")
Back to Journals » OncoTargets and Therapy » Volume 11
Celastrol induces ubiquitin-dependent degradation of mTOR in breast cancer cells
Authors Li X, Zhu G, Yao X, Wang N, Hu R, Kong Q, Zhou D, Long L, Cai J, Zhou W
Received 12 September 2018
Accepted for publication 24 October 2018
Published 11 December 2018 Volume 2018:11 Pages 8977—8985
DOI https://doi.org/10.2147/OTT.S187315
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Takuya Aoki
Xiaoli Li,1–3,* Guangbei Zhu,4,* Xintong Yao,2,3 Ning Wang,5 Ronghui Hu,6 Qingxin Kong,7 Duanfang Zhou,1–3 Liangyuan Long,1–3 Jiali Cai,4 Weiying Zhou1–3
1Department of Pharmacology, College of Pharmacy, Chongqing Medical University, Chongqing 400016, P.R. China; 2Chongqing Key Laboratory of Drug Metabolism, Chongqing 400016, P.R. China; 3Key Laboratory for Biochemistry and Molecular Pharmacology of Chongqing, Chongqing 400016, P.R. China; 4Dapartment of Biophamaceutics, School of Pharmacy and Bioengineering, Chongqing University of Technology, Chongqing 400054, P.R. China; 5First Affiliated Hospital’s Central Laboratory, Army Medical University, Chongqing 400038, P.R. China; 6Department of Radiology, Third Affiliated Hospital of Chongqing Medical University, Chongqing 401120, P.R. China; 7Department of Pharmaceutical Engineering, Jiangsu Food and Pharmaceutical Science College, Jiangsu 223003, P.R. China
*These authors contributed equally to this work
Background: Celastrol is a major active component of the thunder god vine (Tripterygium wilfordii) used in traditional Chinese medicine to treat chronic inflammatory and autoimmune diseases. Celastrol inhibits PI3K–Akt–mTOR signaling, which is frequently dysregulated in tumors and critical for tumor-cell proliferation and survival, but the underlying mechanisms are still not fully understood. In the present study, we investigated detailed mechanisms of celastrol inhibition of mTOR signaling in breast cancer cells.
Methods: First, we evaluated the effect of celastrol on breast cancer-cell growth using MTT assays. Second, we examined the effects of celastrol on mTOR phosphorylation and expression using Western blot. Furthermore, we investigated the cause of mTOR downregulation by celastrol using immunoprecipitation assays. In addition, we evaluated the effect of celastrol on an MDA-MB231 cell-derived xenograft model.
Results: Celastrol suppressed breast cancer cell growth in vitro and in vivo. Celastrol inhibited mTOR phosphorylation and induced mTOR ubiquitination, resulting in its proteasomal degradation. Mechanistically, we found that mTOR is a client of Hsp90–Cdc37 chaperone complex, and celastrol disrupts mTOR interaction with chaperone Hsp90 while promoting mTOR association with cochaperone Cdc37.
Conclusion: Our study reveals that celastrol suppresses mTOR signaling, at least in part through regulating its association with chaperones and inducing its ubiquitination.
Keywords: celastrol, mTOR, Hsp90, Cdc37, ubiquitin, Chinese medicine
Introduction
The PI3K–Akt–mTOR signaling pathway is frequently dysregulated in cancers, including breast cancer, and is critical for tumor-cell proliferation and survival.1 PI3K transmits extracellular signals into a cell by generating phospholipids, which facilitates the phosphorylation and activation of Akt. mTOR is a key component of the PI3K–Akt signaling pathway and exists as two complexes: mTORC1 and mTORC2.2,3 mTORC1 contains mTOR, Raptor, and mLST8, and is activated by growth factors via the PI3K–Akt–TSC1/2 pathway. mTORC2 comprises mTOR, Rictor, mSin1, PRR5 and mLST8, and phosphorylates several AGC kinases, such as Akt and SGK, in response to growth factors.2,3 mTORC2-mediated Akt phosphorylation at Ser473 is critical for full Akt activation and signaling. mTORC1 stimulates protein synthesis, ribosome biogenesis, anabolic metabolism through its downstream targets, such as S6K and 4EBP1, and inhibits autophagy. mTORC2 controls cytoskeleton organization and promotes cell proliferation and survival by activating AGC-kinase family members (i.e., Akt, SGK, and PKC).3
Because it is frequently dysregulated in tumor cells, mTOR signaling has emerged as an attractive target for cancer treatment.4,5 A series of inhibitors targeting the mTOR pathway have recently been evaluated in preclinical and clinical studies as single agent or in combination with the traditional treatment,6,7 e.g., the mTORC1 inhibitor everolimus combined with endocrine therapy has been developed to treat advance hormone-receptor-positive breast cancer patients.8,9
Celastrol is a pharmacologically active compound extracted the from thunder god vine (Tripterygium wilfordii) that has been used in traditional Chinese medicine to treat chronic inflammatory and autoimmune diseases (i.e., rheumatoid arthritis) for hundreds of years.10 Recently, studies have shown that celastrol exhibits therapeutic potential in cancers, including leukemia, melanoma, prostate, gastric, colon, and breast cancers.11,12 Celastrol has been reported to suppress PI3K–Akt–mTOR signaling,13–15 but the underlying mechanisms are still not fully understood. In the current study, we found that celastrol can suppress mTOR signaling by inhibiting mTOR phosphorylation and inducing mTOR ubiquitin-dependent degradation.
Methods
Reagents
MTT, MG132, and chloroquine were purchased from Sigma-Aldrich (St Louis, MO, USA). Rabbit antibodies to mTOR, p-mTOR (S2481), Akt, p-Akt (S473), CDK4 (D9G3E), ubiquitin, Cdc37 (D11A3), and Hsp90, were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-Flag (M2), anti-HA, anti-Flag M2 affinity agarose gel, and anti-HA affinity gel were purchased from Sigma-Aldrich. Celastrol was purchased from Aladdin (Shanghai, China). pcDNA3-Flag-mTOR was a gift from Jie Chen (Addgene plasmid 26603). pcDNA3-HA-Hsp90α and pcDNA3-HA-Cdc37 were gifts from Dr Len Neckers (National Institutes of Health, USA). All inserts in the constructs were verified by DNA sequencing.
Cell culture
MCF7 cells and MDA-MB231 cells were purchased from ATCC and cultured in DMEM supplemented with 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA) and antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin) in 5% CO2 at 37°C. All cells were monitored routinely for Mycoplasma infection, and all cells used in the experiments were Mycoplasma-free.
MTT assay
Cells (104/mL) were seeded into 96-well plates and cultured in normal growth conditions for 1 day, then treated with different doses of celastrol for 24 or 48 hours. Cell viability was measured by MTT assay using a standard protocol, and OD was read at 570 nm using ELISA plate reader (Bio-Rad).
Western blot
Whole-cell proteins were extracted with RIPA lysis buffer and protein concentrations was measured with a BCA protein-assay kit. Subsequently, proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). Immunoblotting was performed as described previously.16
Immunoprecipitation
Cells in a 10 cm dish were lysed in 1 mL of immunoprecipitation (IP) lysis buffer A (50 mM Tris HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, and complete protease-inhibitor mixture) or IP lysis buffer B (20 mM HEPES, pH 7.2, 100 mM NaCl, 1 mM MgCl2, 0.1% Nonidet P40, 10 mM molybdate, and complete protease-inhibitor mixture) on ice for 50 minutes. Lysis buffer B was used for detection of interaction between mTOR and chaperones Hsp90 and Cdc37, and lysis buffer A was used in all the other immunoprecipitation experiments. After centrifugation at 14,000 rpm for 5 minutes, the supernatants were collected and incubated with 30 μL protein A/G agarose for 3 hours at 4°C on a rocking platform for preclearing. An equivalent amount of each precleared lysate was incubated with 50 μL anti-Flag affinity gel or anti-HA affinity gel overnight for IP overnight at 4°C with rotation. Immunoprecipitates were washed with 900 μL IP lysis buffer four times, 15 minutes each time. The final immunoprecipitates were resuspended in 50 μL 2 × SDS loading buffer and boiled for subsequent Western blot analysis.
Immunohistochemistry staining
Tumor sections were dewaxed, soaked in ethanol, and then blocked with 3% H2O2. Aspecific immunoreactivity was blocked with diluted normal rabbit serum at room temperature. Sections were then incubated overnight at 4°C with specific primary antibody diluted in blocking buffer. After being washed with PBS, sections were further incubated with biotinylated secondary antibody (diluted 1:50), stained with a freshly prepared diaminobenzidine solution, and then counterstained with Mayer’s hematoxylin.
Orthotopic implantation model and treatment
Female nude mice 5 weeks old weighing 18–22 g were obtained from HFK Bioscience (Beijing, China), and experiments were performed in accordance with national guidelines for animal care and use and approved by the Animal Care and Use Committee of the Third Military Medical University. An orthotopic implantation model was established in nude mice at the fourth breast with MDA-MB231 cells (4×106 cells/0.2 mL). When tumors had grown to approximately 10 mm3, mice bearing the xenograft were divided randomly into two groups (six mice/group) and were intragastrically administered normal saline (0.1 mL) or celastrol (3 mg/kg). Tumor growth was measured every 4 days after treatment. The inhibition rate of tumor growth was determined until 48 days after treatment. Tumor volume (V) was measured using a slide caliper and calculated using the formula: V (mm3) = 0.5 × ab2, a and b represented the long diameter and perpendicular short diameter (mm) of the tumor, respectively.
Statistical analysis
All results from MTT and tumor size are presented as means ± SD. Data were analyzed by Student’s t-test or ANOVA, and P<0.05 was considered statistically significant.
Results
Celastrol suppresses breast cancer-cell growth in vitro and in vivo
To evaluate the effect of celastrol on breast cancer-cell growth, we treated the breast cancer cell lines MCF7 and MDA-MB231 with different concentrations (0.5–8.0 μM) of celastrol for 24 hours and 48 hours, respectively. MTT assays showed that celastrol suppressed cell growth in both cell lines in dose- and time-dependent manners and that MDA-MB231 cells were more sensitive to celastrol than MCF7 cells (Figure 1A and B).
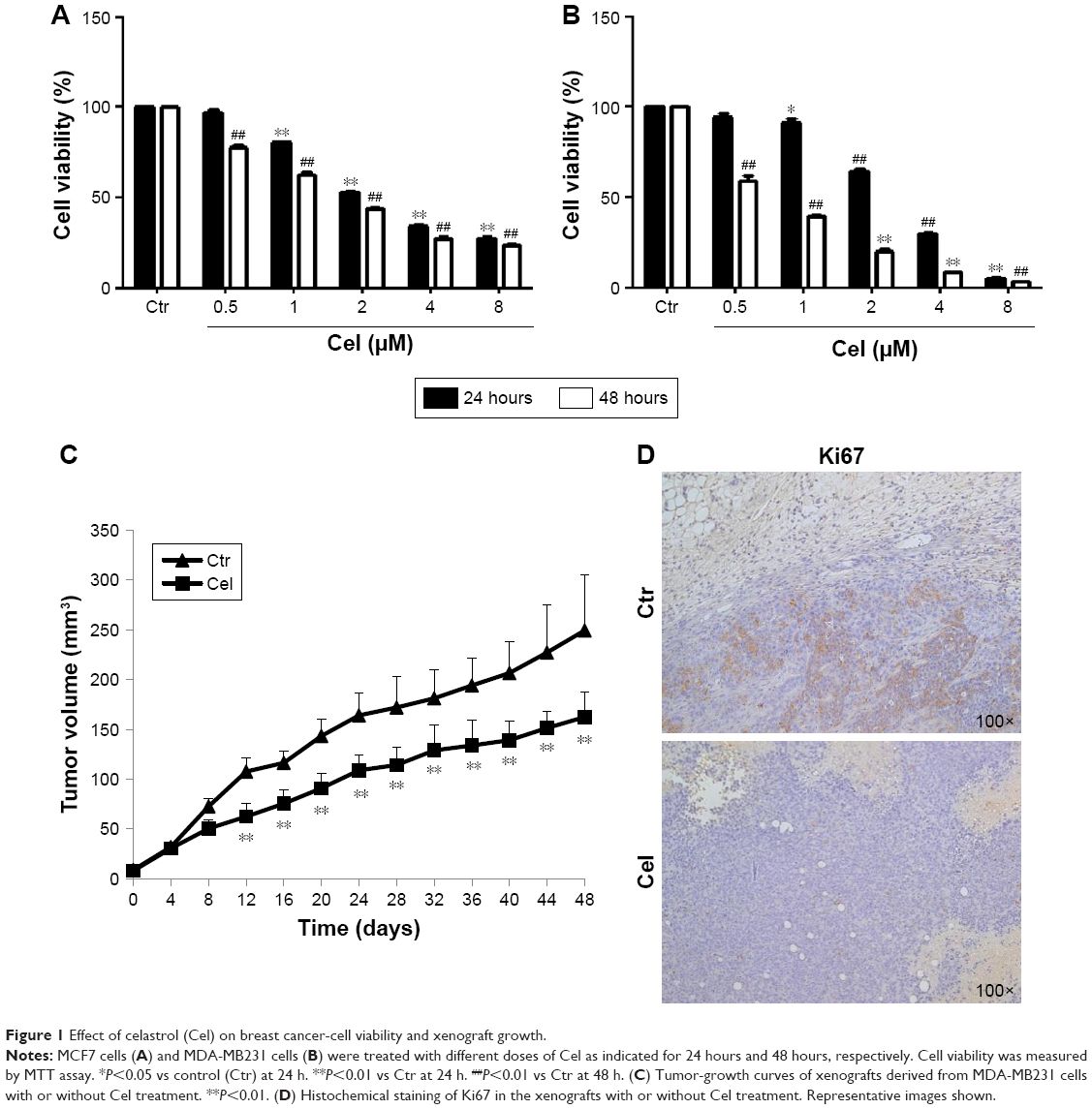 | Figure 1 Effect of celastrol (Cel) on breast cancer-cell viability and xenograft growth. |
We further examined the antitumor effect of celastrol in an MDA-MB231 cell orthotopic implantation mouse model. Celastrol significantly inhibited tumor growth, suggesting that celastrol has an anti-breast cancer effect in vivo (Figure 1C). Analysis of the proliferative index in tumor sections showed that expression of Ki67, a marker of proliferation, was much lower in celastrol-treated mice than control-group mice (Figure 1D).
Celastrol inhibits mTOR phosphorylation and expression
Given that celastrol suppresses PI3K–Akt–mTOR signaling, which plays a pivotal role in breast cancer,13,14 we examined the effects of celastrol on mTOR phosphorylation and expression. As shown in Figure 2A, celastrol inhibited mTOR phosphorylation at Ser2481, which is associated with mTORC2 activity.17 Celastrol inhibition of mTORC2 activity was further supported by the result that celastrol time-dependently decreased Akt Ser473 phosphorylation, which is mediated by mTORC2.18 Interestingly, we repeatedly observed that mTOR expression was reduced by celastrol in MCF7 and MDA-MB231 cells (Figure 2B and C), suggesting that celastrol can also reduce mTOR expression.
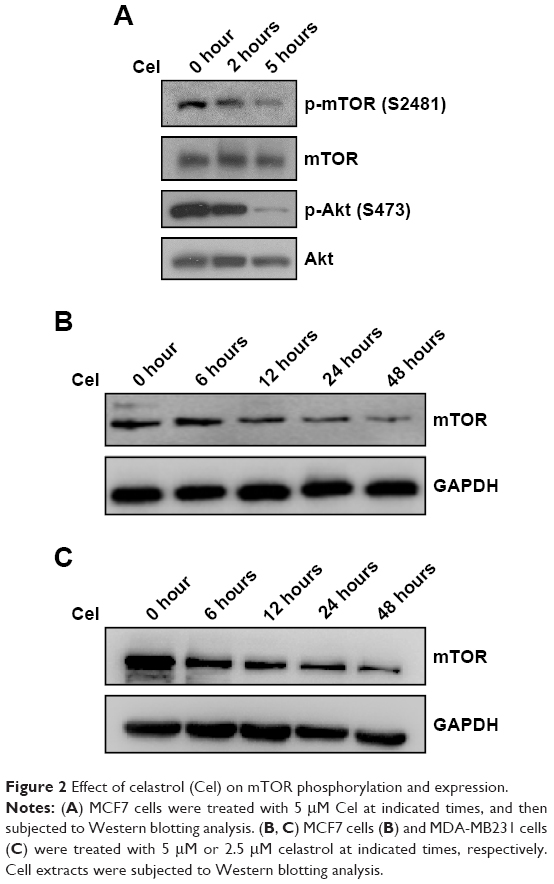 | Figure 2 Effect of celastrol (Cel) on mTOR phosphorylation and expression. |
Celastrol induces mTOR proteasomal degradation
Since celastrol can downregulate mTOR expression, we further tested whether celastrol promoted mTOR degradation. To this end, we first examined the effect of the proteasome inhibitor MG132 or the lysosome inhibitor chloroquine on celastrol-induced mTOR downregulation. The result showed that celastrol-induced mTOR downregulation was attenuated by MG132, but not by chloroquine (Figure 3A–C), suggesting that celastrol may induce mTOR proteasomal degradation.
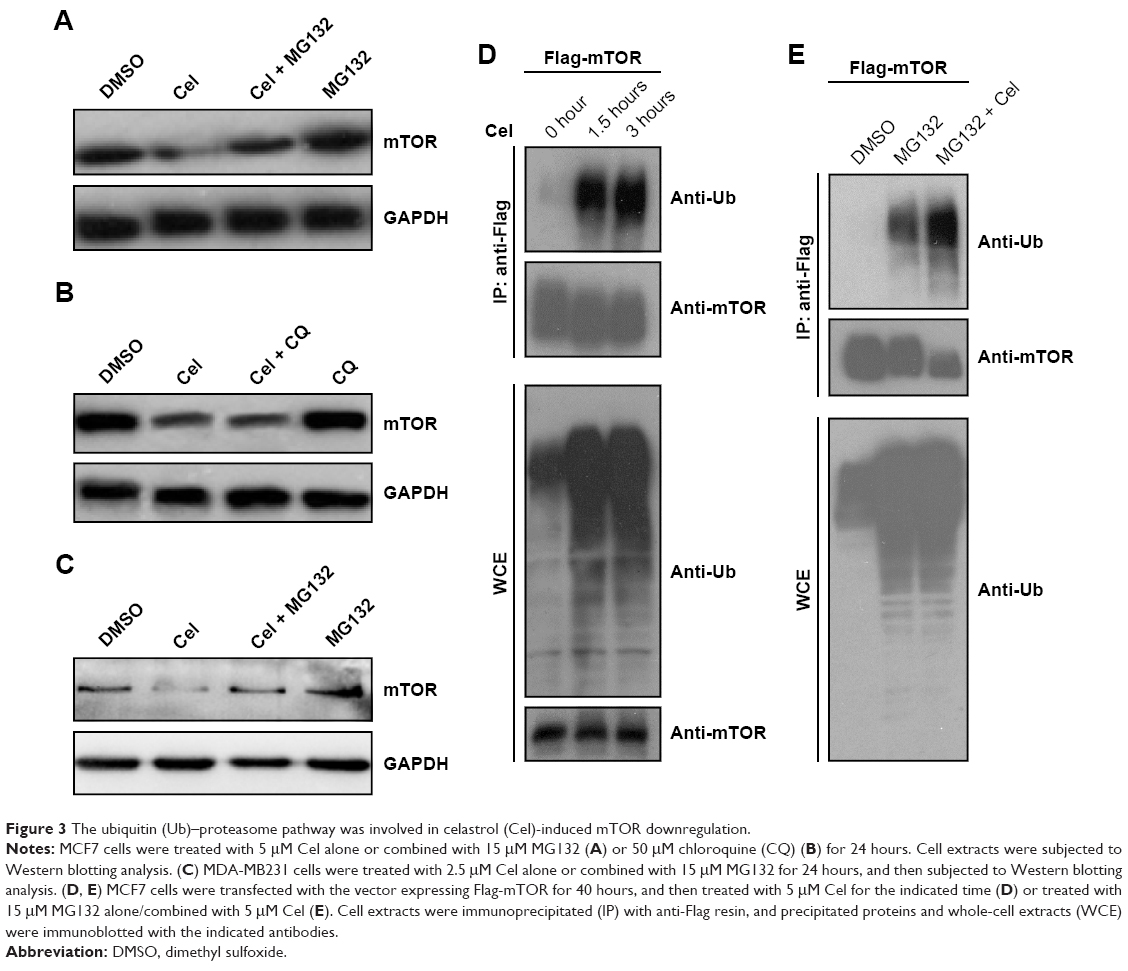 | Figure 3 The ubiquitin (Ub)–proteasome pathway was involved in celastrol (Cel)-induced mTOR downregulation. |
Given proteasomal degradation of a protein is usually ubiquitin-dependent, we further investigated whether celastrol induced mTOR ubiquitination. MCF7 cells were transfected with the Flag-mTOR expression vector, then treated with celastrol. Flag-mTOR was immunoprecipitated from cell extracts using anti-Flag resin, and ubiquitinated Flag-mTOR was detected by Western blotting using an antiubiquitin antibody. As shown in Figure 3D, celastrol treatment dramatically increased ubiquitinated mTOR levels. Since increasing levels of ubiquitinated mTOR can result from induction of mTOR ubiquitination, blocking degradation of ubiquitinated mTOR, or both, we further determined whether celastrol induced mTOR ubiquitination by examining the effect of celastrol on ubiquitinated mTOR levels with preblocked proteasome activity. To this end, we pretreated MCF7 cells expressing Flag-mTOR with 15 μM MG132 to block proteasome activity and then treated the cells with celastrol. As shown in Figure 3E, 15 μM MG132 alone robustly increased levels of ubiquitinated proteins, and its combination with celastrol did not further increase overall ubiquitinated protein levels, but the combination still dramatically increased ubiquitinated Flag-mTOR levels, confirming that celastrol can induce mTOR ubiquitination.
Celastrol alters mTOR association with Hsp90 and Cdc37 chaperones
Hsp90 and its cochaperone Cdc37 interact with and stabilize many kinases.19–21 mTOR is an atypical serine/threonine kinase and has been suggested to be a client of Hsp90.22,23 Surprisingly, there have no reports on the interaction between mTOR and Cdc37.24 To confirm the interaction between mTOR and Hsp90 and test whether mTOR interacts with Cdc37, reciprocal IP assays were performed. As shown in Figure 4A, both HA-Cdc37 and HA-Hsp90 pulled down endogenous mTOR from lysates of MCF7 cells expressing HA-Cdc37 and HA-Hsp90, respectively. Flag-mTOR also pulled down endogenous Hsp90 and Cdc37 in cell extracts of MCF7 cells expressing Flag-mTOR (Figure 4B), suggesting that mTOR interacts with the Hsp90–Cdc37 chaperone complex.
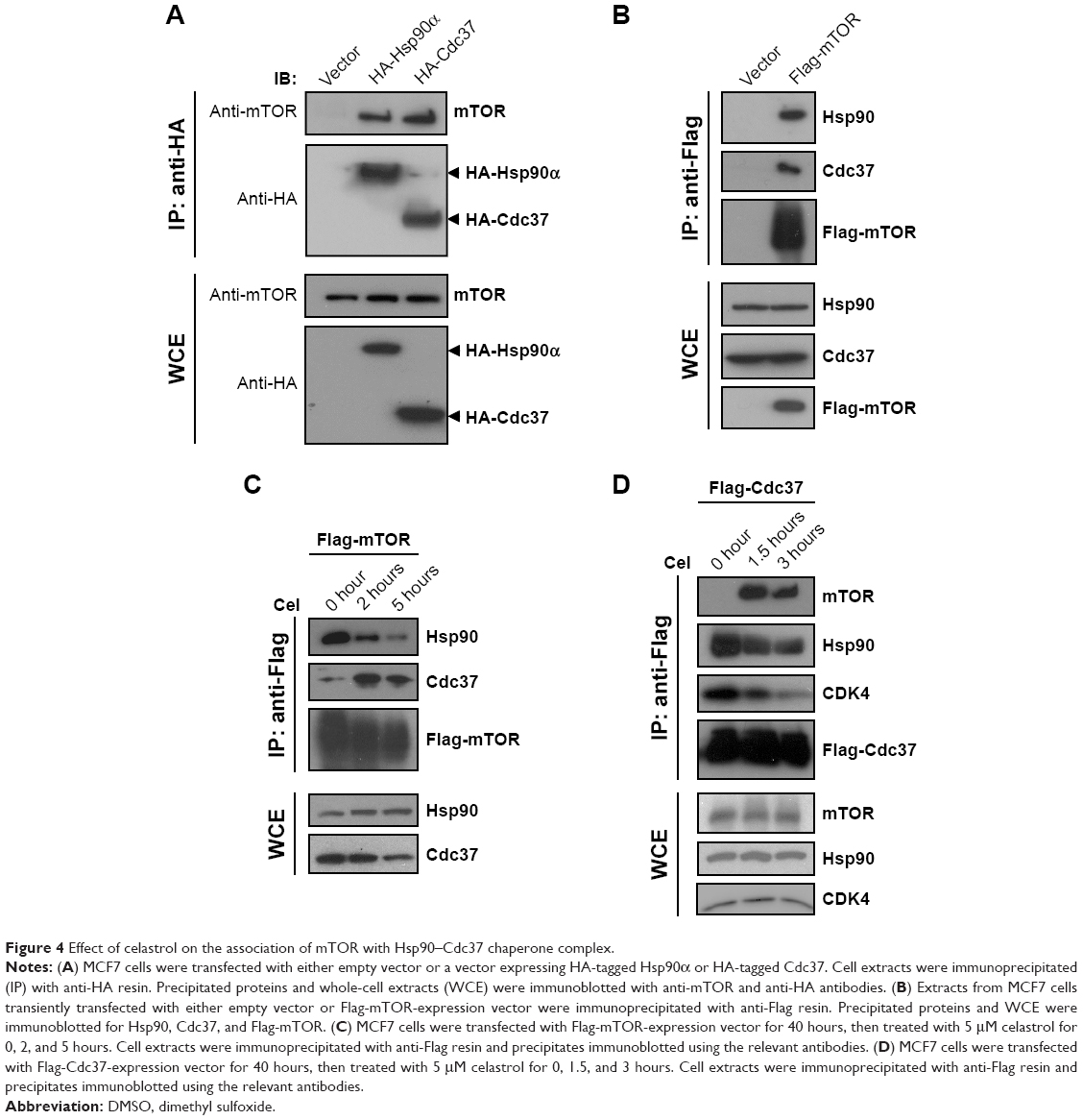 | Figure 4 Effect of celastrol on the association of mTOR with Hsp90–Cdc37 chaperone complex. |
We then examined the effect of celastrol on the interaction between mTOR and the Hsp90–Cdc37 chaperone complex. Celastrol caused a decrease in the interaction between Cdc37 and Hsp90 (Figure 4C and D), which is consistent with previous reports.25–27 Celastrol disrupted the interaction between mTOR and Hsp90 while promoting mTOR association with Cdc37 (Figure 4C and D). Since it was unexpected that celastrol would promote the binding of mTOR to Cdc37, we also examined the effect of celastrol on the interaction between CDK4 and the Hsp90–Cdc37 chaperone complex. In contrast, CDK4, a well-known client of the Hsp90–Cdc37 chaperone complex,28,29 dissociated from both Hsp90 and Cdc37 upon celastrol treatment (Figure 4D), implying that celastrol-induced mTOR association with Cdc37 is kinase-specific.
To examine the effects of Cdc37 on mTOR expression upon celastrol treatment, we knocked down Cdc37 using siRNA in MCF7 cells and then treated the cells with celastrol. As shown in Figure S1 data, knockdown of Cdc37 slightly but significantly attenuated celastrol-induced mTOR downregulation, suggesting that increased interaction with Cdc37 upon celastrol treatment may help promote mTOR degradation.
Discussion
Around 60% of the human kinome interacts with the Hsp90–Cdc37 chaperone complex.30 Cdc37 is an Hsp90 cochaperone that facilitates the transfer of kinase clients to Hsp90 and regulates activity and stability of client kinases.20,21,31 It is established that Hsp90 interacts with mTOR,22,23 an atypical PI3K-family kinase. Surprisingly, there have been no reports on the interaction between mTOR and Cdc37.24 In the current study, we confirmed that mTOR interacted with both Hsp90 and Cdc37, indicating that mTOR is indeed a client of the Hsp90–Cdc37 chaperone complex. Numerous studies have shown that celastrol inhibits Hsp90 activity.10,26,32 Of note, celastrol is different from typical Hsp90 inhibitors (eg, geldanamycin or its derivatives) that inhibit Hsp90 function by binding with high affinity to the ADP–ATP binding pocket of Hsp90.33 Celastrol inhibits Hsp90 ATPase activity without blocking ATP binding and disrupts Hsp90–Cdc37 complex formation.25,26 Unexpectedly, we found that celastrol promoted mTOR binding to Cdc37 while disrupting Hsp90 interactions with mTOR and Cdc37. Since depletion of Cdc37 attenuated celastrol-induced mTOR downregulation, increasing interaction with Cdc37 might promote mTOR degradation. It is not surprising that Cdc37 alone was not able to compensate for loss of Hsp90 function, because emerging evidence suggests that Cdc37 alone in the absence of Hsp90 does not stabilize kinases.30 Interestingly, we observed that knockdown of Cdc37 significantly increased mTOR phosphorylation (Figure S1), implying that increased interaction with Cdc37 upon celastrol treatment may help lock mTOR in kinase-inactivity status. This idea is favored by the fact that celastrol inhibits mTOR phosphorylation at Ser2481 and Akt phosphorylation at Ser473.
In the current study, we found that celastrol induced mTOR ubiquitination, resulting in its proteasomal degradation. Since celastrol has been reported to inhibit the chymotrypsin-like activity of the 20S proteasome,34–36 increased mTOR ubiquitination by celastrol could simply have resulted from inhibition of proteasome activity. To rule out this possibility, we showed that celastrol still robustly induced mTOR ubiquitination after pretreatment with a high concentration of the proteasomal inhibitor MG132, which blocks proteasome activity. Together with the results showing that celastrol downregulated mTOR-expression levels and this effect being attenuated by MG132, our study suggests that celastrol induces ubiquitin-dependent degradation of mTOR. As mentioned, celastrol also has antiproteasome activity, so it is quite possible that proteasomal degradation of mTOR is partially inhibited by celastrol, such that the extent of mTOR reduction upon celastrol treatment may be determined by the balance between celastrol induction of mTOR ubiquitin-dependent degradation and celastrol inhibition of proteasomal activity.
Celastrol has attracted great interest recently, because it is a natural compound and has anti-inflammatory and anticancer activities.10,11,37 In addition, celastrol potentiates the effects of various chemotherapeutic agents, including radiation-induced suppression of melanoma proliferation, oral squamous-cell carcinoma, and prostate cancer.11,38
Our study reveals that celastrol can suppress mTOR signaling through inhibiting mTOR phosphorylation and inducing its ubiquitin-dependent degradation. Since mTOR signaling is frequently dysregulated in cancer, mTOR is a promising therapeutic target for cancer treatment. Rapamycin, an mTOR-specific inhibitor mainly inhibiting mTORC1 activity through interference in mTOR-complex assembly by binding to FK506-binding protein 12, is a US Food and Drug Administration-approved drug for the treatment of certain cancers, most significantly renal cancers. Given that rapamycin efficacy varies by dose in different tumors39,40 and that the currently available mTOR inhibitors used in the clinic can cause severe adverse effects, new mTOR inhibitors are still in high demand. Celastrol is a natural compound extracted from the thunder god vine that has been used in traditional Chinese medicine to treat chronic inflammatory and autoimmune diseases for hundreds of years. Celastrol could be an alternative drug to target mTOR, and may have fewer adverse effects. In addition, some tumor cells that are insensitive to rapamycin might be sensitive to celastrol. As sustained mTOR signaling is often involved in drug resistance in many tumors, including endocrine resistance in breast cancer,8,9,41 we propose the possibility of a combination of endocrine therapy and celastrol as a therapeutic strategy in the treatment of endocrine-resistant breast cancer.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant 81472483). We thank Dr Len Neckers (National Institutes of Health, USA) for providing pcDNA-HA-Hsp90α and pcDNA-HA-Cdc37 plasmids.
Author contributions
WZ conceived the study, designed the experiments, and wrote the manuscript, XL designed the experiments, performed the data analyses, and wrote the manuscript, GZ, XY, DZ, and LL performed the experiments, NW contributed reagents and materials, and RH, QK, and JC helped perform the analysis with constructive discussions. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Lauring J, Park BH, Wolff AC. The phosphoinositide-3-kinase-Akt-mTOR pathway as a therapeutic target in breast cancer. J Natl Compr Canc Netw. 2013;11(6):670–678. | ||
Kim LC, Cook RS, Chen J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene. 2017;36(16):2191–2201. | ||
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. | ||
Owonikoko TK. Inhibitors of mTOR pathway for cancer therapy, moving on from rapalogs to TORKinibs. Cancer. 2015;121(19):3390–3392. | ||
Xie J, Wang X, Proud CG. mTOR inhibitors in cancer therapy. F1000 Res. 2016;5:2078. | ||
Basho RK, Gilcrease M, Murthy RK, et al. Targeting the PI3K/AKT/mTOR pathway for the treatment of mesenchymal triple-negative breast cancer: evidence from a phase 1 trial of mTOR inhibition in combination with liposomal doxorubicin and bevacizumab. JAMA Oncol. 2017;3(4):509–515. | ||
Gordon MA, D’Amato NC, Gu H, et al. Synergy between androgen receptor antagonism and inhibition of mtor and her2 in breast cancer. Mol Cancer Ther. 2017;16(7):1389–1400. | ||
Baselga J, Campone M, Piccart M, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366(6):520–529. | ||
Gnant M, Greil R, Hubalek M, Steger G. Everolimus in postmenopausal, hormone receptor-positive advanced breast cancer: summary and results of an austrian expert panel discussion. Breast Care (Basel). 2013;8(4):293–299. | ||
Salminen A, Lehtonen M, Paimela T, Kaarniranta K. Celastrol: Molecular targets of thunder god vine. Biochem Biophys Res Commun. 2010;394(3):439–442. | ||
Kannaiyan R, Shanmugam MK, Sethi G. Molecular targets of celastrol derived from Thunder of God Vine: potential role in the treatment of inflammatory disorders and cancer. Cancer Lett. 2011;303(1):9–20. | ||
Tozawa K, Sagawa M, Kizaki M. Quinone methide tripterine, celastrol, induces apoptosis in human myeloma cells via NF-κB pathway. Int J Oncol. 2011;39(5):1117–1122. | ||
Pang X, Yi Z, Zhang J, et al. Celastrol suppresses angiogenesis-mediated tumor growth through inhibition of AKT/mammalian target of rapamycin pathway. Cancer Res. 2010;70(5):1951–1959. | ||
Lee JH, Won YS, Park KH, et al. Celastrol inhibits growth and induces apoptotic cell death in melanoma cells via the activation ROS-dependent mitochondrial pathway and the suppression of PI3K/AKT signaling. Apoptosis. 2012;17(12):1275–1286. | ||
Shrivastava S, Jeengar MK, Reddy VS, Reddy GB, Naidu VG. Anticancer effect of celastrol on human triple negative breast cancer: possible involvement of oxidative stress, mitochondrial dysfunction, apoptosis and PI3K/Akt pathways. Exp Mol Pathol. 2015;98(3):313–327. | ||
Zhou W, Fong MY, Min Y, et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell. 2014;25(4):501–515. | ||
Copp J, Manning G, Hunter T. TORC-specific phosphorylation of mammalian target of rapamycin (mTOR): phospho-Ser2481 is a marker for intact mTOR signaling complex 2. Cancer Res. 2009;69(5):1821–1827. | ||
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–1101. | ||
Schopf FH, Biebl MM, Buchner J. The HSP90 chaperone machinery. Nat Rev Mol Cell Biol. 2017;18(6):345–360. | ||
Wang Y, Xu W, Zhou D, Neckers L, Chen S. Coordinated regulation of serum- and glucocorticoid-inducible kinase 3 by a C-terminal hydrophobic motif and Hsp90-Cdc37 chaperone complex. J Biol Chem. 2014;289(8):4815–4826. | ||
Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J Biol Chem. 2002;277(42):39858–39866. | ||
Taipale M, Krykbaeva I, Koeva M, et al. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell. 2012;150(5):987–1001. | ||
Moulick K, Ahn JH, Zong H, et al. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat Chem Biol. 2011;7(11):818–826. | ||
Department of Cell Biology University of Geneva. Didier Picard’s Laboratory website on the list of the reported Cdc37-interacting proteins. Available from: https://www.picard.ch/downloads/Cdc37interactors.pdf | ||
Zhang T, Li Y, Yu Y, Zou P, Jiang Y, Sun D. Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J Biol Chem. 2009;284(51):35381–35389. | ||
Zhang T, Hamza A, Cao X, et al. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol Cancer Ther. 2008;7(1):162–170. | ||
Sreeramulu S, Gande SL, Göbel M, Schwalbe H. Molecular mechanism of inhibition of the human protein complex Hsp90-Cdc37, a kinome chaperone-cochaperone, by triterpene celastrol. Angew Chem Int Ed Engl. 2009;48(32):5853–5855. | ||
Verba KA, Wang RY, Arakawa A, et al. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science. 2016;352(6293):1542–1547. | ||
Dai K, Kobayashi R, Beach D. Physical interaction of mammalian CDC37 with CDK4. J Biol Chem. 1996;271(36):22030–22034. | ||
Verba KA, Agard DA. How Hsp90 and Cdc37 Lubricate Kinase Molecular Switches. Trends Biochem Sci. 2017;42(10):799–811. | ||
Keramisanou D, Aboalroub A, Zhang Z, et al. Molecular Mechanism of Protein Kinase Recognition and Sorting by the Hsp90 Kinome-Specific Cochaperone Cdc37. Mol Cell. 2016;62(2):260–271. | ||
Chadli A, Felts SJ, Wang Q, et al. Celastrol inhibits Hsp90 chaperoning of steroid receptors by inducing fibrillization of the Co-chaperone p23. J Biol Chem. 2010;285(6):4224–4231. | ||
Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell. 1997;89(2):239–250. | ||
Yang H, Chen D, Cui QC, Yuan X, Dou QP. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006;66(9):4758–4765. | ||
Walcott SE, Heikkila JJ. Celastrol can inhibit proteasome activity and upregulate the expression of heat shock protein genes, hsp30 and hsp70, in Xenopus laevis A6 cells. Comp Biochem Physiol A Mol Integr Physiol. 2010;156(2):285–293. | ||
Boridy S, Le PU, Petrecca K, Maysinger D. Celastrol targets proteostasis and acts synergistically with a heat-shock protein 90 inhibitor to kill human glioblastoma cells. Cell Death Dis. 2014;5:e1216. | ||
Kashyap D, Sharma A, Tuli HS, Sak K, Mukherjee T, Bishayee A. Molecular targets of celastrol in cancer: Recent trends and advancements. Crit Rev Oncol Hematol. 2018;128:70–81. | ||
Liu Z, Ma L, Zhou GB. The main anticancer bullets of the Chinese medicinal herb, thunder god vine. Molecules. 2011;16(6):5283–5297. | ||
Mukhopadhyay S, Frias MA, Chatterjee A, Yellen P, Foster DA. The enigma of rapamycin dosage. Mol Cancer Ther. 2016;15(3):347–353. | ||
Mukhopadhyay S, Chatterjee A, Kogan D, Patel D, Foster DA. 5-Aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR) enhances the efficacy of rapamycin in human cancer cells. Cell Cycle. 2015;14(20):3331–3339. | ||
Ilagan E, Manning BD. Emerging role of mTOR in the response to cancer therapeutics. Trends Cancer. 2016;2(5):241–251. |
Supplementary material
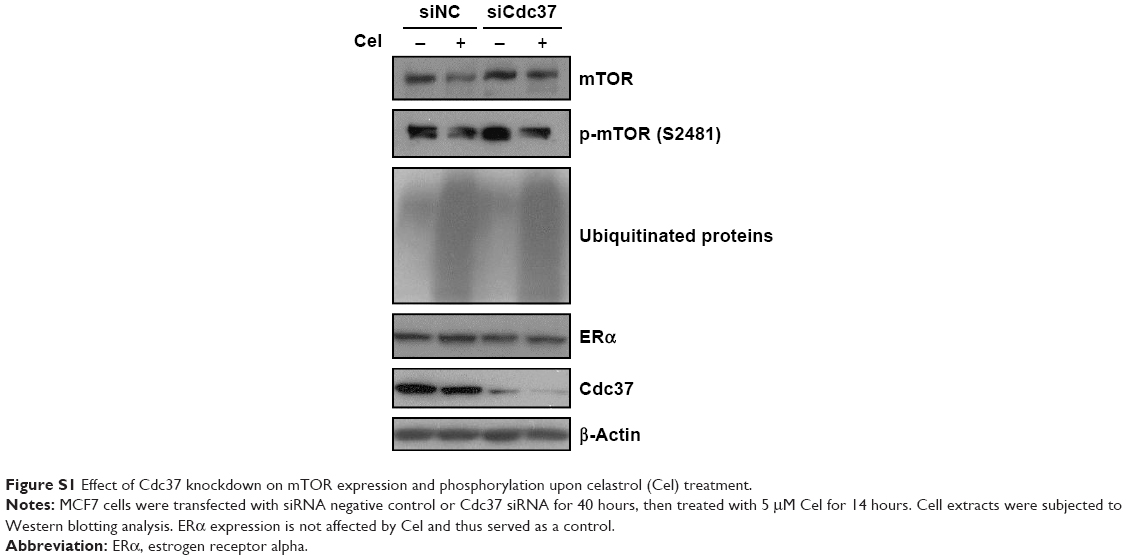 | Figure S1 Effect of Cdc37 knockdown on mTOR expression and phosphorylation upon celastrol (Cel) treatment. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.