad MM., et al. Prognostic Genetic Markers for Thrombosis in COVID-19 Patients: A Focused Analysis on D-Dimer, Homocysteine and Thromboembolism, Front. Pharmacol. 09 December 2020 https://doi.org/10.3389/fphar.2020.587451.
24. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–574.
25. Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020.
26. Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y. Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2. Am J Respir Crit Care Med. 2020;202(5):756–759.
27. Mohammad A, Marafie SK, Alshawaf E, et al. Structural analysis of ACE2 variant N720D demonstrates a higher binding affinity to TMPRSS2. Life Sci. 2020 Aug 5;259:118219. doi:10.1016/j.lfs.2020.118219. Epub ahead of print. PMID: 32768580; PMCID: PMC7405906.
28. Mohammad A, Alshawaf E, Marafie SK, et al. Higher binding affinity of Furin to SARS-CoV-2 spike (S) protein D614G could be associated with higher SARS-CoV-2 infectivity. Int J Infect Dis. 2020 Oct 16:S1201–9712(20)32237–2. doi:10.1016/j.ijid.2020.10.033. Epub ahead of print. PMID: 33075532; PMCID: PMC7567667.
29. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with Pneumonia in China, 2019. N Engl J Med. 2020;382(8):727–733.
30. Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73(4):1907–1916.
31. Zhang B, Zhou X, Qiu Y, et at. Clinical characteristics of 82 death cases with COVID-19. PLoS One. 2020;15(7):e0235458.
32. de Wit E, van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14(8):523–534.
33. Xu Z, Shi L, Wang Y, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8(4):420–422.
34. Tian S, Hu W, Niu L, Liu H, Xu H, Xiao SY. Pulmonary pathology of early-phase 2019 Novel Coronavirus (COVID-19) Pneumonia in two patients with lung cancer. J Thorac Oncol. 2020;15(5):700–704.
35. Tay MZ, Poh CM, Renia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20(6):363–374.
36. Cheung CY, Poon LL, Ng IH, et al. Cytokine responses in severe acute respiratory syndrome coronavirus-infected macrophages in vitro: possible relevance to pathogenesis. J Virol. 2005;79(12):7819–7826.
37. Yilla M, Harcourt BH, Hickman CJ, et al. SARS-coronavirus replication in human peripheral monocytes/macrophages. Virus Res. 2005;107(1):93–101.
38. Tseng CT, Perrone LA, Zhu H, Makino S, Peters CJ. Severe acute respiratory syndrome and the innate immune responses: modulation of effector cell function without productive infection. J Immunol. 2005;174(12):7977–7985.
39. Law HK, Cheung CY, Ng HY, et al. Chemokine up-regulation in SARS-coronavirus-infected, monocyte-derived human dendritic cells. Blood. 2005;106(7):2366–2374.
40. Fuchs TA, Abed U, Goosmann C, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231–241.
41. Schonrich G, Raftery MJ. Neutrophil extracellular traps go viral. Front Immunol. 2016;7:366.
42. Bonaventura A, Liberale L, Carbone F, et al. The pathophysiological role of neutrophil extracellular traps in inflammatory diseases. Thromb Haemost. 2018;118(1):6–27.
43. Fuchs TA, Brill A, Duerschmied D, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107(36):15880–15885.
44. Czaikoski PG, Mota JM, Nascimento DC, et al. Neutrophil extracellular traps induce organ damage during experimental and clinical sepsis. PLoS One. 2016;11(2):e0148142.
45. Lefrancais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight. 2018;3:3.
46. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18(2):134–147.
47. Porto BN, Stein RT. Neutrophil extracellular traps in pulmonary diseases: too much of a good thing? Front Immunol. 2016;7:311.
48. Sorensen OE, Borregaard N. Neutrophil extracellular traps - the dark side of neutrophils. J Clin Invest. 2016;126(5):1612–1620.
49. Manne BK, Denorme F, Middleton EA, et al. Platelet gene expression and function in patients with COVID-19. Blood. 2020;136(11):1317–1329.
50. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J Exp Med. 2020;217:6.
51. Middleton EA, He XY, Denorme F, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136(10):1169–1179.
52. Rossaint J, Herter JM, Van Aken H, et al. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood. 2014;123(16):2573–2584.
53. Yost CC, Schwertz H, Cody MJ, et al. Neonatal NET-inhibitory factor and related peptides inhibit neutrophil extracellular trap formation. J Clin Invest. 2016;126(10):3783–3798.
54. Yost CC, Cody MJ, Harris ES, et al. Impaired neutrophil extracellular trap (NET) formation: a novel innate immune deficiency of human neonates. Blood. 2009;113(25):6419–6427.
55. Mankan AK, Dau T, Jenne D. The NLRP3/ASC/Caspase-1 axis regulates IL-1beta processing in neutrophils. Eur J Immunol. 2012;42(3):710–715.
56. Ryu JC, Kim MJ, Kwon Y, et al. Neutrophil pyroptosis mediates pathology of P. aeruginosa lung infection in the absence of the NADPH oxidase NOX2. Mucosal Immunol. 2017;10(3):757–774.
57. Glowacka I, Bertram S, Herzog P, et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J Virol. 2010;84(2):1198–1205.
58. Wang S, Guo F, Liu K, et al. Endocytosis of the receptor-binding domain of SARS-CoV spike protein together with virus receptor ACE2. Virus Res. 2008;136(1–2):8–15.
59. Jia HP, Look DC, Tan P, et al. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am J Physiol Lung Cell Mol Physiol. 2009;297(1):L84–96.
60. Kuba K, Imai Y, Rao S, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11(8):875–879.
61. Kuba K, Imai Y, Penninger JM. Angiotensin-converting enzyme 2 in lung diseases. Curr Opin Pharmacol. 2006;6(3):271–276.
62. Haga S, Yamamoto N, Nakai-Murakami C, et al. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc Natl Acad Sci U S A. 2008;105(22):7809–7814.
63. Lambert DW, Yarski M, Warner FJ, et al. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem. 2005;280(34):30113–30119.
64. Fu Y, Cheng Y, Wu Y. Understanding SARS-CoV-2-mediated inflammatory responses: from mechanisms to potential therapeutic tools. Virol Sin. 2020.
65. Pan Y, Zhang D, Yang P, Poon LLM, Wang Q. Viral load of SARS-CoV-2 in clinical samples. Lancet Infect Dis. 2020;20(4):411–412.
66. Kim JY, Ko JH, Kim Y, et al. Viral load kinetics of SARS-CoV-2 infection in first two patients in Korea. J Korean Med Sci. 2020;35(7):e86.
67. Zou L, Ruan F, Huang M, et al. SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N Engl J Med. 2020;382(12):1177–1179.
68. Lucas C, Wong P, Klein J, et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020.
69. Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–1062.
70. O’Neill LA, Golenbock D, Bowie AG. The history of Toll-like receptors - redefining innate immunity. Nat Rev Immunol. 2013;13(6):453–460.
71. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461.
72. Channappanavar R, Perlman S. Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol. 2017;39(5):529–539.
73. Mathew D, Giles JR, Baxter AE, et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science. 2020;369:6508.
74. Laing AG, Lorenc A, Del Molino Del Barrio I, et al. A dynamic COVID-19 immune signature includes associations with poor prognosis. Nat Med. 2020;26(10):1623–1635.
75. Vabret N, Britton GJ, Gruber C, et al. Immunology of COVID-19: current State of the Science. Immunity. 2020;52(6):910–941.
76. Blanco-Melo D, Nilsson-Payant BE, Liu WC, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181(5):1036–1045 e1039.
77. Zheng M, Gao Y, Wang G, et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol. 2020;17(5):533–535.
78. Onofrio L, Caraglia M, Facchini G, Margherita V, Placido S, Buonerba C. Toll-like receptors and COVID-19: a two-faced story with an exciting ending. Future Sci OA. 2020;6(8):FSO605.
79. Siu KL, Chan CP, Kok KH, Chiu-Yat Woo P, Jin DY. Suppression of innate antiviral response by severe acute respiratory syndrome coronavirus M protein is mediated through the first transmembrane domain. Cell Mol Immunol. 2014;11(2):141–149.
80. Frieman M, Ratia K, Johnston RE, Mesecar AD, Baric RS. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J Virol. 2009;83(13):6689–6705.
81. Imai Y, Kuba K, Neely GG, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133(2):235–249.
82. McGonagle D, Sharif K, O’Regan A, Bridgewood C. The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun Rev. 2020;19(6):102537.
83. Stone JH, Frigault MJ, Serling-Boyd NJ, et al. Efficacy of tocilizumab in patients hospitalized with Covid-19. N Engl J Med. 2020.
84. Zuo Y, Yalavarthi S, Shi H, et al. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020;5:11.
85. Crouch EC, Hirche TO, Shao B, et al. Myeloperoxidase-dependent inactivation of surfactant protein D in vitro and in vivo. J Biol Chem. 2010;285(22):16757–16770.
86. Agassandian M, Mallampalli RK. Surfactant phospholipid metabolism. Biochim Biophys Acta. 2013;1831(3):612–625.
87. Versteeg GA, Bredenbeek PJ, van den Worm SH, Spaan WJ. Group 2 coronaviruses prevent immediate early interferon induction by protection of viral RNA from host cell recognition. Virology. 2007;361(1):18–26.
88. Sun L, Xing Y, Chen X, et al. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS One. 2012;7(2):e30802.
89. Frieman M, Yount B, Heise M, Kopecky-Bromberg SA, Palese P, Baric RS. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J Virol. 2007;81(18):9812–9824.
90. Narayanan K, Huang C, Lokugamage K, et al. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J Virol. 2008;82(9):4471–4479.
91. Perlman S, Dandekar AA. Immunopathogenesis of coronavirus infections: implications for SARS. Nat Rev Immunol. 2005;5(12):917–927.
92. Zumla A, Hui DS, Perlman S. Middle East respiratory syndrome. Lancet. 2015;386(9997):995–1007.
93. Liao M, Liu Y, Yuan J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med. 2020;26(6):842–844.
94. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020;46(5):846–848.
95. Yonggang Zhou BF, Zheng X, Wang D, Zhao C. Pathogenic T cells and inflammatory monocytes incite inflammatory storm in severe COVID-19 patients. Natl Sci Rev. 2020.
96. de Wilde AH, Snijder EJ, Kikkert M, van Hemert MJ. Host factors in coronavirus replication. Curr Top Microbiol Immunol. 2018;419:1–42.
97. Shin HS, Kim Y, Kim G, et al. Immune responses to middle east respiratory syndrome coronavirus during the acute and convalescent phases of human infection. Clin Infect Dis. 2019;68(6):984–992.
98. Fang Liu AX, Zhang Y, Xuan W, et al. Patients of COVID-19 may benefit from sustained lopinavir-combined regimen and the increase of eosinophil may predict the outcome of COVID-19 progression. Int J Infectious Diseases. 2020.
99. Grifoni A, Weiskopf D, Ramirez SI, et al. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell. 2020.
100. Sallusto F, Lanzavecchia A, Araki K, Ahmed R. From vaccines to memory and back. Immunity. 2010;33(4):451–463.
101. Li CK, Wu H, Yan H, et al. T cell responses to whole SARS coronavirus in humans. J Immunol. 2008;181(8):5490–5500.
102. Choe PG, Perera R, Park WB, et al. MERS-CoV antibody responses 1 year after symptom onset, South Korea, 2015. Emerg Infect Dis. 2017;23(7):1079–1084.
103. Okba NMA, Raj VS, Widjaja I, et al. Sensitive and specific detection of low-level antibody responses in mild middle east respiratory syndrome coronavirus infections. Emerg Infect Dis. 2019;25(10):1868–1877.
104. Zhao J, Alshukairi AN, Baharoon SA, et al. Recovery from the Middle East respiratory syndrome is associated with antibody and T-cell responses. Sci Immunol. 2017;2:14.
105. Callow KA, Parry HF, Sergeant M, Tyrrell DA. The time course of the immune response to experimental coronavirus infection of man. Epidemiol Infect. 1990;105(2):435–446.
106. Crotty S. T follicular helper cell biology: a decade of discovery and diseases. Immunity. 2019;50(5):1132–1148.
107. Zhao J, Zhao J, Mangalam AK, et al. Airway memory CD4(+) T cells mediate protective immunity against emerging respiratory coronaviruses. Immunity. 2016;44(6):1379–1391.
108. Moutaftsi M, Tscharke DC, Vaughan K, et al. Uncovering the interplay between CD8, CD4 and antibody responses to complex pathogens. Future Microbiol. 2010;5(2):21–239.
109. Tian Y, Grifoni A, Sette A, Weiskopf D. Human T cell response to dengue virus infection. Front Immunol. 2019;10:2125.
110. Zheng HY, Zhang M, Yang CX, et al. Elevated exhaustion levels and reduced functional diversity of T cells in peripheral blood may predict severe progression in COVID-19 patients. Cell Mol Immunol. 2020;17(5):541–543.
111. Guo C, Li B, Ma H, et al. Single-cell analysis of two severe COVID-19 patients reveals a monocyte-associated and tocilizumab-responding cytokine storm. Nat Commun. 2020;11(1):3924.
112. Lei L, Qian H, Yang X, et al. The phenotypic changes of gammadelta T cells in COVID-19 patients. J Cell Mol Med. 2020;24(19):11603–11606.
113. Dong P, Ju X, Yan Y, et al. gammadelta T cells provide protective function in highly pathogenic Avian H5N1 Influenza A Virus Infection. Front Immunol. 2018;9:2812.
114. Zheng J, Liu Y, Lau YL, Tu W. gammadelta-T cells: an unpolished sword in human anti-infection immunity. Cell Mol Immunol. 2013;10(1):50–57.
115. Zhao J, Zhao J, Legge K, Perlman S. Age-related increases in PGD(2) expression impair respiratory DC migration, resulting in diminished T cell responses upon respiratory virus infection in mice. J Clin Invest. 2011;21(12):21–4930.
116. Hou H, Zhang B, Huang H, et al. Using IL-2R/lymphocytes for predicting the clinical progression of patients with COVID-19. Clin Exp Immunol. 2020;201(1):76–84.
117. Libraty DH, O’Neil KM, Baker LM, Acosta LP, Olveda RM. Human CD4(+) memory T-lymphocyte responses to SARS coronavirus infection. Virology. 2007;368(2):317–21.
118. Yang LT, Peng H, Zhu ZL, et al. Long-lived effector/central memory T-cell responses to severe acute respiratory syndrome coronavirus (SARS-CoV) S antigen in recovered SARS patients. Clin Immunol. 2006;120(2):171–178.
119. Janice OHL, Ken-En Gan S, Bertoletti A, Tan YJ. Understanding the T cell immune response in SARS coronavirus infection. Emerg Microbes Infect. 2012;1(9):e23.
120. Chen J, Lau YF, Lamirande EW, et al. Cellular immune responses to severe acute respiratory syndrome coronavirus (SARS-CoV) infection in senescent BALB/c mice: CD4+ T cells are important in control of SARS-CoV infection. J Virol. 2010;84(3):1289–1301.
121. Roberts A, Deming D, Paddock CD, et al. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. 2007;3(1):e5.
122. Zhao J, Zhao J, Perlman S. T cell responses are required for protection from clinical disease and for virus clearance in severe acute respiratory syndrome coronavirus-infected mice. J Virol. 2010;84(18):9318–9325.
123. Wilk AJ, Rustagi A, Zhao NQ, et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med. 2020;26(7):1070–1076.
124. Kuri-Cervantes L, Pampena MB, Meng W, et al. Comprehensive mapping of immune perturbations associated with severe COVID-19. Sci Immunol. 2020;5:49.
125. Yu K, He J, Wu Y, et al. Dysregulated adaptive immune response contributes to severe COVID-19. Cell Res. 2020;30(9):814–816.
126. Laing AG, Lorenc A, Del Molino Del Barrio I, et al. A dynamic Covid-19 immune signature includes associations with poor prognosis. Nat Med. 2020;26(10):1623–1635.
127. Irani Thevarajan THON, Koutsakos M, Druce J. Breadth of concomitant immune responses prior to patient recovery: a case report of non-severe COVID-19. Nat Med. 2020.
128. Giamarellos-Bourboulis EJ, Netea MG, Rovina N, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe. 2020;27(6):992–1000 e1003.
129. Wichmann D, Sperhake JP, Lutgehetmann M, et al. Autopsy findings and venous thromboembolism in patients with covid-19: a prospective cohort study. Ann Intern Med. 2020;173(4):268–277.
130. Chua RL, Lukassen S, Trump S, et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat Biotechnol. 2020;38(8):970–979.
131. Wen W, Su W, Tang H, et al. Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. Cell Discov. 2020;6:31.
132. Ni L, Ye F, Cheng ML, et al. Detection of SARS-CoV-2-Specific humoral and cellular immunity in COVID-19 convalescent individuals. Immunity. 2020;52(6):971–977 e973.
133. Peng Y, Mentzer AJ, Liu G, et al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat Immunol. 2020;21(11):1336–1345.
134. Neidleman J, Luo X, Frouard J, et al. SARS-CoV-2-specific T cells exhibit phenotypic features of helper function, lack of terminal differentiation, and high proliferation potential. Cell Rep Med. 2020;1(6):100081.
135. Chen Z, John Wherry E. T cell responses in patients with COVID-19. Nat Rev Immunol. 2020;20(9):529–536.
136. Diao B, Wang C, Tan Y, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol. 2020;11:827.
137. Mazzoni A, Salvati L, Maggi L, et al. Impaired immune cell cytotoxicity in severe COVID-19 is IL-6 dependent. J Clin Invest. 2020;130(9):4694–4703.
138. Weiskopf D, Schmitz KS, Raadsen MP, et al. Phenotype and kinetics of SARS-CoV-2-specific T cells in COVID-19 patients with acute respiratory distress syndrome. Sci Immunol. 2020;5:48.
139. Wang W, Su B, Pang L, et al. High- dimensional immune profiling by mass cytometry revealed immunosuppression and dysfunction of immunity in COVID-19 patients. Cell Mol Immunol. 2020;17:650–652.
140. Zhou Y, Fu B, Zheng X, et al. Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. National Science Review. 2020;7(6):998–1002.
141. Wang F, Nie J, Wang H, et al. Characteristics of peripheral lymphocyte subset alteration in COVID-19 Pneumonia. J Infect Dis. 2020;21(11):1762–1769.
142. Zhang J, Wu Q, Liu Z, et al. Spike-specific circulating T follicular helper cell and cross-neutralizing antibody responses in COVID-19-convalescent individuals. Nat Microbiol. 2020.
143. Wing JB, Tanaka A, Sakaguchi S. Human FOXP3(+) Regulatory T cell heterogeneity and function in autoimmunity and cancer. Immunity. 2019;50(2):302–316.
144. Wang F, Hou H, Luo Y, et al. The laboratory tests and host immunity of COVID-19 patients with different severity of illness. JCI Insight. 2020;5:10.
145. Walter JM, Helmin KA, Abdala-Valencia H, Wunderink RG, Singer BD. Multidimensional assessment of alveolar T cells in critically ill patients. JCI Insight. 2018;3:17.
146. Kalfaoglu B, Almeida-Santos J, Tye CA, Satou Y, Ono M. T-cell hyperactivation and paralysis in severe covid-19 infection revealed by single-cell analysis. Front Immunol. 2020;11:589380.
147. Stephen-Victor E, Das M, Karnam A, Pitard B, Gautier JF, Bayry J. Potential of regulatory T-cell-based therapies in the management of severe COVID-19. Eur Respir J. 2020;56:3.
148. Zhu Z, Chakraborti S, He Y, et al. Potent cross-reactive neutralization of SARS coronavirus isolates by human monoclonal antibodies. Proc Natl Acad Sci U S A. 2007;104(29):12123–12128.
149. Xiao X, Chakraborti S, Dimitrov AS, Gramatikoff K, Dimitrov DS. The SARS-CoV S glycoprotein: expression and functional characterization. Biochem Biophys Res Commun. 2003;312(4):1159–1164.
150. Babcock GJ, Esshaki DJ, Thomas WD
151. Wong SK, Li W, Moore MJ, Choe H, Farzan M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J Biol Chem. 2004;279(5):3197–3201.
152. Wang C, Li W, Drabek D, et al. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat Commun. 2020;11(1):251.
153. Tian X, Li C, Huang A, et al. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg Microbes Infect. 2020;9(1):382–385.
154. Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309(5742):1864–1868.
155. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the sars-cov-2 spike glycoprotein. Cell. 2020;181(2):281–292 e286.
156. Tai W, He L, Zhang X, et al. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol. 2020;17(6):613–620.
157. Liu J, Li S, Liu J, et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine. 2020;55:102763.
158. Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20(6):355–362.
159. Woodruff MC, Ramonell RP, Nguyen DC, et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat Immunol. 2020.
160. Jenks SA, Cashman KS, Woodruff MC, Lee FE, Sanz I. Extrafollicular responses in humans and SLE. Immunol Rev. 2019;288(1):136–148.
161. Zhang W, Zhang H, Liu S, et al. Excessive CD11c(+)Tbet(+) B cells promote aberrant TFH differentiation and affinity-based germinal center selection in lupus. Proc Natl Acad Sci U S A. 2019;116(37):18550–18560.
162. Racine R, Chatterjee M, Winslow GM. CD11c expression identifies a population of extrafollicular antigen-specific splenic plasmablasts responsible for CD4 T-independent antibody responses during intracellular bacterial infection. J Immunol. 2008;181(2):1375–1385.
163. Rubtsov AV, Rubtsova K, Fischer A, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood. 2011;118(5):1305–1315.
164. Mauri C, Menon M. Human regulatory B cells in health and disease: therapeutic potential. J Clin Invest. 2017;127(3):772–779.
165. Wang T, Marken J, Chen J, et al. High TLR7 expression drives the expansion of CD19(+)CD24(hi)CD38(hi) transitional B cells and autoantibody production in SLE patients. Front Immunol. 2019;10:1243.
166. Guo L, Ren L, Yang S, et al. Profiling early humoral response to diagnose novel coronavirus disease (COVID-19). Clin Infect Dis. 2020.
167. Zhao J, Yuan Q, Wang H, et al. Antibody responses to SARS-CoV-2 in patients of novel coronavirus disease 2019. Clin Infect Dis. 2020.
168. Seow J, Graham C, Merrick B, et al. Longitudinal observation and decline of neutralizing antibody responses in the three months following SARS-CoV-2 infection in humans. Nat Microbiol. 2020.
169. Wu F, Liu M, Wang A, et al. Evaluating the association of clinical characteristics with neutralizing antibody levels in patients who have recovered from mild COVID-19 in Shanghai, China. JAMA Intern Med. 2020;180(10):1356–1362.
170. Luchsinger LL, Ransegnola B, Jin D, et al. Serological assays estimate highly variable SARS-CoV-2 neutralizing antibody activity in recovered COVID19 patients. J Clin Microbiol. 2020.
171. Peiris JS, Chu CM, Cheng VC, et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet. 2003;361(9371):1767–1772.
172. Liu L, Wei Q, Lin Q, et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight. 2019;4:4.
173. Iwasaki A, Yang Y. The potential danger of suboptimal antibody responses in COVID-19. Nat Rev Immunol. 2020;20(6):339–341.
174. Robbiani DF, Gaebler C, Muecksch F, et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature. 2020.
175. Tillett RL, Sevinsky JR, Hartley PD, et al. Genomic evidence for reinfection with SARS-CoV-2: a case study. Lancet Infect Dis. 2020.
176. Tomassini S, Kotecha D, Bird PW, Folwell A, Biju S, Tang JW. Setting the criteria for SARS-CoV-2 reinfection - six possible cases. J Infect. 2020.
177. Abolghasemi H, Eshghi P, Cheraghali AM, et al. Clinical efficacy of convalescent plasma for treatment of COVID-19 infections: results of a multicenter clinical study. Transfus Apher Sci. 2020;59(5):102875.
178. Cheng Y, Wong R, Soo YO, et al. Use of convalescent plasma therapy in SARS patients in Hong Kong. Eur J Clin Microbiol Infect Dis. 2005;24(1):44–46.
179. Soo YO, Cheng Y, Wong R, et al. Retrospective comparison of convalescent plasma with continuing high-dose methylprednisolone treatment in SARS patients. Clin Microbiol Infect. 2004;10(7):676–678.
180. Yeh KM, Chiueh TS, Siu LK, et al. Experience of using convalescent plasma for severe acute respiratory syndrome among healthcare workers in a Taiwan hospital. J Antimicrob Chemother. 2005;56(5):919–922.
181. Halstead SB, O’Rourke EJ. Antibody-enhanced dengue virus infection in primate leukocytes. Nature. 1977;265(5596):739–741.
182. Haslwanter D, Blaas D, Heinz FX, Stiasny K. A novel mechanism of antibody-mediated enhancement of flavivirus infection. PLoS Pathog. 2017;13(9):e1006643.
183. Takada A, Kawaoka Y. Antibody-dependent enhancement of viral infection: molecular mechanisms and in vivo implications. Rev Med Virol. 2003;13(6):387–398.
184. Takada A, Feldmann H, Ksiazek TG, Kawaoka Y. Antibody-dependent enhancement of Ebola virus infection. J Virol. 2003;77(13):7539–7544.
185. Ochiai H, Kurokawa M, Matsui S, et al. Infection enhancement of influenza A NWS virus in primary murine macrophages by anti-hemagglutinin monoclonal antibody. J Med Virol. 1992;36(3):217–21.
186. Yip MS, Leung NH, Cheung CY, et al. Antibody-dependent infection of human macrophages by severe acute respiratory syndrome coronavirus. Virol J. 2014;11:82.
187. Jaume M, Yip MS, Cheung CY, et al. Anti-severe acute respiratory syndrome coronavirus spike antibodies trigger infection of human immune cells via a pH- and cysteine protease-independent FcgammaR pathway. J Virol. 2011;85(20):10582–10597.
188. Yoshikawa T, Hill T, Li K, Peters CJ, Tseng CT. Severe acute respiratory syndrome (SARS) coronavirus-induced lung epithelial cytokines exacerbate SARS pathogenesis by modulating intrinsic functions of monocyte-derived macrophages and dendritic cells. J Virol. 2009;83(7):3039–3048.
189. Channappanavar R, Fehr AR, Vijay R, et al. Dysregulated type i interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. 2016;19(2):181–193.
190. Tetro JA. Is COVID-19 receiving ADE from other coronaviruses? Microbes Infect. 2020;22(2):72–73.
191. Bastard P, Rosen LB, Zhang Q, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370:6515.
192. Zhang Q, Bastard P, Liu Z, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370:6515.
193. Wang TT, Ravetch JV. Functional diversification of IgGs through Fc glycosylation. J Clin Invest. 2019;129(9):3492–3498.
194. Kam KQ, Yung CF, Cui L, et al. A well infant with coronavirus disease 2019 (COVID-19) with high viral load. Clin Infect Dis. 2020.
195. Wu Z, McGoogan JM Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) Outbreak in China: summary of a report of 72314 cases from the chinese center for disease control and prevention. JAMA. 2020.
196. Yuanyuan Dong XM, Yabin H, Xin Q, Jiang F, Jiang Z, Tong S. Epidemiology of COVID-19 Among Children in China. Pediatrics. 2020;145(6):e20200702.
197. Yuanyuan Dong XM, Yabin H, Xin Q, Jiang F, Jiang Z, Tong S. Epidemiological characteristics of 2143 pediatric patients with 2019 coronavirus disease in China. J Emerg Med. 2020;58(4):712–713.
198. Locati M, Curtale G, Mantovani A. Diversity, mechanisms, and significance of macrophage plasticity. Annu Rev Pathol. 2020;15:123–147.
199. Mantovani A, Netea MG. Trained innate immunity, epigenetics, and Covid-19. N Engl J Med. 2020;383(11):1078–1080.
200. de Laval B, Maurizio J, Kandalla PK, et al. C/EBPbeta-dependent epigenetic memory induces trained immunity in hematopoietic stem cells. Cell Stem Cell. 2020;26(5):657–674 e658.
201. Netea MG, Dominguez-Andres J, Barreiro LB, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol. 2020;20(6):375–388.
202. Cirovic B, de Bree LCJ, Groh L, et al. BCG vaccination in humans elicits trained immunity via the hematopoietic progenitor compartment. Cell Host Microbe. 2020;28(2):322–334 e325.
203. Castagnoli R, Votto M, Licari A, et al. Severe acute respiratory syndrome coronavirus 2 (sars-cov-2) infection in children and adolescents: a systematic review. JAMA Pediatr. 2020;174(9):882–889.
204. Escobar LE, Molina-Cruz A, Barillas-Mury C. BCG vaccine protection from severe coronavirus disease 2019 (COVID-19). Proc Natl Acad Sci U S A. 2020;117(30):17720–17726.
205. Kim JH, Liepkalns J, Reber AJ, et al. Prior infection with influenza virus but not vaccination leaves a long-term immunological imprint that intensifies the protective efficacy of antigenically drifted vaccine strains. Vaccine. 2016;34(4):495–502.
206. Schmidt AG, Do KT, McCarthy KR, et al. Immunogenic stimulus for germline precursors of antibodies that engage the influenza hemagglutinin receptor-binding site. Cell Rep. 2015;13(12):2842–2850.
207. Andrews SF, Kaur K, Pauli NT, Huang M, Huang Y, Wilson PC. High preexisting serological antibody levels correlate with diversification of the influenza vaccine response. J Virol. 2015;89(6):3308–3317.
208. Angeletti D, Gibbs JS, Angel M, et al. Defining B cell immunodominance to viruses. Nat Immunol. 2017;18(4):456–463.
209. Gostic KM, Ambrose M, Worobey M, Lloyd-Smith JO. Potent protection against H5N1 and H7N9 influenza via childhood hemagglutinin imprinting. Science. 2016;354(6313):722–726.
210. Lee J, Boutz DR, Chromikova V, et al. Molecular-level analysis of the serum antibody repertoire in young adults before and after seasonal influenza vaccination. Nat Med. 2016;22(12):1456–1464.
211. Lee J, Paparoditis P, Horton AP, et al. Persistent antibody clonotypes dominate the serum response to influenza over multiple years and repeated vaccinations. Cell Host Microbe. 2019;25(3):367–376 e365.
212. Yang ZY, Werner HC, Kong WP, et al. Evasion of antibody neutralization in emerging severe acute respiratory syndrome coronaviruses. Proc Natl Acad Sci U S A. 2005;102(3):797–801.
213. Ho MS, Chen WJ, Chen HY, et al. Neutralizing antibody response and SARS severity. Emerg Infect Dis. 2005;11(11):1730–1737.
214. Donnelly CA, Fisher MC, Fraser C, et al. Epidemiological and genetic analysis of severe acute respiratory syndrome. Lancet Infect Dis. 2004;4(11):672–683.
215. Wu Z, Yang L, Ren X, et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 2016;10(3):609–620.
216. Alba Grifoni DW, Ramirez SI, Jose M. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell. 2020.
217. Andrews SF, Huang Y, Kaur K, et al. Immune history profoundly affects broadly protective B cell responses to influenza. Sci Transl Med. 2015;7(316):316ra192.
218. Ali H, Alshukry A, Marafie SK, et al. Outcomes of COVID-19: Disparities by ethnicity. Infect Genet Evol. 2020 Nov 24:104639. doi:10.1016/j.meegid.2020.104639. Epub ahead of print. PMID: 33246086; PMCID: PMC7683943.
219. Alahmad B, Al-Shammari AA, Bennakhi A, et al. Fasting Blood Glucose and COVID-19 Severity: Nonlinearity Matters. Diabetes Care. 2020 October. Epub. https://doi.org/10.2337/dc20-1941.
220. Deming D, Sheahan T, Heise M, et al. Vaccine efficacy in senescent mice challenged with recombinant SARS-CoV bearing epidemic and zoonotic spike variants. PLoS Med. 2006;3(12):e525.
221. Yasui F, Kai C, Kitabatake M, et al. Prior immunization with severe acute respiratory syndrome (SARS)-associated coronavirus (SARS-CoV) nucleocapsid protein causes severe pneumonia in mice infected with SARS-CoV. J Immunol. 2008;181(9):6337–6348.
222. Bolles M, Deming D, Long K, et al. A double-inactivated severe acute respiratory syndrome coronavirus vaccine provides incomplete protection in mice and induces increased eosinophilic proinflammatory pulmonary response upon challenge. J Virol. 2011;85(23):12201–12215.
223. Lurie N, Saville M, Hatchett R, Halton J. Developing Covid-19 vaccines at pandemic speed. N Engl J Med. 2020;382(21):1969–1973.
224. Li XY, Du B, Wang YS, et al. [The keypoints in treatment of the critical coronavirus disease 2019 patient(2)]. Zhonghua Jie He He Hu Xi Za Zhi. 2020;43(4):277–281.
225. National Library of Medicine (NLM) at the National Institutes of Health (NIH). Avaialable from: https://clinicaltrials.gov/ct2/home.
226. Klein SL, Pekosz A, Park HS, et al. Sex, age, and hospitalization drive antibody responses in a COVID-19 convalescent plasma donor population. J Clin Invest. 2020;130(11):6141–6150.
227. Johnson RF, Bagci U, Keith L, et al. 3B11-N, a monoclonal antibody against MERS-CoV, reduces lung pathology in rhesus monkeys following intratracheal inoculation of MERS-CoV Jordan-n3/2012. Virology. 2016;490:49–58.
228. Kleine-Weber H, Elzayat MT, Wang L, et al. Mutations in the spike protein of middle east respiratory syndrome coronavirus transmitted in korea increase resistance to antibody-mediated neutralization. J Virol. 2019;93:2.
229. Rockx B, Donaldson E, Frieman M, et al. Escape from human monoclonal antibody neutralization affects in vitro and in vivo fitness of severe acute respiratory syndrome coronavirus. J Infect Dis. 2010;201(6):946–955.
230. Qiu M, Shi Y, Guo Z, et al. Antibody responses to individual proteins of SARS coronavirus and their neutralization activities. Microbes Infect. 2005;7(5–6):882–889.
231. Berry JD, Hay K, Rini JM, et al. Neutralizing epitopes of the SARS-CoV S-protein cluster independent of repertoire, antigen structure or mAb technology. MAbs. 2010;2(1):53–66.
232. Zhang L, Zhang F, Yu W, et al. Antibody responses against SARS coronavirus are correlated with disease outcome of infected individuals. J Med Virol. 2006;78(1):1–8.
233. Arabi YM, Hajeer AH, Luke T, et al. Feasibility of Using Convalescent Plasma Immunotherapy for MERS-CoV Infection, Saudi Arabia. Emerg Infect Dis. 2016;22(9):1554–1561.
234. Drosten C, Meyer B, Muller MA, et al. Transmission of MERS-coronavirus in household contacts. N Engl J Med. 2014;371(9):828–835.
235. Park WB, Perera RA, Choe PG, et al. Kinetics of Serologic Responses to MERS Coronavirus Infection in Humans, South Korea. Emerg Infect Dis. 2015;21(12):2186–2189.
236. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8(1):34–47.
237. Bournazos S, DiLillo DJ, Ravetch JV. The role of Fc-FcgammaR interactions in IgG-mediated microbial neutralization. J Exp Med. 2015;212(9):1361–1369.
238. Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313(5787):670–673.
239. Zatterale F, Longo M, Naderi J, et al. Chronic adipose tissue inflammation linking obesity to insulin resistance and type 2 diabetes. Front Physiol. 2019;10:1607.
240. Hanley B, Roufosse CA, Osborn M, Naresh KN. Convalescent donor SARS-COV-2-specific cytotoxic T lymphocyte infusion as a possible treatment option for COVID-19 patients with severe disease has not received enough attention till date. Br J Haematol. 2020;189(6):1062–1063.
241. Ganji A, Farahani I, Khansarinejad B, Ghazavi A, Mosayebi G. Increased expression of CD8 marker on T-cells in COVID-19 patients. Blood Cells Mol Dis. 2020;83:102437.
242. Moon C. Fighting COVID-19 exhausts T cells. Nat Rev Immunol. 2020;20(5):277.
243. Finelli C. Obesity, COVID-19 and immunotherapy: the complex relationship! Immunotherapy. 2020;12(15):1105–1109.
244. Nimmerjahn F, Ravetch JV. Analyzing antibody-Fc-receptor interactions. Methods Mol Biol. 2008;415:151–162.
245. van Mirre E, van Royen A, Hack CE. IVIg-mediated amelioration of murine ITP via FcgammaRIIb is not necessarily independent of SHIP-1 and SHP-1 activity. Blood. 2004;103(5):1973.
246. Veri MC, Gorlatov S, Li H, et al. Monoclonal antibodies capable of discriminating the human inhibitory Fcgamma-receptor IIB (CD32B) from the activating Fcgamma-receptor IIA (CD32A): biochemical, biological and functional characterization. Immunology. 2007;21(3):392–404.
247. Nimmerjahn F, Ravetch JV. Anti-inflammatory actions of intravenous immunoglobulin. Annu Rev Immunol. 2008;26:513–533.
248. Kurlander RJ, Hall J. Comparison of intravenous gamma globulin and a monoclonal anti-Fc receptor antibody as inhibitors of immune clearance in vivo in mice. J Clin Invest. 1986;77(6):2010–2018.
249. Roivant announces development of anti-GM-CSF monoclonal antibody to prevent and treat acute respiratory distress syndrome (ARDS) in patients with COVID-19; 2020. Avaialable from: https://roivant.com/roivant.
250. Bonaventura A, Vecchie A, Wang TS, et al. Targeting GM-CSF in COVID-19 Pneumonia: rationale and Strategies. Front Immunol. 2020;11:1625.
251. Humanigen partners with CTI, a leading contract research organization, for planned Phase III study for lenzilumab for coronavirus treatment; 2020. Avaialable from: https://www.humanigen.com/press/Humanigen-Partners-With-CTI%2C-A-Leading-Contract-Research-Organization%2C-For-Planned-Phase-III-Study-For-Lenzilumab-For-Coronavirus-Treatment.
252. Initiation of two- centre compassionate use study involving namilumab in the treatment of individual patients with rapidly worsening COVID-19 infection in Italy. Izana Bioscience; 2020. Avaialable from: bio.com/initiation.
253. Rokni M, Ghasemi V, Tavakoli Z. Immune responses and pathogenesis of SARS-CoV-2 during an outbreak in Iran: comparison with SARS and MERS. Rev Med Virol. 2020;30(3):e2107.
254. Rokni M, Hamblin MR, Rezaei N. Cytokines and COVID-19: friends or foes? Hum Vaccin Immunother. 2020;1–3.
255. CytoSorb, the Wuhan coronavirus, and cytokine storm. CytoSorbents Corporation; 2020. Avaialable from: https://www.prnewswire.com/news-releases/cytosorb-the-wuhan-coronavirus-and-cytokine-storm-300994196.html.
256. Chen C, Qi F, Shi K, et al. Thalidomide combined with low-dose short-term glucocorticoid in the treatment of critical Coronavirus Disease 2019. Clin Transl Med. 2020.
257. Gautret P, Lagier JC, Parola P, et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int J Antimicrob Agents. 2020;105949.
258. Horiuchi T, Tsukamoto H. Complement-targeted therapy: development of C5- and C5a-targeted inhibition. Inflamm Regen. 2016;36:11.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.