")
Back to Journals » OncoTargets and Therapy » Volume 10
CDKL1 promotes tumor proliferation and invasion in colorectal cancer
Authors Qin C, Ren L, Ji M, Lv S, Wei Y , Zhu D, Lin Q, Xu P, Chang W, Xu J
Received 23 January 2017
Accepted for publication 23 February 2017
Published 16 March 2017 Volume 2017:10 Pages 1613—1624
DOI https://doi.org/10.2147/OTT.S133014
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
Chunzhi Qin,1,* Li Ren,1,* Meiling Ji,1,* Shixu Lv,2,* Ye Wei,1 Dexiang Zhu,1 Qi Lin,1 Pingping Xu,1 Wenju Chang,1 Jianmin Xu1
1Department of General Surgery, Zhongshan Hospital, Fudan University, Shanghai, 2Department of Surgical Oncology, First Affiliated Hospital of Wenzhou Medical University, Wenzhou, People’s Republic of China
*These authors contributed equally to this work
Background: CDKL1 is a member of the cell division cycle 2 (CDC2)-related serine threonine protein kinase family and is overexpressed in malignant tumors such as melanoma, breast cancer, and gastric cancer.
Objective: This study aimed to evaluate whether CDKL1 can serve as a potential molecular target for colorectal cancer therapy.
Materials and methods: Expression of CDKL1 in colorectal cancer tissues and cell lines was measured by immunohistochemistry and Western blot, respectively. To investigate the role of CDKL1 in colorectal cancer, CDKL1-small hairpin RNA-expressing lentivirus was constructed and infected into HCT116 and Caco2 cells. The effects of RNA interference (RNAi)-mediated CDKL1 downregulation on cell proliferation and invasion were assessed by CCK-8, colony formation, transwell, and tumorigenicity assays in nude mice. The effects of CDKL1 downregulation on cell cycle and apoptosis were analyzed by flow cytometry. Furthermore, microarray method and data analysis elucidated the molecular mechanisms underlying the phenomenon.
Results: CDKL1 protein was overexpressed in colorectal cancer tissues compared with paired normal tissues. Knockdown of CDKL1 in HCT116 and Caco2 significantly inhibited cell growth, colony formation ability, tumor invasion, and G1–S phase transition of the cell cycle. The knockdown of CDKL1 stimulated the upregulation of p15 and retinoblastoma protein.
Conclusion: CDKL1 plays a vital role in tumor proliferation and invasion in colorectal cancer in vitro and in vivo and, thus, may be considered as a valuable target for therapeutic intervention.
Keywords: CDKL1, cell cycle, protein kinases, colorectal cancer
Introduction
Colorectal cancer (CRC) is one of the most common malignancies worldwide and the leading cause of cancer-related deaths.1 With current therapeutic approaches, the prognosis of CRC is yet dismal. The pathogenesis of CRC is complex and occurs through the successive aberrant expression of tumor suppressor genes or oncogenes in the adenoma–carcinoma sequence.2 Throughout the process, the most important genes for CRC development are p53, RAS, and APC.2 Thus, identifying these biomarkers, defining them, and elaborating their functions will be the key to curing the disease.
The cyclin-dependent kinase-like 1 (CDKL1) gene, also known as P42 or KKIALRE, located on chromosome 14q21.3, encodes 2 isoforms (with 276 and 358 amino acids) of a protein kinase with a putative cell division cycle 2 (CDC2)-related serine/threonine domain.3,4 The CDKL family consists of 5 members. The mammalian CDKL1 is predominantly expressed in the brain, lungs, kidneys, and ovaries.3,4 Zebrafish CDKL1, sharing a high degree of homology with the mammalian ortholog, plays an essential role in zebrafish development, and interference of its expression results in brain malformation and body axis curvature.5 Human CDKL1 and CDKL5 were reported to play important functions in the pathogenesis of neurodevelopmental disorder.4,6 Recently, CDKL1 has been reported to be upregulated in melanoma and breast and gastric cancers.7–9
Dysregulation of human kinases has frequently been linked to a variety of human malignancies, including cancer. As we know, cyclin-dependent kinases (CDKs) can trigger cell cycle progression, and their dysregulation contributes to unscheduled proliferation during tumor development.10 High nuclear/cytoplasmic ratio of CDK1 expression can predict poor prognosis in CRC patients.11 CDK8 is regarded as an oncogene of CRC that regulates β-catenin activity.12 Moreover, P57/Kip2 as an inhibitor of several cyclin–CDK complexes has been proven to be a tumor suppressor,13 and a variety of medications targeting CDKs have undergone clinical trials for gastrointestinal cancer, breast cancer, and leukemia.14,15 As a mimic of CDKs, whether CDKL1 is involved in the progression of human CRCs is not clear, and although some studies have shown associations between CDKL1 expression and cancers,7–9 the signaling pathway of CDKL1 remains elusive.
In this study, we have found that CDKL1 is overexpressed in CRC and its suppression by RNA interference (RNAi) inhibits cell proliferation and invasion of CRC cells by impeding the cell cycle. Moreover, analysis of gene expression profiling after CDKL1 knockdown found several gene sets putatively involved in CRC progressions, such as transforming growth factor (TGF)-β signaling, RNA transcription, enzyme inhibitor activity, and chromosome maintenance. Furthermore, we confirmed that the P15-Rb (retinoblastoma tumor suppressor protein) pathway is related to CDKL1 knockdown. Taken together, our findings suggest that CDKL1 might be a potent oncogene in CRC and, therefore, may represent a new target for therapeutic intervention.
Materials and methods
Clinical samples and immunohistochemistry (IHC) staining
The investigated specimens (tumors and paired normal tissues) were collected randomly from 100 CRC patients with radical colectomy between January 2008 and December 2009 conducted at the Department of General Surgery, Zhongshan Hospital, People’s Republic of China. IHC was performed as described previously.16 All sections were scored blindly by 2 investigators under a light microscope and recorded. CDKL1 expression was estimated using the criterion specified on The Human Protein Atlas website,17 based on the multiplication of intensity and percentage of the stained cells. The tissue staining was scored as negative (−), slightly positive (+), moderately positive (++), and strongly positive (+++). Ethical approval was obtained from the clinical research ethics committee of Zhongshan Hospital, Fudan University (Shanghai, People’s Republic of China). Written informed consent for the acquisition and use of tissue samples was obtained from all patients.
Cell culture
The CRC cell lines HT29, Caco2, SW480, SW620, RKO, Lovo, and DLD1 were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, People’s Republic of China), and HCT116 was obtained from KeyGen Biotech (Nanjing, People’s Republic of China). The HCT116 and HT29 cells were maintained in McCoy’s 5A medium (KeyGen Biotech); Dulbecco’s Modified Eagle’s Medium (DMEM) (HyClone, Logan, UT, USA) was used for Lovo, SW480, SW620, RKO, and Caco2, whereas DLD1 cell line was maintained in RPMI-1640 (Corning, Manassas, VA, USA). The media were supplemented with 10% fetal bovine serum (FBS; Gibco BRL, Grand Island, NY, USA) in an incubator with 5% CO2 at 37°C.
Small hairpin RNAs (shRNAs), plasmids, and cell transfection
The shRNA target sequence (5′-AGCAAGTGTTTAGCACGAA-3′) for CDKL1 isotypes (NM_004196 and NM_001282236) was designed and constructed. Nonsense sequence (5′-TTCTCCGAACGTGTCACGT-3′) was designed and used as the shRNA control. CDKL1-shRNA and control-shRNA were cloned into GV115 plasmid vectors (GeneChem, Shanghai, People’s Republic of China) containing the green fluorescent protein (GFP) gene. Next, the plasmid vectors were packed into lentivirus particles. When the cells were confluent to 104 cells per well in 6-well plates, lentivirus particles with a multiplicity of infection (MOI) of 20 were added. The stably transfected GFP-expressing cells were detected using fluorescence microscopy (Olympus, Tokyo, Japan), and Western blotting was performed with CDKL1 antibody.
CCK-8 assay
Lentivirus-infected cells in the logarithmic phase were seeded into 96-well plates at the density of 2,000 cells per well (HCT116) or 6,000 cells per well (Caco2), and the plates were incubated in 5% CO2 incubator at 37°C. The cell viability was assessed by the CCK-8 assay at several time points in the next 2 days. Briefly, 10 μL CCK-8 solution was added to the cells and incubated. The optical density (OD) of each well was measured at 450 nm using the CCK-8 plate reader (Epoch™; BioTek, Winooski, VT, USA) according to the manufacturer’s instructions.
Colony formation assay
The cells in the logarithmic phase were inoculated into 6-well plates at densities of 200 and 800 cells per well for HCT116 and Caco2, respectively. The medium was replaced every week. When the cell numbers in most of the single colonies were >50, the cells were fixed with paraformaldehyde (Sangon Biotech, Shanghai, People’s Republic of China) for 30 min, washed with phosphate-buffered saline (PBS), and stained with 0.1% crystal violet (BioSharp, Hefei, People’s Republic of China). After washing the cells with double-distilled water, the clusters were imaged and the number of colonies were counted.
Transwell assay
The assay was performed with precoated cell invasion kit (pore size 8.0 μm; Corning Inc, New York, NY, USA) and Matrigel (250 μg/mL; BD Biosciences, Bedford, MA, USA) in transwell chambers. Cells at the density of 5×104 for HCT116 and 2×105 for Caco2 were then allowed to migrate from the medium chamber to that containing medium with 30% FBS in the lower chambers. The invaded cells were fixed with paraformaldehyde after 60 h of incubation and stained with 0.1% crystal violet. The cells that invaded through the membrane were counted under a light microscope (4 random fields per well).
Flow cytometry analysis
Cell culture supernatants and adherent cells were harvested and washed with PBS by centrifugation at 1,000 rpm. Then, the cells were fixed with 70% ethanol at −20°C for 2 h and stained with propidium iodide (PI) (BD Biosciences, San Jose, CA, USA). After incubation for 15 min at room temperature in the dark, the cells were analyzed for DNA content using flow cytometry (FACS Aria; BD Biosciences). For the determination of apoptotic cell death, the cells were stained with Annexin V–APC (BD Biosciences) and PI, followed by fluorescence-activated cell sorting (FACS) analysis according to the manufacturer’s instructions (BD Pharmingen). Fluorescence compensation was conducted to exclude the overlapping of the emission spectra. A total of 50,000 events were acquired for analysis using ModFit LT 3.2 software (Verity Software House, Topsham, ME, USA).
Microarray and data analysis
In order to detect the potential signaling pathways altered while knocking down CDKL1, we performed a gene expression profiling experiment using GeneChip Hybridization Oven 645, containing 22,000 probe sets (Affymetrix, Santa Clara, CA, USA). Total RNA was prepared from control and RNAi DLD1 cells with TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. The RNA samples were quantified using NanoDrop 2000 (NanoDrop Technologies, Montchanin, DE, USA) and Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). The arrays were performed on GeneChip fluidics 450 workstation (Affymetrix) and GeneChip scanner 3000 (Affymetrix) according to the manufacturer’s instructions. Robust multichip analysis (RMA) was used for normalization to assess the level of background variability between hybridizations. In all the analyses, fold change (|FC|) was set as >2 and P-value as <0.01. Pathway and gene ontology analyses were carried out next. All microarray data, along with the design parameters, have been submitted to National Center for Biotechnology Information (NCBI)’s Gene Expression Omnibus database (GEO accession GSE94887).
Western blot analysis
Harvested cells were lysed, and the total protein was quantitated with the bicinchoninic acid (BCA) kit (Beyotime Biotechnology, Shanghai, People’s Republic of China). An equivalent of 40 μg protein extract was resolved using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. Then, the blots were blocked with 5% milk, followed by incubation with the respective antibodies. The immunoreactive bands were detected using an enhanced chemiluminescence kit (Thermo Fisher) and visualized with the Tanon-4500 Gel Imaging System (Tanon, Shanghai, People’s Republic of China). Tubulin, H3, and LaminB1 were used as internal controls. The antibodies used were as follows: anti-CDKL1 (1:1,000, Abcam, Cambridge, UK); anti-CDK4, anti-CDK6, anti-cyclinD1, and anti-LaminB1 (1:2,000, Proteintech, Rosemont, IL, USA); horseradish peroxidase (HRP)-conjugated anti-mouse and anti-rabbit secondary antibodies (1:10,000, Abbkine, Wuhan, Hubei, People’s Republic of China); and anti-H3, anti-P15, anti-Rb, anti-phospho-Rb (Ser807/811), and anti-Phospho-Rb (Ser780) (1:2,000, Cell Signaling Technology, Danvers, MA, USA).
In vivo tumorigenicity assay
Four-week-old male BALB/c mice were purchased from Shanghai Sippr-BK Laboratory Animal Co Ltd, Shanghai, People’s Republic of China. Six animals per group (4 groups: control and RNAi groups for HCT116 or Caco2 cells) were used in each experiment. Briefly, HCT116 or Caco2 cells (expressing control-shRNA or CDKL1-shRNA) in 100 μL PBS at 1×108 cells/mL were inoculated into the armpit of the mice. The tumors were measured weekly using a Vernier caliper, and the tumor volume was calculated using the formula π/6× length × width2.18 Tumors were collected 4 weeks after inoculation. All studies were approved by the Animal Care Committee of Zhongshan Hospital, Fudan University. All mice were handled according to the Use of Laboratory Animals and the National Institutes of Health Guidelines for Care.
Statistical analysis
CDKL1 expression in human tissues was analyzed by nonparametric analysis using the Wilcoxon test. All data were expressed as mean ± SD of 3 independent experiments, wherein each assay was performed in at least triplicates. The Student’s t-test was used to evaluate the differences between the control cells and CDKL1-knockdown cells using SPSS 17.0 software (IBM Corp., Armonk, NY, USA). P-values (2-sided) <0.05 were considered significant.
Results
CDKL1 is overexpressed in human CRC
IHC was used for the detection of CDKL1 expression in the paraffin-embedded tissue samples of CRC patients (Figure 1). The results showed that CDKL1 protein was overexpressed in CRC tissue as compared to the adjacent normal tissue (P<0.01) (Table 1), primarily in the nucleus and less in the plasma. These results indicate that high levels of CDKL1 are present in CRC. Next, we searched for CRC cell lines that displayed high levels of CDKL1 expression. The expression of CDKL1 protein was confirmed by Western blotting (Figure 2A). Thus, we selected HCT116 and Caco2 cells for subsequent assays.
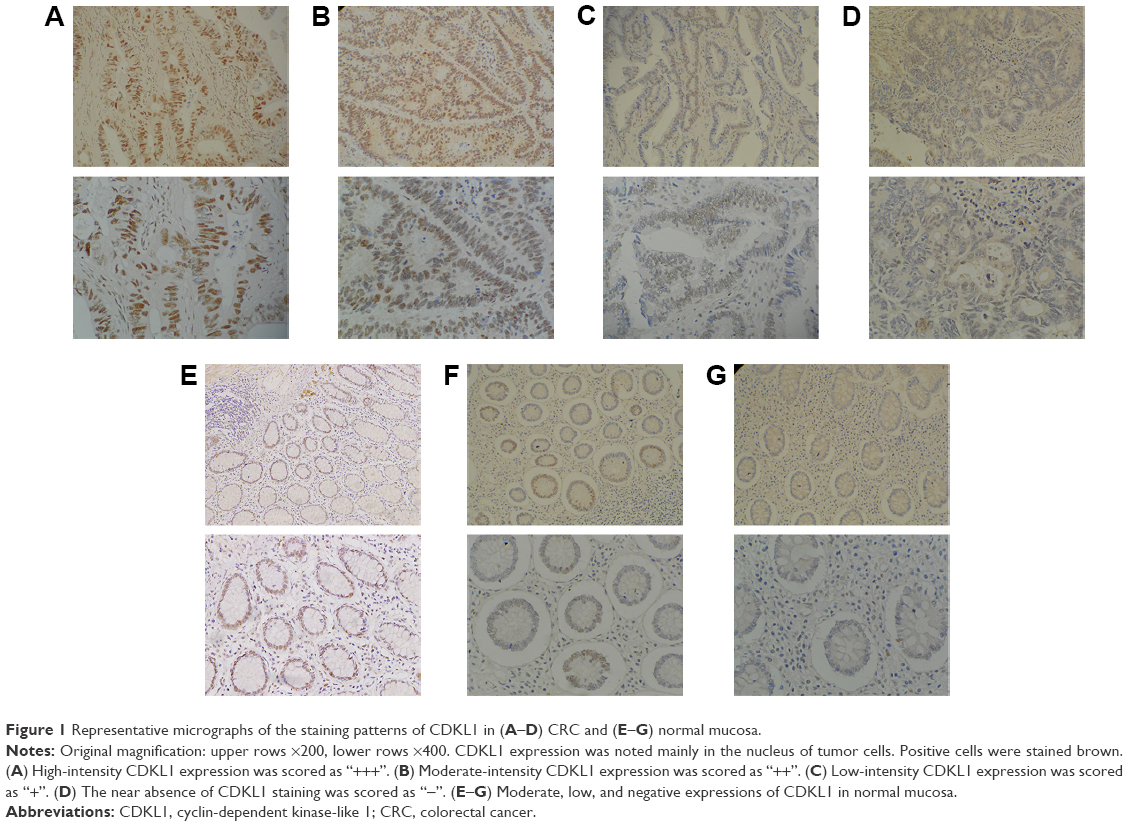 | Figure 1 Representative micrographs of the staining patterns of CDKL1 in (A–D) CRC and (E–G) normal mucosa. |
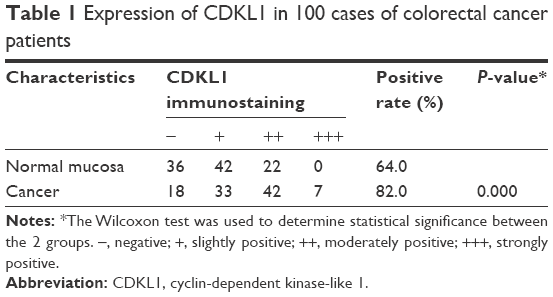 | Table 1 Expression of CDKL1 in 100 cases of colorectal cancer patients |
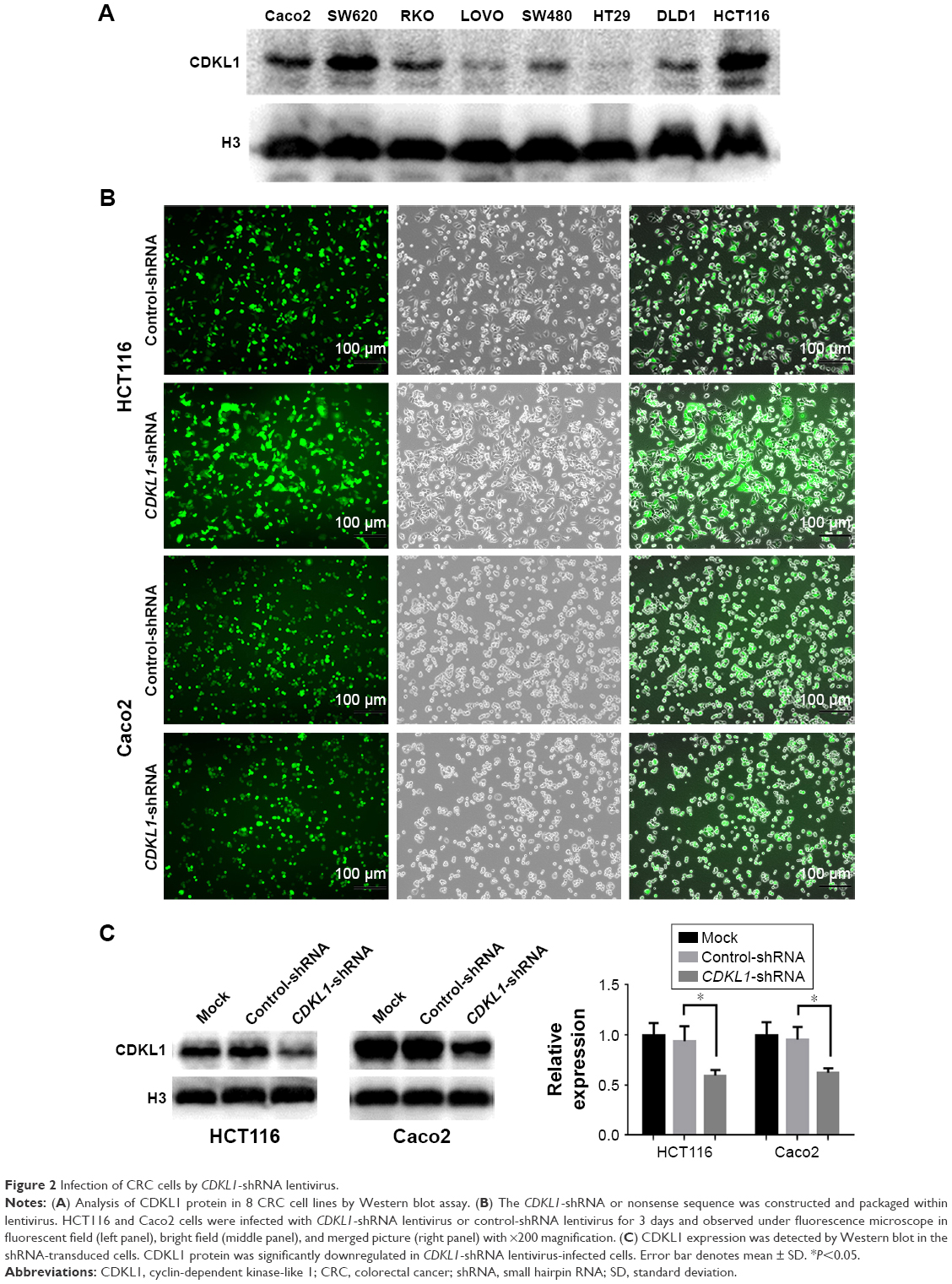 | Figure 2 Infection of CRC cells by CDKL1-shRNA lentivirus. |
CDKL1 knockdown affects proliferation and invasion of CRC cells
As a kinase, CDKL1 overexpression in CRC suggested that it might be required for cancer proliferation. First, we successfully carried out infection of HCT116 and Caco2 with lentivirus, as evident by the fluorescence microscopy results and the protein levels (Figure 2B and C). The CCK-8 assay revealed that the number of viable cells in the CDKL1-shRNA group decreased significantly as compared to the control group (P<0.01, Figure 3A and B). In addition, the colony formation assay was performed to determine the tumorigenesis in vitro. The cells were grown and allowed to form colonies 14 days after infection. Compared to the control group, the number of colonies in HCT116 and Caco2 cells of the CDKL1-shRNA group was decreased significantly (P<0.01, Figure 3C and D). Furthermore, by the transwell assay, we observed that the migratory potential of the cells was decreased significantly when endogenous CDKL1 expression was knocked down by CDKL1-shRNA vectors in both cell lines (Figure 3E and F). Following these results, we conclude a critical role for CDKL1 in the proliferation and migratory potential of CRC cells.
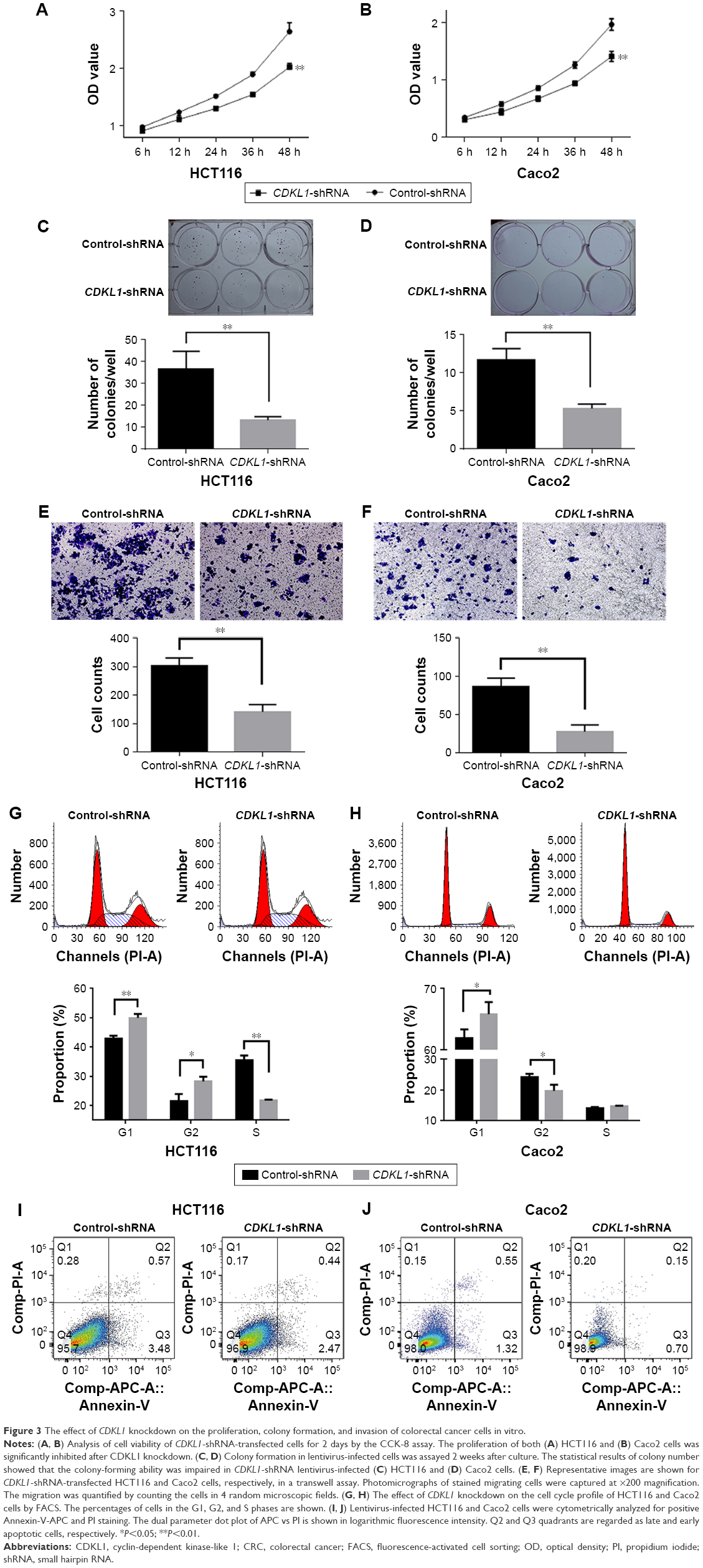 | Figure 3 The effect of CDKL1 knockdown on the proliferation, colony formation, and invasion of colorectal cancer cells in vitro. |
CDKL1 facilitates the transition from G1 to S phase in CRC cells
Considering that CDKL1 is related to CDKs, we examined the effect of CDKL1 silencing on cell cycle regulation in CRC cell lines. The cell cycle profiling by FACS indicated that silencing of CDKL1 expression with shRNA resulted in an increased number of cells in the G1 phase in HCT116 and Caco2 cells (Figure 3G and H). However, no consistency was observed in the distribution of G2 and S stages of the 2 cell lines. These results suggest that CDKL1 promotes the proliferation of CRC cells by facilitating the transition from G1 to S phase. Moreover, the apoptosis assay revealed that CDKL1-shRNA lentivirus exerts little influence on apoptosis in both cell lines (Figure 3I and J).
CDKL1 knockdown inhibits colon tumorigenesis in vivo
To examine the effects of the silencing of CDKL1 on tumor growth in vivo, CRC cells that stably expressed control-shRNA or CDKL1-shRNA were inoculated into the armpit of BALB/c mice. All the animals developed tumors 7 days after inoculation. Moreover, 28 days after inoculation, the tumors of the control-shRNA animals were significantly larger than those of the CDKL1-shRNA group (Figure 4A and B). The mean tumor volume was 725.53±200.02 mm3 in mice inoculated with HCT116 cells that expressed control-shRNA as compared to 149.44±155.10 mm3 in the CDKL1-shRNA group (Figure 4C). On the other hand, the mean tumor volume in the Caco2 group was 1,200.33±436.99 mm3 (control-shRNA) as compared to 307.73±76.83 mm3 (CDKL1-shRNA) (Figure 4D). These data demonstrate the important role of CDKL1 in promoting the growth of colon cancer cells.
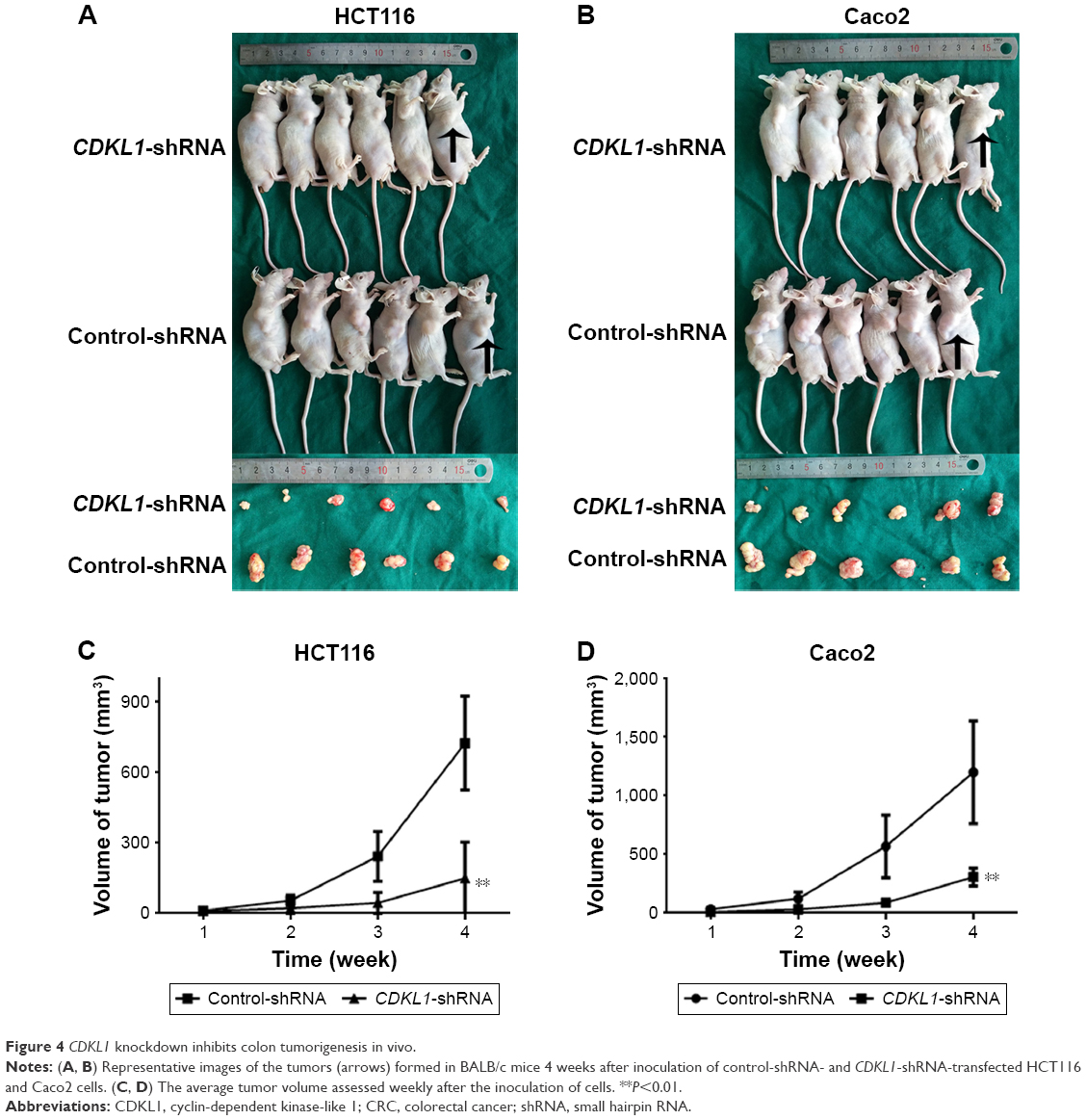 | Figure 4 CDKL1 knockdown inhibits colon tumorigenesis in vivo. |
Multiple signaling proteins are activated by CDKL1-shRNA vector
Complementary DNA (cDNA) microarray identified that expression of 32 genes was upregulated while 21 genes were downregulated under the condition of |FC| >2 and P-value <0.01 (Figure 5A–C). Further analysis found that the gene sets involved in CDKL1 knockdown are components of TGF-β receptor signaling, cellular defense response, secretory pathway, RNA polymerase II transcription factor activity, enzyme inhibitor activity, chromosomes, membrane fractions, Toll pathway, pathogenic Escherichia coli infection, and spliceosomes. (Table 2) Next, Western blotting assay verified that knockdown of CDKL1 yielded a variably increased protein level of P15 (CDKN2B). Furthermore, examining the P15 cell cycle pathway molecules such as CDK4, CDK6, cyclinD1, Rb, and phosphorylated Rb revealed that total Rb was significantly upregulated after CDKL1 knockdown in both cell lines irrespective of CDK4/6 status, and cyclinD1 was upregulated in Caco2 (Figure 5D). However, the 2 cells shared no consistency on specific phosphorylation sites of Rb, such as Ser780 and Ser807/811.
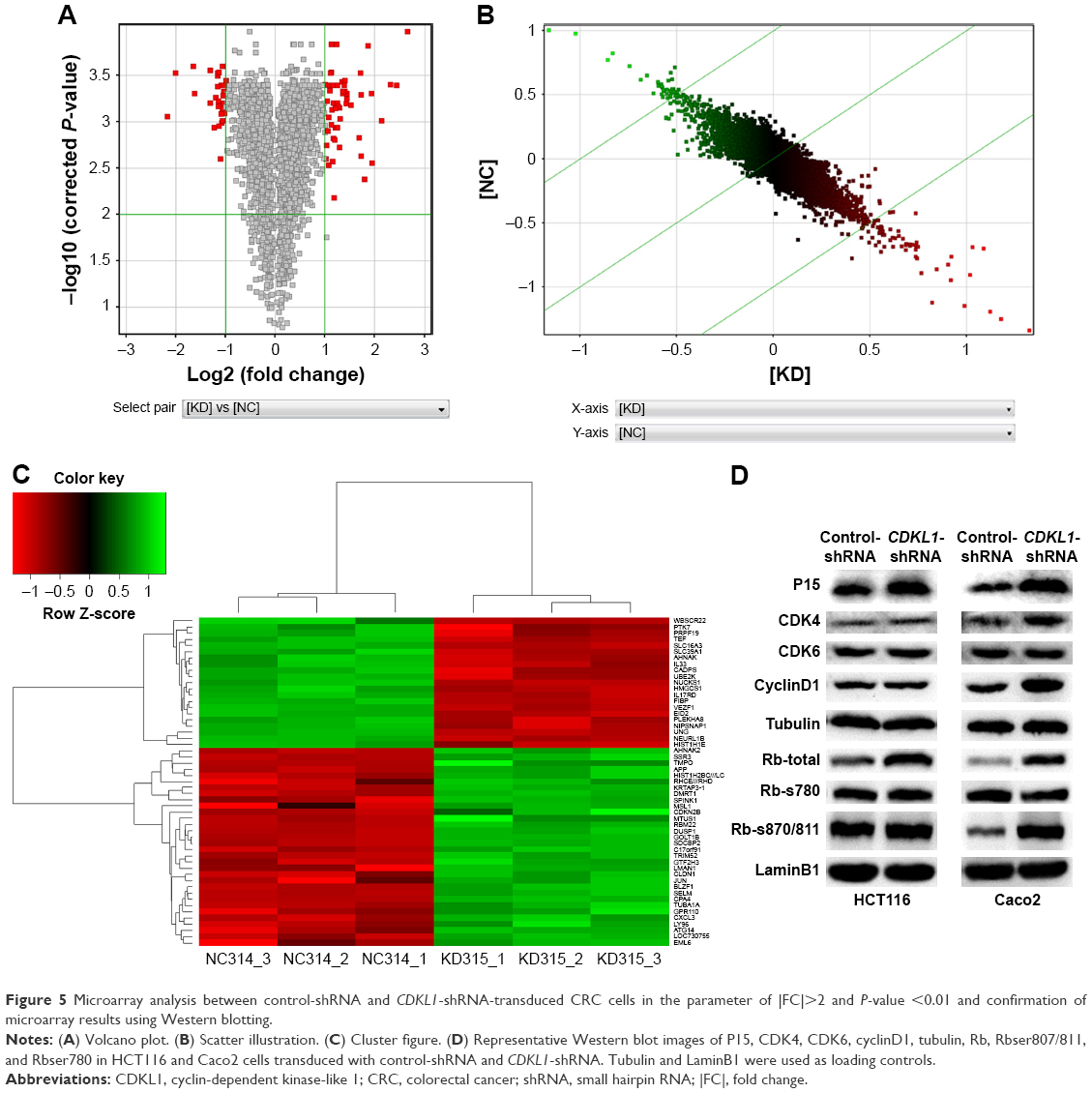 | Figure 5 Microarray analysis between control-shRNA and CDKL1-shRNA-transduced CRC cells in the parameter of |FC|>2 and P-value <0.01 and confirmation of microarray results using Western blotting. |
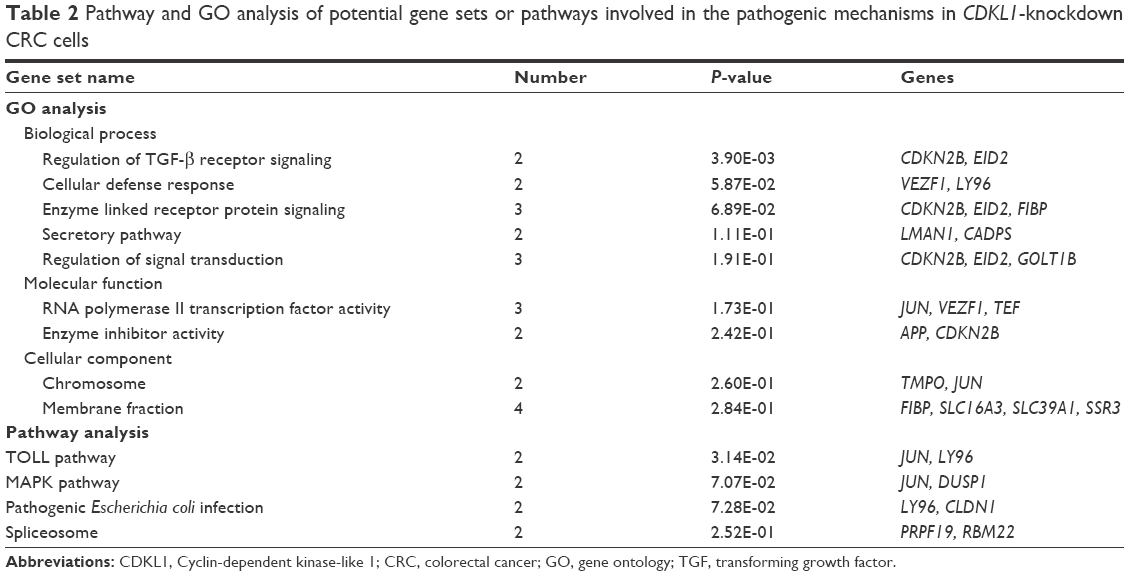 | Table 2 Pathway and GO analysis of potential gene sets or pathways involved in the pathogenic mechanisms in CDKL1-knockdown CRC cells |
Discussion
In the current study, the expression of CDKL1 protein increased significantly in tumor tissues as compared to the paired normal mucosa in CRC patients. It indicated that the high expression of CDKL1 might be correlated with CRC. Thus, to further explore the molecular function of CDKL1, we chose RNAi to disrupt the gene in human CRC cells and investigate the functional consequences. We found that the knockdown of CDKL1 led to inhibited proliferation, impaired cloning, lowered invasion ability, and stalled G1–S transition. Our study identified that CDKL1 may be a potential driver gene in CRC.
Human cells harbor a large protein family composed of protein kinases, most of which are poorly studied, except CDKs.19 CDKs modulate the progression of the cell cycle by its sequential activation and inactivation.10,12 They are critical in regulating metabolism, cell division, and apoptosis by phosphorylation of diverse protein substrates. Their dysregulations directly or indirectly mediate unscheduled proliferation of most tumor cells. Human cells contain multiple CDKs, including CDK1, CDK2, and CDK4/6, several regulatory cyclins, as well as CDK inhibitors.20,21 For instance, CDK4/6 associates with cyclinD to regulate the cell cycle progression in the G1 phase; the complex senses cell mitogenic signals and prompts the cells to initiate DNA synthesis.10,14,20,22 The deregulation of these complexes has been implicated in a variety of cancers. Currently, in clinical practice, palbociclib is an orally administered cell cycle inhibitor, highly specific against CDK4/6.23 It can cause G1 cell cycle arrest and produce marked tumor regression in mice bearing human colon carcinoma.24 CDKL1 shares the same functional domains structurally as the CDKs, such as the cdc2 sequence features.3,5 The phylogenetic conservation of this molecule underscores their potential physiological similarity. Herein, we revealed that CDKL1 depletion led to an obvious arrest at the G0/G1 phase of CRC. Moreover, FACS results excluded the possibility that CDKL1 knockdown impedes CRC cell growth through apoptosis.
To maintain G1 regulation, the activity of the CDK4/cyclinD complex is antagonized by CDK inhibitors such as the INK4 family, including INK4A, INK4B, INK4C, and INK4D, and the Cip and Kip family, composed of p21, p27, and p57.10,13,14 These cell cycle inhibitors have been shown to block proliferation of adult stem cells in multiple tissues. Among them, INK4B (P15) specifically inhibits the catalytic subunits of CDK4/6 involved in the regulation of G1–S phase transition.10,25 It is capable of inducing cell cycle arrest in the G1 phase and has been identified as a critical tumor suppressor.26,27 Thus, we decided to lay emphasis on P15 based on our microarray results after CDKL1 knockdown. Furthermore, we demonstrated that depletion of CDKL1 notably upregulated the protein expression of P15. P15 is shown to be a target of CDKL1 in CRC, either direct or indirect. However, further studies are essential for finding and verifying the partner or direct inhibitor of CDKL1. We also proved that as a downstream molecule of the P15 pathway, Rb is inhibited after CDKL1 knockdown. Rb is a well-known tumor suppressor in cell cycle regulation.28 Phosphorylation of Rb negatively regulates the cell cycle through E2F repression.29–31 However, Rb contains 13 conserved sites that are phosphorylated by the CDK–P15 complex in cycling cells and phosphorylation will cause site-specific and diverse conformational changes in the complex.32,33 Further studies need to be conducted to decipher the phosphorylated site of Rb related to CDKL1 knockdown. We found that the downstream molecules of P15 pathway such as cyclinD1 were inconsistent among HCT116 and Caco2, which may explain the different cell cycle distribution (S and G2 phases) between the 2 cell lines; however, further investigation is imperative. The MAPK pathway regulates diverse cellular functions including cell migration and is dysregulated in a variety of cancers.34 Our microarray results showed that DUSP1 and JUN, as 2 important molecules of the pathway, were dysregulated; this may lead to the attenuation of the invasion of CRC cells after CDKL1 knockdown. In addition, in the literature, there are no data relating CDKL1 with tumor invasion, so further experiments are required.
Conclusion
CDKL1 is a molecule of the complex cell cycle regulation network. Our study proves that CDKL1 expression contributes to CRC progression. The underlying mechanisms of CDKL1 activity and its substrates may lead to its use as a potential therapeutic target for CRC patients.
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. | ||
Meyerson M, Enders GH, Wu CL, et al. A family of human cdc2-related protein kinases. EMBO J. 1992;11(8):2909–2917. | ||
Yen SH, Kenessey A, Lee SC, Dickson DW. The distribution and biochemical properties of a Cdc2-related kinase, KKIALRE, in normal and Alzheimer brains. J Neurochem. 1995;65(6):2577–2584. | ||
Hsu LS, Liang CJ, Tseng CY, Yeh CW, Tsai JN. Zebrafish cyclin-dependent protein kinase-like 1 (zcdkl1): identification and functional characterization. Int J Mol Sci. 2011;12(6):3606–3617. | ||
Tao J, Van Esch H, Hagedorn-Greiwe M, et al. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am J Hum Genet. 2004;75(6):1149–1154. | ||
Tang L, Gao Y, Yan F, Tang J. Evaluation of cyclin-dependent kinase-like 1 expression in breast cancer tissues and its regulation in cancer cell growth. Cancer Biother Radiopharm. 2012;27(6):392–398. | ||
Sun W, Yao L, Jiang B, Shao H, Zhao Y, Wang Q. A role for Cdkl1 in the development of gastric cancer. Acta Oncol. 2012;51(6):790–796. | ||
Song Z, Lin J, Sun Z, Ni J, Sha Y. RNAi-mediated downregulation of CDKL1 inhibits growth and colony-formation ability, promotes apoptosis of human melanoma cells. J Dermatol Sci. 2015;79(1):57–63. | ||
Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9(3):153–166. | ||
Sung WW, Lin YM, Wu PR, et al. High nuclear/cytoplasmic ratio of Cdk1 expression predicts poor prognosis in colorectal cancer patients. BMC Cancer. 2014;14:951. | ||
Firestein R, Bass AJ, Kim SY, et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature. 2008;455(7212):547–551. | ||
Vlachos P, Joseph B. The Cdk inhibitor p57(Kip2) controls LIM-kinase 1 activity and regulates actin cytoskeleton dynamics. Oncogene. 2009;28(47):4175–4188. | ||
Mikhail S, Albanese C, Pishvaian MJ. Cyclin-dependent kinase inhibitors and the treatment of gastrointestinal cancers. Am J Pathol. 2015;185(5):1185–1197. | ||
Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14(2):130–146. | ||
Gao XH, Liu QZ, Chang W, et al. Expression of ZNF148 in different developing stages of colorectal cancer and its prognostic value: a large Chinese study based on tissue microarray. Cancer. 2013;119(12):2212–2222. | ||
Knut and Alice Wallenberg Foundation [webpage on the Internet]. Assays and Annotation. The Human Protein Atlas. Available from: http://www.proteinatlas.org/about/assays+annotation. Accessed January 16, 2015. | ||
Chen Y, Shi L, Zhang L, et al. The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. J Biol Chem. 2008;283(26):17969–17978. | ||
Varjosalo M, Keskitalo S, Van Drogen A, et al. The protein interaction landscape of the human CMGC kinase group. Cell Rep. 2013;3(4):1306–1320. | ||
Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014;15(6):122. | ||
Poole AJ, Heap D, Carroll RE, Tyner AL. Tumor suppressor functions for the Cdk inhibitor p21 in the mouse colon. Oncogene. 2004;23(49):8128–8134. | ||
Bendris N, Lemmers B, Blanchard JM. Cell cycle, cytoskeleton dynamics and beyond: the many functions of cyclins and CDK inhibitors. Cell Cycle. 2015;14(12):1786–1798. | ||
DeMichele A, Clark AS, Tan KS, et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: phase II activity, safety, and predictive biomarker assessment. Clin Cancer Res. 2015;21(5):995–1001. | ||
Fry DW, Harvey PJ, Keller PR, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3(11):1427–1438. | ||
Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432(7015):316–323. | ||
Rosu-Myles M, Taylor BJ, Wolff L. Loss of the tumor suppressor p15Ink4b enhances myeloid progenitor formation from common myeloid progenitors. Exp Hematol. 2007;35(3):394–406. | ||
Wolff L, Bies J. p15Ink4b Functions in determining hematopoietic cell fates: implications for its role as a tumor suppressor. Blood Cells Mol Dis. 2013;50(4):227–231. | ||
Cobrinik D. Pocket proteins and cell cycle control. Oncogene. 2005;24(17):2796–2809. | ||
Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8(9):671–682. | ||
Burke JR, Deshong AJ, Pelton JG, Rubin SM. Phosphorylation-induced conformational changes in the retinoblastoma protein inhibit E2F transactivation domain binding. J Biol Chem. 2010;285(21):16286–16293. | ||
Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98(6):859–869. | ||
Rubin SM. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem Sci. 2013;38(1):12–19. | ||
Lees JA, Buchkovich KJ, Marshak DR, Anderson CW, Harlow E. The retinoblastoma protein is phosphorylated on multiple sites by human cdc2. EMBO J. 1991;10(13):4279–4290. | ||
Friday BB, Adjei AA. Advances in targeting the Ras/Raf/MEK/Erk mitogen-activated protein kinase cascade with MEK inhibitors for cancer therapy. Clin Cancer Res. 2008;14(2):342–346. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.