")
Back to Journals » International Journal of Nanomedicine » Volume 15
Celastrol Self-Stabilized Nanoparticles for Effective Treatment of Melanoma
Authors Li J, Jia Y, Zhang P, Yang H, Cong X , An L, Xiao C
Received 26 September 2019
Accepted for publication 1 February 2020
Published 19 February 2020 Volume 2020:15 Pages 1205—1214
DOI https://doi.org/10.2147/IJN.S232603
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Jinran Li,1 Yuxi Jia,1 Peng Zhang,2 Huailin Yang,2 Xianling Cong,1 Lin An,3 Chunsheng Xiao2,4
1Department of Dermatology, China-Japan Union Hospital, Jilin University, Changchun 130033, People’s Republic of China; 2Key Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, People’s Republic of China; 3Department of Hand Surgery, China-Japan Union Hospital of Jilin University, Changchun 130033, People’s Republic of China; 4Jilin Biomedical Polymers Engineering Laboratory, Changchun 130022, People’s Republic of China
Correspondence: Xianling Cong; Lin An Tel +86-431-89876626; +86-431-84995523
Email [email protected]; [email protected]
Background: Celastrol (CEL), a triterpene extracted from the Chinese herb tripterygium wilfordii, has been reported to have profound anticancer activities. However, poor water solubility and high side toxicities have severely restricted the clinical applications of CEL.
Purpose: We proposed a facile “in situ drug conjugation-induced self-assembly” strategy to prepare CEL-loaded nanoparticles (CEL-NPs) that exhibited enhanced antitumor activity against melanoma.
Methods: First, the CEL was chemically conjugated onto a methoxyl poly(ethylene glycol)-b-poly(L-lysine) (mPEG-PLL) backbone, resulting in the conversion of the double hydrophilic mPEG-PLL polymer into an amphiphilic polymer prodrug, mPEG-PLL/CEL. The obtained mPEG-PLL/CEL could self-assemble into stable micelles in aqueous solution due to the hydrophobic association of CEL moieties in the side chains and the possible electrostatic interaction between the carboxyl group in CEL and the residue amine group in the PLL segment. Thus, the obtained mPEG-PLL/CEL nanoparticles were named CEL self-stabilized nanoparticles (CEL-NPs), which were then characterized by dynamic light scattering and transmission electron microscopy. Furthermore, the antitumor effects of the CEL-NPs were investigated by an MTT assay in vitro and in a B16F10 tumor-bearing mice model.
Results: The CEL-NPs exhibited sustained drug release behavior and were effectively endocytosed by B16F10 cells. Furthermore, the in vivo antitumor evaluation demonstrated that the CEL-NPs had remarkably higher tumor growth inhibition rates and lower systemic side effects than free CEL.
Conclusion: In summary, our present work not only demonstrates the generation of stable CEL-loaded nanoparticles for the efficient treatment of melanoma but also describes a general way to prepare drug self-stabilized nanomedicine for anticancer therapy.
Keywords: celastrol, self-assembly, self-stabilization, nanomedicine, melanoma
Introduction
Chemotherapy is one of the leading treatments for cancer therapy. However, many drugs for chemotherapy possess poor water-solubility, rapid blood clearance, and lack targeting ability, resulting in unsatisfactory therapeutic outcomes. These drugs are also associated with the development of drug-resistance and serious side toxicities.1–4 Thus, innovative and effective cancer treatments are in great demand. Over the past four decades, the application of nanotechnology to cancer treatments has attracted increasing attention. The incorporation of a chemotherapeutic drug into nanocarriers to form nanomedicines has some well-known advantages, such as improved drug dispersion, prolonged blood circulation, and increased accumulation at the tumor site due to the enhanced permeability and retention (EPR) effect.5–11 Based on these improvements, nanomedicines are expected to have better antitumor effects and reduced side toxicities compared to traditional small-molecule chemotherapeutic drugs.5,10–12
Melanoma is the most lethal skin tumor. It has few durable therapies and a low survival rate.13 The treatment for melanoma consists of local therapy and systemic therapy, such as surgery, radiotherapy, chemotherapy, immunotherapy, and tumor-targeted gene therapy.14–16 In systemic therapy, conventional chemotherapy for melanoma has shown very low levels of efficacy and a high incidence of recurrence.17 Recently, the identification of new bioactive molecules from Chinese traditional herbs has gained great momentum. Increasingly, the ingredients derived from natural plants have been shown to exert therapeutic effects on various diseases, including inflammation and cancer.18–21 Celastrol (CEL) is a triterpene molecule isolated from the Chinese herb tripterygium wilfordii that has been shown to inhibit tumor growth in various tumor models, such as prostate cancer,22 osteosarcoma,23 hepatocellular carcinoma,24 pancreatic cancer,25 breast cancer26 and melanoma.27 CEL can effectively inhibit tumor cell proliferation and induce apoptotic cell death through the inhibition of proteasomes and/or NF-κB activity.28–31 Nevertheless, the poor water solubility of CEL usually leads to low bioavailability and thus, larger doses of CEL have to be administrated to improve the therapeutic effects.32 As a result, side effects, such as hepatotoxicity and nephrotoxicity have occurred at the increased dosages.33–35 Therefore, the development of new drug delivery technologies to overcome the serious adverse effects and improve the therapeutic outcomes is critical for the clinical application of CEL.36–40
To this end, we herein report the preparation of a novel type of CEL self-stabilized nanoparticles (CEL-NPs) for the effective treatment of melanoma. The CEL-NPs consisting of CEL and methoxyl poly (ethylene glycol)-b-poly (L-lysine) (mPEG-PLL) polymer were prepared by a facile “in situ chemical conjugation-induced self-assembly” strategy. The mPEG segment can form a hydrophilic shielding layer on the surface of the nanoparticles to ensure the stability of the CEL-NPs during blood circulation. Meanwhile, the PLL segment was used to conjugate with CEL through imine bonds and also to generate electrostatic interaction with CEL, leading to the formation of a CEL self-cross-linked core in the CEL-NPs. The resulting CEL-NPs were well-dispersed in water and exhibited sustained release behavior. The antitumor efficacy against B16F10 melanoma cells was systematically investigated both in vitro and in vivo.
Materials and Methods
Materials
Methoxyl poly(ethylene glycol)-b-poly(L-lysine) (mPEG-PLL) was synthesized and characterized in our previous report.36 The molecular weight of methoxyl poly(ethylene glycol) is 5000 Da and the degree of polymerization of PLL is about 50. Celastrol (≥ 98%) was purchased from Yuanye Biological Science and Technology Ltd. (Shanghai, China). 3-(4,5-Dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) was purchased from Sigma-Aldrich (Shanghai, China). Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco) and fetal bovine serum (FBS, Gibco) were acquired from Thermo Fisher Scientific (Shanghai, China). All of the above-mentioned reagents were used directly without pretreatment. Dimethyl sulfoxide (DMSO) and other chemicals were purchased from Sinopharm Chemical Reagent Co. Ltd (Shanghai, China).
Cell Lines and Animals
B16F10 murine melanoma cells were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Male C57BL mice at 5–6 weeks of age (approximately 20 g in weight) were purchased from the Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). All of the animals were treated with care according to the guidelines outlined in the Guide for the Care and Use of Laboratory Animals. All procedures were implemented after approval from the Animal Care and Use Committee of Jilin University.
Characterization
1H NMR spectra were recorded on a Bruker AV-500NMR spectrometer in deuterated DMSO. Dynamic light scattering (DLS) was detected on a Wyatt QELS instrument with a vertically polarized He-Ne laser (DAWN EOS, Wyatt Technology Co., Santa Barbara, California, USA). The ultraviolet (UV) absorption spectrum was acquired on a UV-2401PC spectrophotometer (Shimadzu, Japan). Transmission electron microscopy (TEM) measurements were made on a JEOL JEM-1011 transmission electron microscope (Tokyo, Japan) with an accelerating voltage of 100 kV. Flow cytometry analysis (FCA) was conducted on a Guava EasyCyte™ 12 Flow Cytometer (Millipore, Billerica, MA, USA) and confocal laser scanning microscopy (CLSM) was performed on a CLSM 780 (Carl Zeiss). High-performance liquid chromatography (HPLC) was performed using a binary HPLC pump with a C18 column (250 mm × 4.6 mm; made in Ireland).
Preparation and Characterization of Celastrol-Loaded mPEG-PLL Nanoparticles (CEL-NPs)
CEL-NPs were prepared by the following approach. mPEG-PLL (50 mg) and CEL (10 mg) were dissolved in 5 mL of DMSO, then 20 mL of phosphate-buffered saline (PBS, pH 7.4) was added dropwise to the solution under stirring in the dark. The mixture was stirred at room temperature for 2 h in the dark, followed by dialysis (MWCO 3500 Da) against deionized water for 12 h to remove the free CEL and DMSO. The CEL-NPs were obtained as a yellow solid after lyophilization. The drug-loading content (DLC) and drug loading-efficiency (DLE) of CEL were detected by UV–Vis spectrophotometry and calculated by the following formulas:
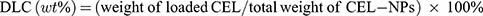
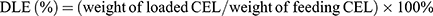
Stability of the CEL-NPs
The stability of the CEL-NPs was monitored by DLS in pH 7.4 PBS solution containing 10% fetal bovine serum. The particle sizes were measured at predetermined times.
In vitro Release of CEL
The CEL release behavior of the CEL-NPs was studied in a pH 7.4 phosphate-buffered (PB) solution. Briefly, CEL-NPs were suspended in 3.0 mL of the PB solution and transferred into a dialysis bag (MWCO 3500 Da). Then, the dialysis bag was placed in the PB solution (50 mL) and shaken at 60 rpm in the dark. At preselected times, 2.0 mL of media into which CEL was released was removed and 2.0 mL of fresh, original solution was added to each sample. The concentration of the released CEL was determined by high-performance liquid chromatography (HPLC) measurements. The mobile phase consisted of a mixture of 0.2% (v/v) phosphoric acid in methanol and water (9:1, v/v) was set to a flow rate of 1 mL min−1. The detection wavelength was 425 nm and the injection volume was 20 μL.
Cellular Uptake
The cellular uptake and intracellular drug distribution behavior of the CEL-NPs in B16F10 cells were detected by FCA and CLSM. Cy5-labeled CEL-NPs (CEL-NPs-Cy5) were used for the FCA and CLSM measurements.
For the FCA measurements, B16F10 cells were seeded into 6-well plates at a density of 2.0 ×105 cells per well in 1.8 mL of prepared DMEM media and incubated for 24 h. After culturing, the cells were treated with CEL-NPs-Cy-5 at a final concentration of 27 μg mL−1 and cultured for another 2 or 6 h. Cells without drug treatment were used as the control group. The medium was removed and the cells were washed with PBS. Then, the cells were harvested in 0.5 mL of PBS and analyzed by FCA.
For the CLSM observations, B16F10 cells were cultivated in 6-well glass-bottom culture dishes at a seeding density of 1.0 ×105 cells per well in 1.8 mL of prepared DMEM media and cultured for 24 h. Then, CEL-NPs-Cy-5 was added to the wells and the cells were cultured for another 2 or 6 h. The cells were washed five times with PBS and fixed in buffered formaldehyde (1.0 mL, 4% (w/v)) for 20 min. Subsequently, the cells were washed five times again with PBS to remove the fixative, followed by staining the cell nuclei with 4ʹ,6-diamidino-2-phenylindole (DAPI) for 4 min (blue). The cells were harvested after being washed five times with PBS to remove the free dye material and then were examined by CLSM.
Cytotoxicity Assays
The cell viability after CEL or CEL-NPs treatment was evaluated by the MTT assay. B16F10 cells were seeded in a 96-well plate at a density of 4000 cells per well in 0.18 mL of prepared DMEM media and cultured for 24 h. Subsequently, the cells were treated with the indicated concentrations of drugs for 48 h. Then, 20 μL of MTT solution (5.0 mg mL−1) was added to each well. The medium was replaced by DMSO (150 μL per well) 4 hrs later. After shaking for 5 min, 96-well plates were read using a microplate reader (Bio-Rad 680 microplate reader) (490 nm) to obtain the absorbance values.
Ex vivo Fluorescence Imaging
To analyze the in vivo distribution of the CEL-NPs in the B16F10 tumor-bearing mice, an ex vivo fluorescence imaging (Maestro Imaging System) experiment was carried as follows. First, CEL-NPs-Cy5 was injected into B16F10 tumor-bearing mice through the tail vein. After 3 and 10 h, the mice were sacrificed and the tumor and major organs (heart, liver, spleen, lung, and kidney) were harvested for fluorescence imaging.
In vivo Antitumor Efficiency
The mice were inoculated subcutaneously into the right upper abdomen with 8.0 × 105 cells/100 μL of B16F10 cells. Four days later, the tumors grew to about 50 mm3 and the treatment was initiated. For treatment, the mice were randomly divided into six groups (five mice for each group), which were treated with PBS, mPEG-PLL (22 mg kg−1, the amount of blank nanoparticles equal to the amount of mPEG-PLL in the CEL-NPs containing 4 mg kg−1 CEL), free CEL (2 or 4 mg kg−1), and CEL-NPs (containing 2 or 4 mg kg−1 CEL). The free CEL was dissolved in a medium mixture (1% DMSO, 7% Cremophor/ethanol (3:1) and 92% PBS) and was injected intraperitoneally every two days at doses of 2 and 4 mg kg−1. PBS and CEL-NPs in PBS were injected via the tail vein every two days. The tumor sizes and body weights were measured every other day and the tumor volume was calculated using the formula, V= length×width2/2. The tumor inhibition rate (%) = (Vcontrol-Vsample)/Vcontrol×100%, where the Vcontrol and Vsample represented the tumor volumes in the control and sample groups on day 12, respectively.
Histological Analyses
For histological analysis, the tumors and the major organs (heart, lung, liver, spleen, and kidney) were excised and fixed in buffered formaldehyde (4%, w/v) for one day at 4 °C. Then, all of the above tissues were embedded in paraffin and sliced for hematoxylin and eosin (H&E) staining.
Statistical Analysis
The data are shown as mean ± standard deviation (SD). Comparisons among the groups were performed by Student’s t-tests. Levels of *p < 0.05, **p < 0.01, and ***p < 0.001 were considered statistically significant.
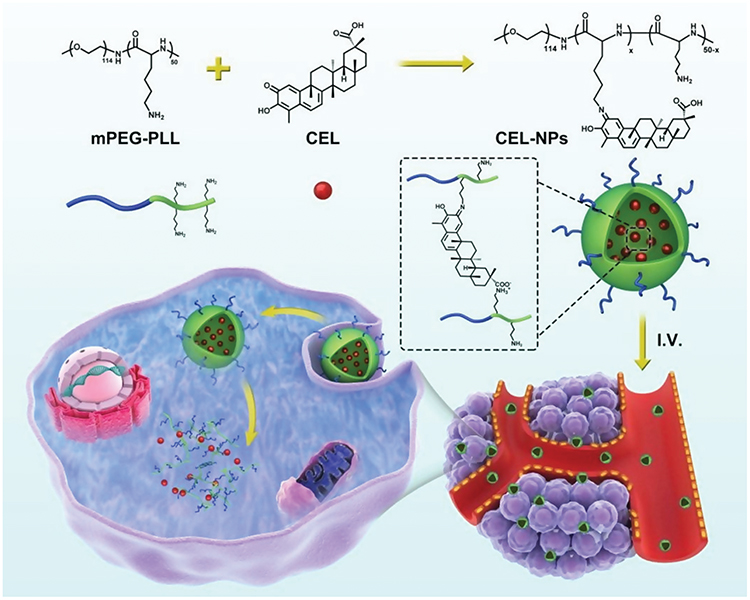 |
Scheme 1 Illustration of the preparation of CEL-NPs for drug delivery in vivo. |
Results and Discussion
Preparation and Characterization of CEL-NPs
In this work, we proposed a facile method for the preparation of CEL-loaded nanoparticles via an “in situ chemical conjugation-induced self-assembly” strategy (Scheme 1). First, CEL was conjugated onto an mPEG-PLL backbone through the in situ formation of an imine bond between CEL and an amino group in the PLL segment, which resulted in the conversion of the double hydrophilic polymer mPEG-PLL into an amphiphilic polymeric prodrug, mPEG-PLL/CEL. The resultant mPEG-PLL/CEL could self-assemble into nanoparticles with mPEG as the shell and a CEL-conjugated PLL backbone as the core. At the same time, the carboxyl group in CEL also created a weak electrostatic interaction with the residual amino groups in the PLL segment (Scheme 1). Therefore, the CEL-NPs formed by the chemical conjugation-induced self-assembly and electrostatic interaction were called CEL self-stabilized nanoparticles. The formation of the CEL-NPs was first verified by DLS and TEM characterization (Figure 1A). The hydrodynamic radius (Rh) of the CEL-NPs was 65 ± 6.4 nm according to the DLS measurement and the TEM image showed that the CEL-NPs were spherical structures about 103.1 ± 10.7 nm in size. In addition, CEL-NPs at a concentration of 1.6 mg mL−1 were readily dispersed in water and formed a clear yellow solution, whereas the free CEL was not soluble in water (Figure 1B), which again suggested the successful incorporation of CEL by mPEG-PLL to form stable CEL-NPs. Additionally, the scattered light beam could be clearly seen when the laser was passing through the CEL-NPs solution and the light intensity increased with increasing concentrations of CEL-NPs (Fig. S1), further confirming the prepared CEL-NPs as colloidal nanoparticles. The formation of the CEL-NPs was also characterized by 1H NMR spectroscopy. As shown in Fig. S2, only the proton peak corresponding to -CH2- in mPEG and diminished proton peaks corresponding to the PLL segments were observed in the 1H NMR spectrum of the CEL-NPs in D2O, suggesting that the CEL-NPs had core-shell structures in aqueous media.36,41 The incorporation of CEL into the CEL-NPs was further confirmed by UV–Vis spectrometry (Fig. S3), which revealed that the DLC and DLE of the CEL in the CEL-NPs were 9.95 wt% and 53.9%, respectively. To evaluate the stability of the CEL-NPs in complex physiological media, we monitored the particle size changes of the CEL-NPs by DLS at predetermined times. There was no obvious change in the CEL-NP particle sizes up to 24 h (Fig S4), indicating the high stability of the CEL-NPs under physiological conditions. Then, the in vitro drug release behavior of the CEL-NPs was investigated at pH 7.4. The results are shown in Figure 2. The release of CEL from the CEL-NPs showed a sustained release pattern and about 60% of the CEL was released after 48 h. The drug release behavior of the CEL-NPs was also tested at acidic pHs, such as 6.8 and 5.3. Surprisingly, the release of CEL was slower at acidic pHs than that at pH 7.4 (data not shown), which might be attributed to the decreased solubility of CEL at acidic pHs.
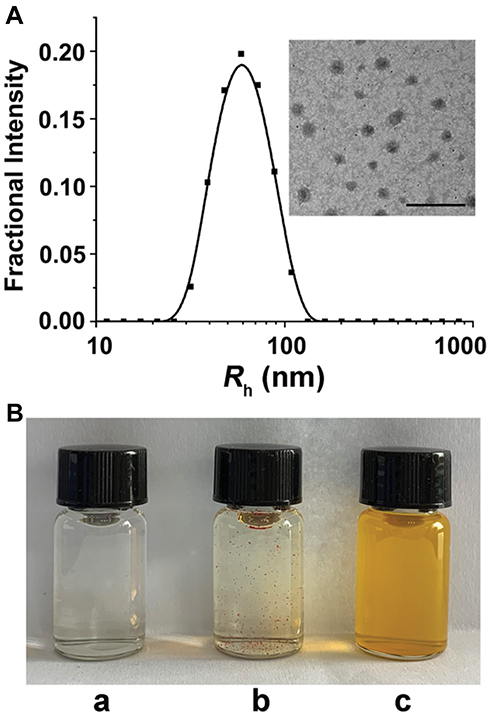 |
Figure 1 (A) DLS measurements and TEM image of the CEL-NPs. Scale bar: 500 nm. (B) Photographs of mPEG-PLL (a), CEL (b), and CEL-NPs (c) dissolved/dispersed in water. |
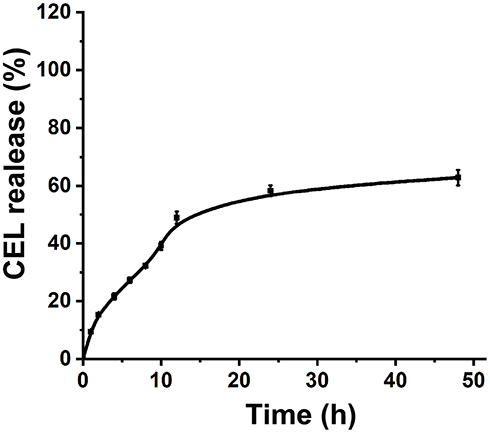 |
Figure 2 The CEL release behavior from CEL-NPs in PB at pH 7.4. The data are presented as mean ± standard deviation (n = 3). |
Cellular Uptake of the CEL-NPs
The cellular uptake of the CEL-NPs was tested by FCA and CLSM. Cy5-labeled CEL-NPs, denoted CEL-NPs-Cy5, were used for these experiments. As shown in (Figure 3 and Fig. S5), the cells treated with CEL-NPs-Cy5 for 6 hrs showed a higher fluorescence intensity than the cells treated with the same drug for 2 hrs, indicating that the CEL-NPs could be internalized by the B16F10 cells. This result was further confirmed by CLSM observations as illustrated in Figure 4. The nucleus was stained blue by DAPI and the Cy5 fluorescence was red. The red fluorescence could be observed in the cancer cells after incubation for 2 h and became more notable as the incubation time was extended to 6 h (Figure 4 and Fig. S6). These results further confirmed the successful internalization of CEL-NPs into B16F10 cells, consistent with the FCA results.
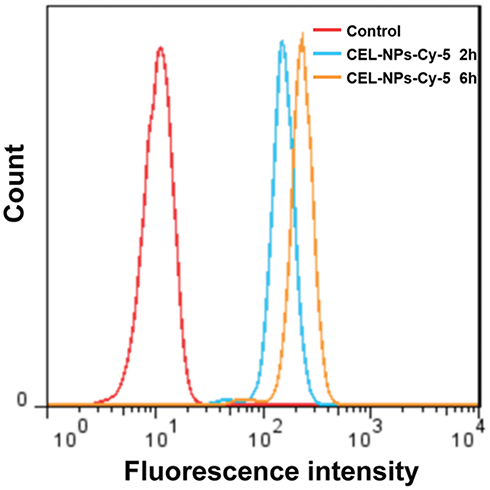 |
Figure 3 Flow cytometry analysis of B16F10 cells cultured with CEL-NPs-Cy5 for 2 or 6 h. |
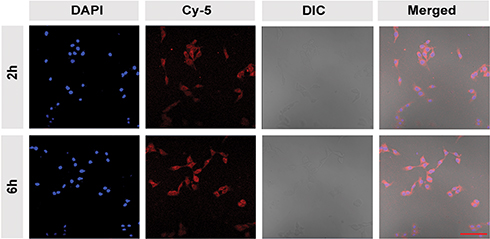 |
Figure 4 Confocal laser scanning microscopy images of B16F10 cells after treatment with CEL-NPs-Cy5 for 2 or 6 h. Scale bar: 100 µm. |
In vitro Cytotoxicity Assays
Cell viability after the treatment of B16F10 cells with CEL, mPEG-PLL, and CEL-NPs were determined by the MTT assay. After incubation for 48 h, the cytotoxicity of the CEL-NPs was close to that of free CEL (Figure 5A), indicating that the CEL-loaded nanoparticles displayed good antitumor ability. The IC50 values for free CEL and CEL-NPs were 3.56 μM (1.60 μg mL−1) and 2.81 μM (1.26 μg mL−1), respectively. In addition, it should be noted that the mPEG-PLL polymer showed negligible toxicities at all the tested concentrations (Figure 5B). The tested polymer concentrations were consistent with the corresponding concentrations of mPEG-PLL in the CEL-NPs. Taken together, these results demonstrate that the as-prepared CEL-NPs represent a promising nanomedicine for the effective treatment of melanoma cancer.
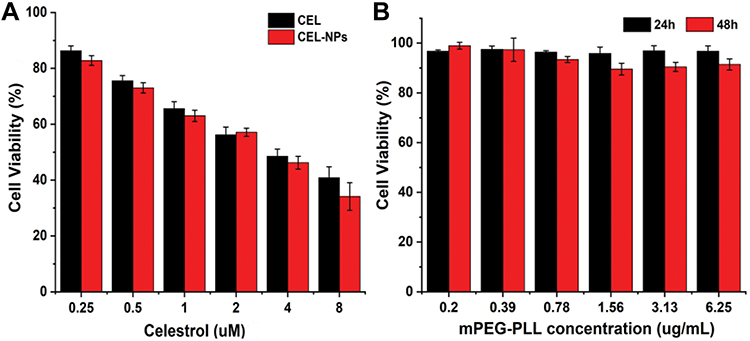 |
Figure 5 In vitro cytotoxicity assays. (A) Cell viability of B16F10 cells incubated with CEL-NPs or CEL for 48 h. (B) Cell viability of B16F10 cells after treatment with mPEG-PLL for 24 or 48 h. The data are presented as mean ± standard deviation (n = 3). |
Ex vivo Fluorescence Imaging
To assess the in vivo distribution of CEL-NPs-Cy5, major organs and the tumors of B16F10 tumor-bearing mice were excised at 3 h and 10 h post-injection for ex vivo fluorescence imaging. As shown in Figure 6, a strong fluorescence signal was observed in the tumor site and the fluorescence intensity in the tumor site was almost unchanged from 3 h to 10 h. This data suggests that the CEL-NPs-Cy5 effectively accumulated in the tumor tissue through enhanced permeability and the retention (EPR) effect, which is critical for improving the therapeutic efficacy of the CEL-NPs.
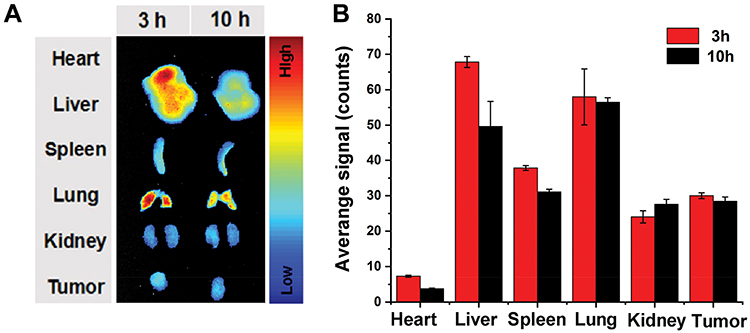 |
Figure 6 (A) Biodistribution studies of the CEL-NPs-Cy5 in B16F10 tumor-bearing mice. (B) Average signals were counted from the major organs (heart, liver, spleen, lung, and kidney) and tumors from B16F10 tumor-bearing mice after the injection of CEL-NPs-Cy5. The data are presented as mean ± standard deviation (n = 3). |
In vivo Anticancer Efficacy
Given the excellent anti-cancer activity of the CEL-NPs on B16F10 cells in vitro, the in vivo tumor inhibitory effect of the CEL-NPs was then evaluated by using the B16F10 mouse melanoma model. The tumor-bearing mice were randomly divided into six groups, which were separately treated with PBS, mPEG-PLL, free CEL (2 or 4 mg kg−1), and CEL-NPs (containing 2 or 4 mg kg−1 CEL) every two days. As shown in Figure 7A–C, the tumor growth was suppressed in the groups treated with CEL-containing formulations compared to the control PBS-treated group. In the 2 and 4 mg kg−1 CEL-NPs-treated groups, the tumor sizes were significantly smaller than in the free CEL-treated group. Furthermore, the average tumor sizes in the groups treated with higher concentrations of CEL (or CEL-NPs) were smaller than those in mice treated with lower concentrations of CEL (or CEL-NPs), indicating that the treatment effect was closely associated with the drug concentration. The weight changes in the mice during the treatment course were also monitored to evaluate the systematic toxicities of the drug formulations. As shown in Figure 7D, the body weights in the CEL-NPs-treated groups were slightly lower than those in the PBS and mPEG-PLL-treated groups, indicating that the CEL-NPs had little systemic toxicities. In contrast, obvious weight losses occurred in the groups treated with free CEL compared to the CEL-NPs groups (p < 0.05), indicating that the free CEL had high side toxicities. The improved antitumor efficacy and reduced systemic toxicity of the CEL-NPs may be ascribed to the formation of a nanomedicine that may have prolonged blood circulation and the EPR effect to improve drug accumulation at the tumor site.
Histological Analyses
The in vivo antitumor efficacy was further investigated by histological analysis. As shown in Figure 7E, significant tumor necrosis was observed in the groups treated with the CEL-containing formulations and the largest necrotic area appeared in the CEL-NPs-treated group, demonstrating that the CEL-NPs was more effective in inhibiting tumor growth than the other treatments. Then, the systemic toxicities for the tested formulations were evaluated by H&E staining of the main organs. As shown in Figure 8, the group treated with free CEL exhibited varying degrees of degeneration and edema in the liver and shrinkage or disappearance of the renal capsule cavity in the kidney. In contrast, no ultrastructural changes were observed in the organs of the control and CEL-NPs-treated groups. Taken together, these results and those shown in Figure 7A–D indicate that the CEL-NPs have great potential for the treatment of melanoma with reduced systemic toxicity.
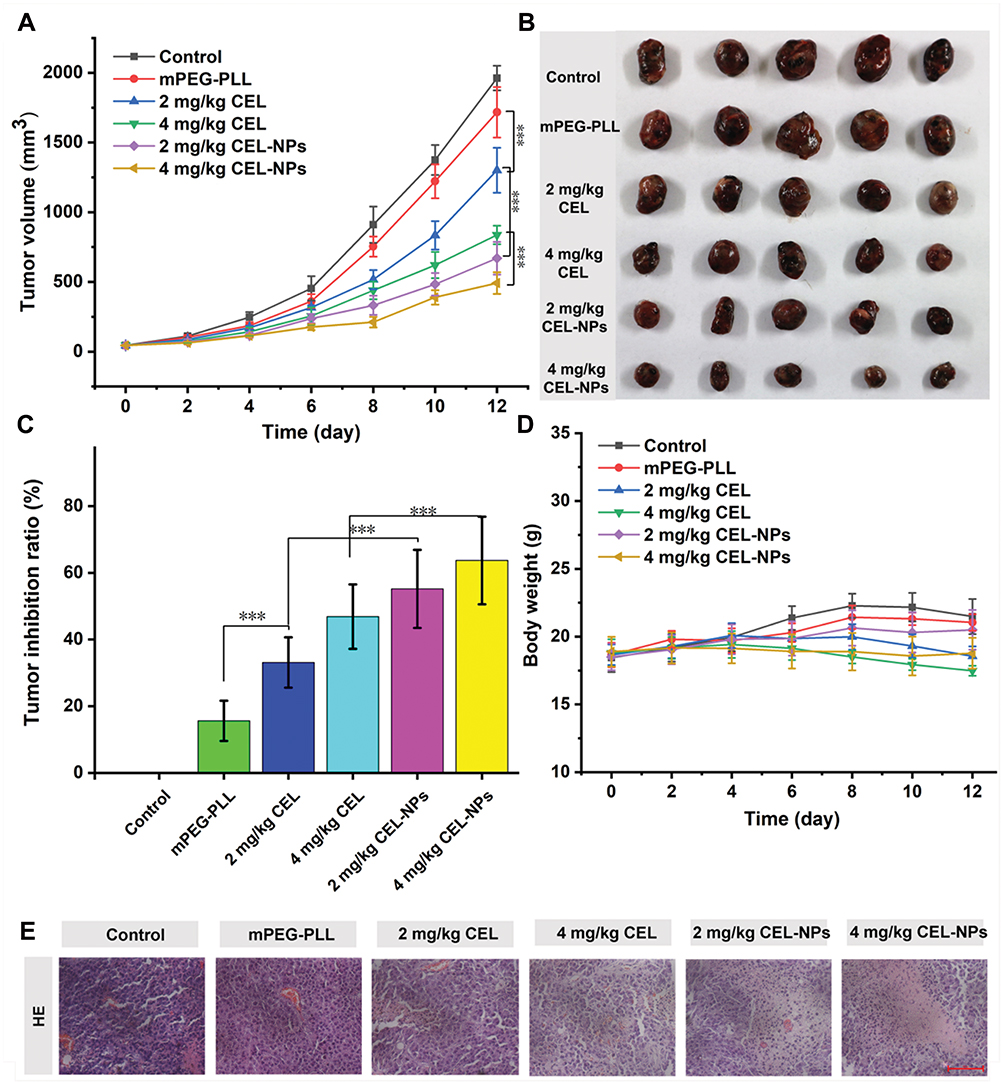 |
Figure 7 In vivo antitumor efficacy and body weight changes in mice. (A) Changes in tumor volume in B16F10 tumor-bearing mice after receiving different treatments (***p < 0.001). (B) Images of the tumors from different groups on day 12. (C) Tumor inhibition rates in the mPEG-PLL, free CEL (2 and 4 mg kg−1), and CEL-NPs (containing 2 and 4 mg kg−1 CEL)-treated groups (***p < 0.001). (D) Changes in the body weights during treatment. (E) H&E staining of tumors from the mice at the end of treatment. Scale bar:100 μm. The data are presented as mean ± standard deviation (n = 5). |
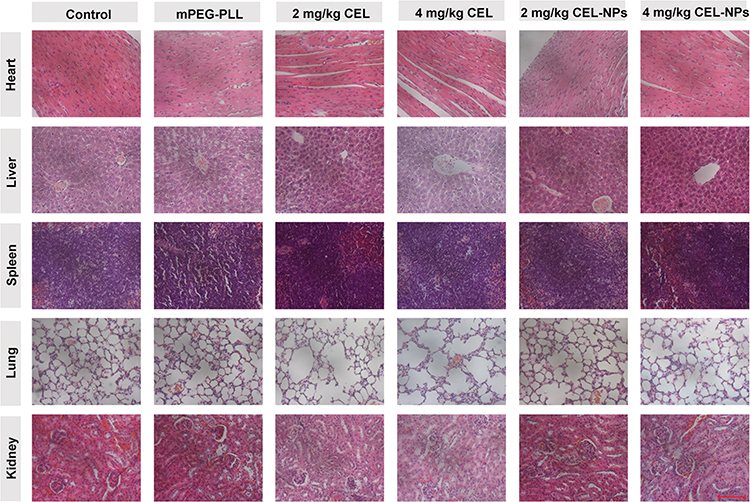 |
Figure 8 H&E staining of major organs excised from the mice at the end of the treatment periods. Scale bar: 100 µm. |
Conclusions
In summary, we successfully prepared a novel type of CEL self-stabilized nanoparticles, CEL-NPs, through a facile “in situ drug conjugation-induced self-assembly” strategy. The CEL-NPs formed by the chemical conjugation of CEL onto mPEG-PLL induced self-assembly and possible electrostatic interactions between mPEG-PLL and CEL. The resultant CEL-NPs were spherically shaped and well-dispersed in water medium. Moreover, the CEL-NPs were effectively internalized into cancer cells and thus, displayed excellent antitumor activity against mouse melanoma cells (B16F10 cells). Also, the CEL-NPs showed enhanced antitumor efficacy and reduced side toxicities in B16F10 tumor-bearing mice compared to free CEL. In general, our present work has provided a new CEL-based nanomedicine for the effective treatment of melanoma, as well as a general route for preparing drug self-stabilized nanomedicines for anticancer therapy.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (51573184, 51803209, 51520105004 and 51833010), Jilin Province Science and Technology Development Program (20190201205JC and 20190103038JH), and the Youth Innovation Promotion Association of Chinese Academy and Sciences (2017266).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Duncan R. The dawning era of polymer therapeutics. Nat Rev Drug Discovery. 2003;2:347–360. doi:10.1038/nrd1088
2. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–284. doi:10.1038/nrc1590
3. Haag R, Kratz F. Polymer therapeutics: concepts and applications. Angew Chem Int Ed. 2006;45:1198–1215. doi:10.1002/(ISSN)1521-3773
4. Trédan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99:1441–1454. doi:10.1093/jnci/djm135
5. Duncan R. Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer. 2006;6:688–701. doi:10.1038/nrc1958
6. Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751. doi:10.1038/nnano.2007.387
7. Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv Drug Deliv Rev. 2012;64:37–48. doi:10.1016/j.addr.2012.09.013
8. Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65:71–79. doi:10.1016/j.addr.2012.10.002
9. Sun T, Zhang YS, Pang B, Hyun DC, Yang M, Xia Y. Engineered nanoparticles for drug delivery in cancer therapy. Angew Chem Int Ed. 2014;53:12320–12364. doi:10.1002/anie.201403036
10. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2016;17:20. doi:10.1038/nrc.2016.108
11. Sun Q, Zhou Z, Qiu N, Shen Y. Rational design of cancer nanomedicine: nanoproperty integration and synchronization. Adv Mater. 2017;29:1606628. doi:10.1002/adma.v29.14
12. Lian H, Du Y, Chen X, et al. Core cross-linked poly(ethylene glycol)-graft-Dextran nanoparticles for reduction and pH dual responsive intracellular drug delivery. J Colloid Interface Sci. 2017;496:201–210. doi:10.1016/j.jcis.2017.02.032
13. Gloster HM, Brodland DG. The epidemiology of skin cancer. Dermatol Surg. 1996;22:217−226. doi:10.1111/j.1524-4725.1996.tb00312.x
14. Luo L, Yang YP, Du T, et al. Targeted nanoparticle-mediated gene therapy mimics oncolytic virus for effective melanoma treatment. Adv Funct Mater. 2018;28:1800173. doi:10.1002/adfm.v28.29
15. van Zeijl MC, van den Eertwegh AJ, Haanen JB, Wouters MW. (Neo)adjuvant systemic therapy for melanoma. Eur J Surg Oncol. 2017;43:534–543. doi:10.1016/j.ejso.2016.07.001
16. Raigani S, Cohen S, Boland GM. The role of surgery for melanoma in an era of effective systemic therapy. Curr Oncol Rep. 2017;19:17. doi:10.1007/s11912-017-0575-8
17. Azijli K, Stelloo E, Peters GJ, van den Eertwegh AJ. New developments in the treatment of metastatic melanoma: immune checkpoint inhibitors and targeted therapies. Anticancer Res. 2014;34:1493−1505.
18. Efferth T, Li PCH, Konkimalla VSB, Kaina B. From traditional Chinese medicine to rational cancer therapy. Trends Mol Med. 2007;13:353–361. doi:10.1016/j.molmed.2007.07.001
19. Man S, Gao W, Wei C, Liu C. Anticancer drugs from traditional toxic chinese medicines. Phytother Res. 2012;26:1449–1465. doi:10.1002/ptr.4609
20. Shah U, Shah R, Acharya S, Acharya N. Novel anticancer agents from plant sources. Chin J Nat Med. 2013;11:16–23. doi:10.1016/S1875-5364(13)60002-3
21. Xu L, Zhao W, Wang D, Ma X. Chinese medicine in the battle against obesity and metabolic diseases. Front Physiol. 2018;9:850. doi:10.3389/fphys.2018.00850
22. Yang H, Chen D, Cui QC, Yuan X, Dou QP. Celastrol, a triterpene extracted from the chinese “thunder of god vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006;66:4758–4765. doi:10.1158/0008-5472.CAN-05-4529
23. Li HY, Zhang J, Sun LL, et al. Celastrol induces apoptosis and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells: an in vitro and in vivo study. Cell Death Dis. 2015;6:e1604. doi:10.1158/1078-0432.CCR-07-1536
24. Rajendran P, Li F, Shanmugam MK, et al. Celastrol suppresses growth and induces apoptosis of human hepatocellular carcinoma through the modulation of STAT3/JAK2 signaling cascade in vitro and in vivo. Cancer Prev Res. 2012;5:631–643. doi:10.1158/1940-6207.CAPR-11-0420
25. Yadav VR, Sung B, Prasad S, et al. Celastrol suppresses invasion of colon and pancreatic cancer cells through the downregulation of expression of CXCR4 chemokine receptor. J Mol Med. 2010;88:1243–1253. doi:10.1007/s00109-010-0669-3
26. Kim JH, Lee JO, Lee SK, et al. Celastrol suppresses breast cancer MCF-7 cell viability via the AMP-activated protein kinase (AMPK)-induced p53–polo like kinase 2 (PLK-2) pathway. Cell Signal. 2013;25:805–813. doi:10.1016/j.cellsig.2012.12.005
27. Abbas S, Bhoumik A, Dahl R, Vasile S, Krajewski S, Cosford NDP. Preclinical studies of celastrol and acetyl isogambogic acid in melanoma. Clin Cancer Res. 2007;13:6769–6778. doi:10.1158/1078-0432.CCR-07-1536
28. Kannaiyan R, Shanmugam MK, Sethi G. Molecular targets of celastrol derived from Thunder of God Vine: potential role in the treatment of inflammatory disorders and cancer. Cancer Lett. 2011;303:9–20. doi:10.1016/j.canlet.2010.10.025
29. Kashyap D, Sharma A, Tuli HS, Sak K, Mukherjee T, Bishayee A. Molecular targets of celastrol in cancer: recent trends and advancements. Crit Rev Oncol Hematol. 2018;128:70–81. doi:10.1016/j.critrevonc.2018.05.019
30. Lan G, Zhang J, Ye W, et al. Celastrol as a tool for the study of the biological events of metabolic diseases. Sci China Chem. 2019;62:409–416. doi:10.1007/s11426-018-9404-9
31. Ng SW, Chan Y, Chellappan DK, et al. Molecular modulators of celastrol as the keystones for its diverse pharmacological activities. Biomed Pharmacother. 2019;109:1785–1792. doi:10.1016/j.biopha.2018.11.051
32. Li ZR, Yao L, Li JG, et al. Celastrol nanoparticles inhibit corneal neovascularization induced by suturing in rats. Int J Nanomed. 2012;7:1163–1173.
33. Cascao R, Fonseca JE, Moita LF. Celastrol: a spectrum of treatment opportunities in chronic diseases. Front Med-Lausanne. 2017;4:69. doi:10.3389/fmed.2017.00069
34. Niemela E, Desai D, Nkizinkiko Y, Eriksson JE, Rosenholm JM. Sugar-decorated mesoporous silica nanoparticles as delivery vehicles for the poorly soluble drug celastrol enables targeted induction of apoptosis in cancer cells. Eur J Pharm Biopharm. 2015;96:11–21. doi:10.1016/j.ejpb.2015.07.009
35. Zhang XW, Zhang TP, Zhou XT, et al. Enhancement of oral bioavailability of tripterine through lipid nanospheres: preparation, characterization, and absorption evaluation. J Pharm Sci-Us. 2014;103:1711–1719. doi:10.1002/jps.23967
36. Du Y, Yan W, Lian H, Xiang CY, Duan LJ, Xiao CS. 2,2 ‘-Dithiodisuccinic acid-stabilized polyion complex micelles for pH and reduction dual-responsive drug delivery. J Colloid Interf Sci. 2018;522:74–81. doi:10.1016/j.jcis.2018.03.049
37. Shan WG, Wang HG, Wu R, Zhan ZJ, Ma LF. Synthesis and anti-tumor activity study of water-soluble PEG-celastrol coupling derivatives as self-assembled nanoparticles. Bioorg Med Chem Lett. 2019;29:685–687. doi:10.1016/j.bmcl.2019.01.042
38. Muhamad N, Plengsuriyakarn T, Na-Bangchang K. Application of active targeting nanoparticle delivery system for chemotherapeutic drugs and traditional/herbal medicines in cancer therapy: a systematic review. Int J Nanomedicine. 2018;13:3921–3935. doi:10.2147/IJN
39. Xiao YT, Liu J, Guo MY, et al. Synergistic combination chemotherapy using carrier-free celastrol and doxorubicin nanocrystals for overcoming drug resistance. Nanoscale. 2018;10:12639–12649. doi:10.1039/C8NR02700E
40. Ma Z, Fan YQ, Wu YM, et al. Traditional Chinese medicine-combination therapies utilizing nanotechnology-based targeted delivery systems: a new strategy for antitumor treatment. Int J Nanomedicine. 2019;14:2029–2053. doi:10.2147/IJN.S197889
41. Zhang Y, Ding J, Li M, et al. One-step “click chemistry”-synthesized cross-linked prodrug nanogel for highly selective intracellular drug delivery and upregulated antitumor efficacy. ACS Appl Mater Interfaces. 2016;8:10673–10682. doi:10.1021/acsami.6b00426
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.