")
Back to Journals » ImmunoTargets and Therapy » Volume 4
CD28 co-stimulation in T-cell homeostasis: a recent perspective
Authors Beyersdorf N, Kerkau T, Hünig T
Received 29 January 2015
Accepted for publication 26 March 2015
Published 28 May 2015 Volume 2015:4 Pages 111—122
DOI https://doi.org/10.2147/ITT.S61647
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Michael Shurin
Niklas Beyersdorf, Thomas Kerkau, Thomas Hünig
Institute for Virology and Immunobiology, University of Würzburg, Würzburg, Germany
Abstract: T-cells play a key role within the adaptive immune system mediating cellular immunity and orchestrating the immune response as a whole. Their activation requires not only recognition of antigen/major histocompatibility complexes by the T-cell receptor but in addition co-stimulation via the CD28 molecule through binding to CD80, CD86, or as recently discovered, inducible co-stimulator ligand expressed by antigen-presenting cells. Apart from tight control of the co-stimulatory signal by the T-cell receptor complex, expression of the inhibitory receptor cytotoxic T-lymphocyte antigen-4 (CTLA-4) sharing its ligands with CD28 is required to avoid inappropriate or prolonged T-cell activation. CD4+ Foxp3+ regulatory T (Treg) cells, which are crucial inhibitors of autoimmunity, add another level of complexity in that they differ from conventional non-regulatory CD4+ T-cells by strongly depending on CD28 signaling for their generation and homeostasis. Moreover, CTLA-4 is constitutively expressed by Treg cells where it serves as a key mediator of suppression, while conventional CD4+ T-cells express CTLA-4 only after activation. Here, we discuss recent insights into the molecular events underlying CD28-mediated co-stimulation, its impact on gene regulation, and the differential role of CD28 expression on Treg cells versus conventional CD4+ and CD8+ T-cells. Moreover, we summarize the exciting therapeutic options which have arisen from our current understanding of T-cell co-stimulation. Some of these have already been translated into the clinic, while others are expected to follow soon due to promising preclinical results. In particular, we discuss the failed 2006 trial of the CD28 superagonist TGN1412, and the return of this potent T-cell activator to clinical development.
Keywords: CTLA-4, mAbs, rheumatoid arthritis, Treg cells, CD28 superagonist, TGN1412
Introduction: T-cell responses and the role of CD28 co-stimulation
CD28 is a homodimeric stimulatory cell surface receptor of the Ig superfamily. It is expressed on virtually all T-cells in rodents, and on the vast majority of CD4+ but only about half of circulating human CD8+ T-cells. The control of T-cell responses by CD28 co-stimulation provides a means of preventing unwanted (anti-self) and triggering wanted (antimicrobial) immunity. Thus, antigen-presenting cells (APCs), in particular dendritic cells, are not only uniquely able to capture, transport, and (cross-)present microbial antigens but are also equipped with pattern recognition receptors that provide information about the captured antigen. If this is an infectious agent, CD80 and CD86, the ligands for CD28, the main co-stimulator of primary T-cell responses, are upregulated (Figure 1). If T-cells inspecting the surface of dendritic cells in lymphoid tissues detect major histocompatibility complex molecules loaded with cognate peptide and simultaneously engage CD80/86 with CD28, they are fully activated to proliferate, and under the guidance of additional, cytokine-mediated signals, differentiate into the various types of effector cells; without co-stimulation, they become refractory to further stimulation (a situation called anergy) or even undergo apoptosis. CD28 engagement alone, on the other hand, is without apparent consequence for the T-cells. As we shall see, however, very strong CD28 signals can synergize with the weak tonic signals generated by the process of antigen search itself to trigger T-cell activation. Compared to other, activation-induced, molecules with co-stimulatory properties such as CD27, OX40, and 4-1BB,1 CD28’s constitutive expression even on resting T-cells explains its central and prominent role in T-cell activation.
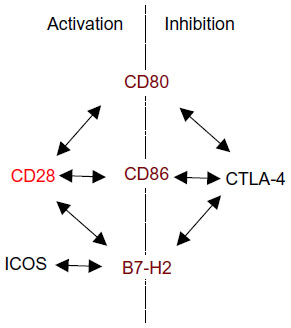 | Figure 1 Ligand sharing by CD28, CTLA-4, and ICOS. |
The power of co-stimulated T-cell responses, and the potential damage they can do if misguided, warrants effective control. Cytotoxic T-lymphocyte antigen-4 (CTLA-4) (CD152), which is constitutively expressed on regulatory T (Treg) cells and upregulated on conventional T-cells after activation, is one key element providing this control (Figure 1). This is illustrated by the catastrophic autoimmune-lymphoproliferative disease experienced by CTLA-4-deficient mice2 and by autoimmunity observed in cancer patients undergoing CTLA-4 blockade.3 The intimate interrelation of CD28-mediated co-stimulation and CTLA-4-mediated inhibition is apparent not only from their use of the same ligands, for which they compete at the surface of APCs (Figure 1), but also from the absence of autoimmunity in mice lacking both, CD28 and CTLA-4.4
Here, we briefly review the structural and functional interactions of CD28, CTLA-4, and their shared ligands, and describe established and emerging therapeutic approaches that manipulate the stimulatory and inhibitory components of this system to either suppress unwanted immunopathology or boost immunity to self- or near-self-antigens, which can be targeted for T-cell-mediated attack on tumor cells.
Molecular mechanisms and signaling pathways
As already mentioned, CD28 induces a co-stimulatory signal in T-cells recognizing cognate antigen/major histocompatibility complexes via their T-cell receptor (TCR). In fact, there are at least three mechanisms to ensure that CD28 is only capable of generating a co-stimulatory signal when the TCR is engaged but does not activate the T-cell by itself.
The first mechanism concerns the binding of CD28 to its ligands CD80 and CD86 on the surface of the APCs. TCR ligation has only recently been shown to induce a conformational change in the CD28 molecule5 (Figure 2B). It was further shown by the same group that this conformational change within the CD28 homodimer allows for bivalent enhanced avidity binding to CD80.6 Without TCR stimulation, CD28 is only capable of binding monovalently, that is, with low affinity (Figure 2A).7 For the inhibitory receptor CTLA-4, a close structural relative of CD28, bivalent binding has been recognized much earlier than for CD28 (Figure 2C). Accordingly, it was assumed that the high avidity of CTLA-4 for the shared ligands would explain why CTLA-4-mediated inhibition dominates over CD28-induced co-stimulation (reviewed in Walker and Sansom8). How the avidity of CD28 after the activation-induced conformational changes compares to that of CTLA-4 has, however, not yet been addressed. It may, however, be speculated that it is still below that of CTLA-4 as monovalent binding of CTLA-4 is of higher affinity than monovalent binding of CD28.8
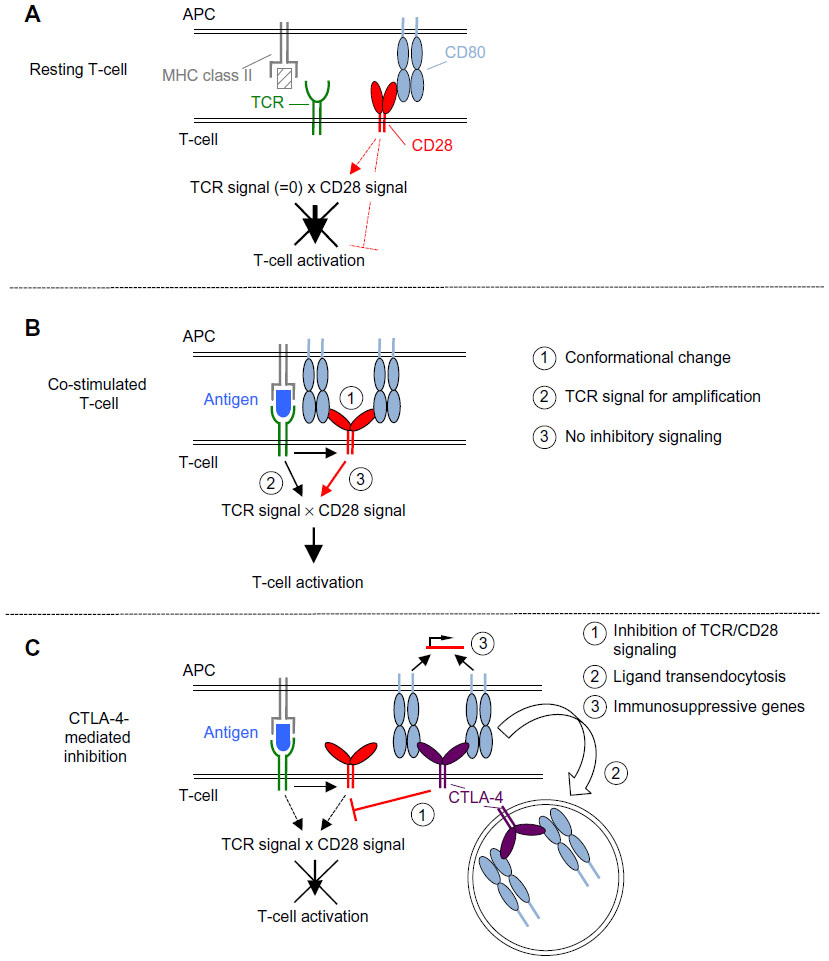 | Figure 2 Scheme of the molecular mechanisms underlying CD28-mediated co-stimulation and CTLA-4-mediated inhibition. |
The second mechanism is the strict dependence of even supraphysiological CD28-derived signals, as those induced by superagonistic anti-CD28 monoclonal antibodies (mAbs) (CD28 superagonist [CD28SA], see The power of CD28SAs section), on the provision of at least a “tonic” signal from the TCR (Figure 2A and B).9 This means that there are signaling molecules in the T-cells like the zeta-associated protein of 70 kDa (ZAP-70) which are stimulated by the TCR complex but not by CD28 and which control the activation status of the SH2 domain-containing leukocyte protein of 76 kDa (SLP76)/VAV1/interleukin-2-inducible T-cell kinase (ITK) signalosome integrating the TCR and CD28 signaling pathways (Figure 3).9
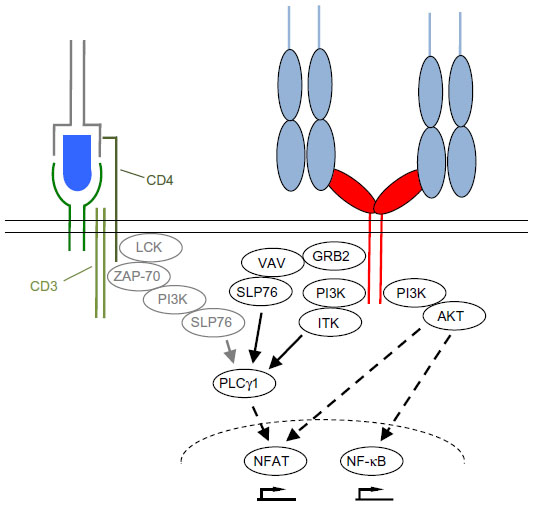 | Figure 3 Simplified scheme of downstream signaling events initiated upon CD28 and TCR ligation. |
Third, ligation of CD28 in the absence of TCR stimulation may lead to the induction of inhibitory signals in the T-cell (Figure 2A). This is exemplified by the induction of acid sphingomyelinase activity upon CD28 ligation in the absence of TCR stimulation,10,11 which leads to the generation of ceramide from sphingomyelin. Accumulation of ceramide in the cell membrane is, in turn, associated with inhibition of the T-cell.12,13 At least under the in vitro conditions tested so far using mAbs, the induction of this inhibitory signal is prevented upon simultaneous stimulation of the TCR together with CD28, thus allowing for full T-cell activation.11 This very interesting observation, however, still awaits confirmation in assays studying natural CD28–ligand interactions.
Upon TCR and CD28 stimulation, the T-cell forms an immunological synapse (IS) with the APC (reviewed in Kumari et al14 and Saito et al15). The IS consists of distinct supramolecular activation clusters (SMACs) arranged like a bull’s eye with TCR complexes and CD28 in the center (cSMAC) and molecules like lymphocyte function-associated antigen-1 or CD45 in the peripheral or distal SMAC (pSMAC/dSMAC). The CD28 molecules within the SMAC transduce activating signals into the cell via the YNMN and the PYAP motifs contained in the intracellular part of CD28 (reviewed in Chen and Flies1 and Boomer and Green16) (Figure 3). As a consequence, phosphoinositide 3-kinase (PI3K), protein kinase Cθ, growth factor receptor-bound protein 2, RAS guanyl nucleotide-releasing protein, and lymphocyte cell-specific protein-tyrosine kinase are recruited to CD28.1 Particularly as a result of PI3K recruitment, CD28 stimulation strongly enhances signaling along the AKT/mammalian target of rapamycin pathway (Figure 3). TCR complex signaling is also quantitatively enhanced, for example, by the CD28-induced production of the substrate for both PI3K and phospholipase Cγ1 (PLCγ1), namely, phosphatidylinositol 4,5-biphosphate (PIP2), through phosphatidylinositol 4-phosphate 5-kinase activation.17 Supplying PIP2 enhances signaling molecule recruitment via PI3K-mediated phosphatidylinositol 3,4,5-triphosphate generation and the generation of inositol triphosphate by PLCγ1 resulting in prolonged Ca2+ flux into the T-cell through calcium release-activated calcium channels18 – a hallmark of CD28-mediated co-stimulation.17,19
Apart from the TCR complex and CD28, the inhibitory receptor CTLA-4 is also very efficiently recruited from intracellular vesicles to the IS (Figure 2C).20 At the IS, CTLA-4, via its cytoplasmic tail, efficiently recruits phosphatases like protein phosphatase 2A and SH2 domain-containing phosphatase 2 to the IS where they dephosphorylate the CD3 chains of the TCR complex and recruit signaling molecules like ZAP-70 and the linker of activated T-cells (reviewed in Walker and Sansom8 and Chen and Flies1). As a consequence of these dephosphorylation events and/or due to molecular pathways yet to be discovered, CTLA-4 counteracts the formation of lipid microdomains, the so-called lipid rafts, induced upon TCR and CD28-mediated co-stimulation which are required for efficient protein clustering (reviewed in Chen and Flies,1 Rudd,21 and Rudd et al22). As a result, CTLA-4 ligation inhibits the formation of the so-called ZAP-70 microclusters at the cell membrane, again interfering with proximal TCR signaling.1,8,21 Beyond interfering with the signaling process itself, CTLA-4 prevents the induction of the integrin-mediated “stop signal” in T-cells upon contact with APCs.21 This “stop signal” contributes to the formation of a stable IS, is induced by TCR ligation, and probably amplified by CD28 co-ligation.21
Due to its capacity to block CD80 and CD86, to transmit immunosuppressive signals into APCs via these ligands, and even capture them from the surface of APCs by transendocytosis (reviewed in Walker and Sansom8), CTLA-4 is also a potent inhibitor of T-cell activation in trans. In bone marrow chimeric mice,23 it has been shown that CTLA-4 expression by Treg cells24 is necessary and sufficient to prevent the catastrophic lymphoproliferative syndrome induced by CTLA-4 knockout T-cells. Together with evidence from studies on T-cell responses toward foreign antigens, it appears that the key function of CTLA-4 in vivo is to mediate suppression by Treg cells in trans.8 Modulation of immune responses by Treg cells through CTLA-4, however, does not necessarily compromise immunity but may even be crucial for the development of efficient memory responses requiring the so-called multifunctional effector CD4+ T-cells.25
Apart from CD80 and CD86, the inducible co-stimulator ligand, B7-H2, has recently been identified to also bind to CD28 and CTLA-4 expressed by human T-cells.26 Functionally, binding of B7-H2 to CD28 appears to induce a very similar, if not identical, co-stimulatory signal upon TCR co-ligation as the interaction of CD80/CD86 and CD28, which is distinct from the signal induced in the T-cell upon B7-H2-inducible co-stimulator interaction.26
In summary, CD28 induces a co-stimulatory signal in the T-cell upon co-ligation together with the TCR, thus amplifying the TCR complex-derived signal and by recruiting additional signaling molecules. The negative regulator CTLA-4 counteracts CD28 signaling in cis by recruiting inhibitory phosphatases to the IS, and in trans by transmitting inhibitory signals into APCs, as well as by competing for the binding and removal of co-stimulatory ligands.
CD28 co-stimulation: gene regulation and biological effects
In-depth gene expression analysis demonstrated that CD28 co-stimulation modulates TCR-induced gene expression rather than regulating a distinct cluster of genes.27,28 Within the scope of this review, we focus on the impact of CD28 co-stimulation on the expression of genes that are involved in T-cell proliferation, differentiation, and survival.
Following TCR/CD28 stimulation, resting T-cells enter into the cell cycle on the one hand by activation of cyclin D2 and CDK4/6 complexes mediating progression into the G1 phase.29,30 On the other hand, CD28 signaling antagonizes the effects of CDK inhibitors like INK4C that negatively control G0/G1 transition.31 T-cell proliferation is sustained by production of interleukin-2 (IL-2) that acts in an autocrine fashion (Figure 4A). The role of CD28 co-stimulation in the induction of IL-2 gene expression has been extensively studied. CD28 has been shown to upregulate the activity of several transcription factors including nuclear factor of activated T-cells (NFAT), nuclear factor κ-light-chain enhancer of activated B cells (NF-κB), and activator protein-1 (AP-1).32–34 Activation of IL-2 transcription is initiated by binding of these transcription factors to specific regions in the IL-2 promoter: the CD28 responsive element and the adjacent NF-IL-2B AP-1 site.35 Besides transcriptional control of the IL-2 gene, CD28 signals have been shown to enhance the stability of the IL-2 mRNA.34 IL-2 exerts its function on T-cells by binding to the high-affinity trimeric IL-2 receptor (IL-2R) composed of CD122 (IL-2Rβ), CD132 (the common cytokine receptor γ-chain) and CD25 (IL-2Rα) (Figure 4A). CD25 gene expression in T-cells is rapidly induced upon TCR/CD28 co-stimulation by activation of NF-κB, NFAT, AP-1, and cAMP response element-binding protein/activating transcription factor (reviewed in Kim et al36). Thereupon, IL-2 binds to the high-affinity receptor leading to further enhancement of CD25 expression by a Stat5-dependent mechanism.36 Taken together, CD28 co-stimulation affects T-cell activation and proliferation at multiple levels, including amplification of signals in response to antigen recognition, activation/translocation of transcription factors promoting expression of proteins required for cell-cycle entry, and of the growth-promoting cytokine IL-2 along with its high-affinity receptor.
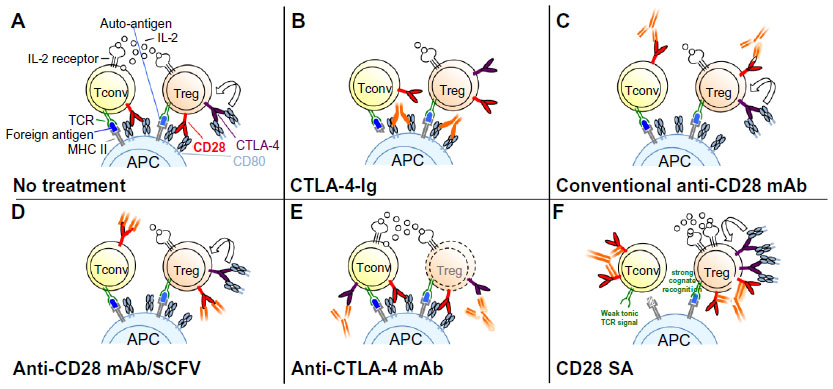 | Figure 4 Schematic modes of action of drugs targeting CD28 and/or CTLA-4. |
CD28 co-stimulation is not only a booster of T-cell proliferation and cytokine production but in addition influences the survival of T-cells. Apoptosis in T-cells is mediated by two major pathways: I) The extrinsic pathway is initiated by binding of a ligand to a death receptor (eg, Fas) expressed on the cell surface leading to the activation of the caspase cascade. II) The intrinsic apoptotic pathway is activated by several stress stimuli like DNA damage or growth factor withdrawal and leads to mitochondrial outer membrane permeabilization and release of proapoptotic factors. It has been shown that interactions between CD28 and CD80/86 result in the intracellular accumulation of the anti-apoptotic protein Bcl-XL rendering T-cells resistant to extrinsic and intrinsic death signals.37 Further studies revealed that the NF-κB pathway is involved in CD28-induced expression of Bcl-XL.38 In addition, co-stimulation via CD28 can reduce TCR-induced cell death through repression of CD95L expression.39
Upon T-cell activation, several metabolic pathways are activated that increase the amount of energy provided for proliferation and effector functions. In this regard, CD28 co-stimulation leads to enhanced glucose uptake that is regulated at multiple levels.40,41 As an example, the expression of Glut1, the primary glucose transporter in hematopoietic cells,42 is modulated by CD28 through both AKT-dependent and AKT-independent pathways.41
After several rounds of cell division, T-cells gain different effector functions depending on the presence of distinct polarizing cytokines. In this context, it has been shown that CD28 co-stimulation induces the production of the TH2-type cytokine IL-4 via an IL-4-dependent mechanism.43 In addition, CD28 stimulation is required for the activation of the transcription factor GATA-3 that promotes T-cell differentiation into the TH2 subset,44 while coincidental ligation of CTLA-4 and the TCR favors TH1 cell differentiation.45
Chronic antigenic stimulation (which is supported by CD28 co-stimulation) can influence the function of T-cells in several ways. Thus, repeated stimulation of T-cells in environments of high antigenic load can result in aging of the cell that is accompanied by functional exhaustion and cell death. Repeated stimulation of T-cells can also result in replicative senescence. While exhausted cells eventually undergo apoptosis, senescent T-cells usually are resistant to apoptosis and persist (reviewed in Akbar and Henson46). Moreover, they are characterized by loss of CD28 expression, reduced telomerase activity, and enhanced production of pro-inflammatory cytokines as compared to non-senescent cells (reviewed in Weng et al47). Furthermore, T-cells lacking CD28 show several changes in gene expression leading to altered expression of molecules like co-stimulatory proteins, chemokine receptors, and natural killer (NK) cell receptors.47 Of note, CD4+ CD28− T-cells emerge in several immune disorders including rheumatoid arthritis (RA), which often affects elderly people.48,49 Signaling via CD28 is essential for induction of telomerase activity,50 and prolonged expression of CD28 delays functional changes associated with T-cell senescence.51 Taken together, senescent T-cells losing expression of CD28 acquire – at least under certain conditions – effector functions leading to autoimmunity. In this context, it has to be mentioned that loss of CD28 is also a feature of terminally differentiated CD8+ T-cells which represent a functionally heterogeneous cell population (cytotoxic versus regulatory) and are commonly found in individuals with chronic immune activation (reviewed in Strioga et al52).
Role of CD28 in Treg cell generation, homeostasis, and function
The activation and effector function of immune cells is controlled by a specialized subset of CD4+ T-cells, the CD4+ CD25+ Foxp3+ Treg cells.53 Expression of the transcription factor forkhead box P3 (Foxp3) is essential for Treg cell development in the thymus and its suppressive functions in the periphery (Figure 4A). It is well established that co-stimulation by CD28 is required for induction of Foxp3 expression via nuclear translocation of RelA and efficient development of Treg cells.54
This particular dependence of Treg cells on CD28-mediated co-stimulation is illustrated by an 80% reduced Treg compartment in CD28-deficient mice.55 While this reduction is already apparent in the thymus, interruption of peripheral provision of CD80/86 also results in a rapid decline of Treg cell numbers.56 Within the CD28/CTLA-4/CD80/86 triangle, this effect has recently been mapped by induced CD28 deletion to the loss of co-stimulation during the continuous turnover of Treg cells in response to self-antigens.57 Thus, upon loss of CD28, Treg cells stop proliferating and drastically fall in numbers. In addition, their functional capacity declines as read out in suppressor assays, and by a reduction in CTLA-4 levels. These effects are cell intrinsic, that is, independent of the provision of IL-2 by conventional CD4 T-cells, which is also under co-stimulatory control57 (Table 1). Not too surprisingly, similar effects have very recently been observed when TCR expression by Treg cells was interrupted.58
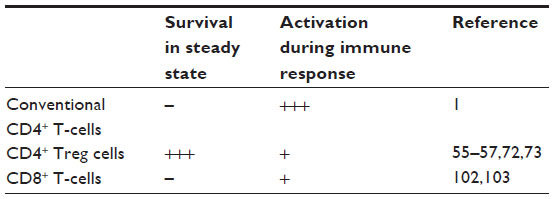 | Table 1 Impact of CD28 ligation on different mouse T-cell subsets on their activation and differentiation |
Besides Treg cells that differentiate to that phenotype in the thymus (natural or thymic Treg cells), they can also be generated by conversion from conventional CD4 T-cells activated by antigen recognition and provided with transforming growth factor beta and IL-2 (induced Treg [iTreg] cells). In T-cells from conventional CD28 knockout mice, iTreg induction by anti-CD3 plus transforming growth factor beta is defective but can be restored by adding IL-2.59 However, since CD4+ T-cells in such mice are adapted to CD28 deficiency, the conclusion that the observed CD28 dependence of this differentiation process is fully explained by the requirement for CD28 co-stimulation in the production of IL-2 by conventional CD4+ T-cells is shaky. For a definitive answer, in vivo experiments using induced CD28 deletion in mixed bone marrow chimeras where wild type and inducibly CD28-deleting T-cells coexist and can be identified, along with a physiological TCR stimulus will be required.
Taken together, CD28 is a key player not only in the activation of T-cell effector responses but also in their control by Treg cells, which recognize self-antigens in a co-stimulation-dependent manner, and sense ongoing effector T-cell responses via the CD28-dependent production of IL-2.
Targeting CD28 co-stimulatory pathways
Inhibition of CD28-mediated co-stimulation
CTLA-4 linked to the Fc domain of human IgG1
As described above, CTLA-4 binding to CD80 or CD86 inhibits T-cell activation and proliferation. Linsley et al exploited this property of CTLA-4 to generate a fusion protein consisting of the extracellular domain of human CTLA-4 linked to the Fc domain of human IgG1 (CTLA-4-Ig; Abatacept) (Figure 4B).60 Ig Fc domains of such fusion proteins are helpful for several reasons as they not only facilitate purification and detection but also markedly increase the in vivo half-life of such recombinant proteins. Further studies showed that Abatacept – similar to CTLA-4 itself – has a high affinity for CD80 and CD86, blocks CD28-dependent co-stimulation, and inhibits T-cell proliferation in vitro (reviewed in Linsley and Nadler61) (Figure 4B). Based on these results, CTLA-4-Ig was successfully tested in numerous preclinical animal models of autoimmune diseases.61 For example, application of CTLA-4-Ig in collagen-induced arthritis of the rat prevented the onset of the disease indicating that CTLA-4-Ig-mediated blockade of T-cell co-stimulation in vivo inhibits the activation of autoreactive T-cells in the collagen-immunized animals.62
Likewise, autoreactive T-cells play an important role in the immune pathology of many human autoimmune diseases including RA, and thus, their co-stimulation-dependent activation presents a potential target of CTLA-4-Ig. Indeed, several clinical trials demonstrated safety and efficacy of Abatacept in RA, leading to the approval of this drug in the US in 2006 for the treatment of moderate-to severe RA,63 and in 2008 for the treatment of juvenile idiopathic arthritis.61 In contrast, Abatacept failed to protect graft survival in an animal model of renal transplantation due to incomplete blockade of T-cell activation.64 In order to overcome this deficit, a second-generation CTLA-Ig protein – Belatacept – was generated, which differs from Abatacept in two amino acids resulting in tenfold higher in vitro potency.64 After promising results in nonhuman primate models of renal transplantation, Belatacept was successfully tested in Phase II and III clinical trials and approved in 2011 by the US Food and Drug Administration for rejection prophylaxis in adult kidney transplant recipients (reviewed in Wojciechowski and Vincenti65).
Blocking CD28 with mAbs and recombinant single-chain Fv fragments
Apart from inhibiting CD28-mediated co-stimulation, CTLA-4-Ig also abrogates the interaction of CTLA-4 on T-cells with its ligands expressed by APCs (Figure 4B).1 Therefore, CTLA-4-Ig also prevents negative signaling into the T-cell or the APC, which diminishes the effect of the CD28 blockade achieved by this reagent.
A solution to this dilemma is to interrupt the interaction of CD28 with its ligands by directly targeting the CD28 molecule. Seemingly paradoxically, CD28-specific mAbs, which in vitro are capable of inducing a potent co-stimulatory signal in T-cells inhibit unwanted T-cell responses in vivo (Figure 4C).66,67 However, in the absence of TCR ligation, the signal induced by these anti-CD28 mAbs is not suitable to activate the T-cell but rather results in complete down-modulation of CD28 expression at the cell surface (Figure 4C).67 Upon subsequent encounter of antigen, the T-cell is functionally CD28 deficient and will, thus, not receive an adequate co-stimulatory signal. Apart from down-modulating CD28 expression at the cell surface, conventional anti-CD28 mAbs may also protect from immunopathology, particularly acute graft-versus-host disease, by increasing interferon-γ expression, which in turn increases apoptosis in alloreactive pathogenic T-cells.68 As already mentioned, induction of acid sphingomyelinase activity by anti-CD28 mAbs also inhibits subsequent T-cell activation.10–13
The most straightforward way to inhibit CD28 signaling is to generate CD28-specific mAbs that block the interaction of CD28 with its ligands (Figure 4D).69,70 For the mouse, mAb E18, itself raised in CD28-deficient mice, was the first mouse-anti-mouse CD28 mAb capable of fully inhibiting ligand binding to CD28.71,72 Despite its capacity to strongly co-stimulate T-cells in vitro, application of E18 as an intact antibody blocked CD28 stimulation upon injection into healthy mice in vivo leading to a gradual depletion of Treg cells72 due to their strong dependence on CD28 signaling for survival.55–57 Under conditions of pathological T-cell activation such as during a superantigen response or during acute graft-versus-host disease, this is different, however. Here, pathogenic conventional CD4+ T-cells are more dependent on CD28-mediated co-stimulation for their activation and expansion than Treg cells (Table 1 and Figure 4D).72 Therefore, under these inflammatory conditions, application of mAb E18 inhibited the expansion of the pathogenic T-cells more strongly than that of Treg cells leading to an increase in Treg cell frequencies among CD4+ T-cells.72 This shift in the balance between the Treg cells and the pathogenic T-cells crucially contributed to the suppression of the pathogenic T-cell response.72
To definitely avoid the generation of an agonistic signal, it is, however, necessary to derive monovalent reagents that block ligand binding, either in the form of Fab fragments derived by enzymatic cleavage or as genetically engineered single-chain Fv (SCFV) fragments, derived from ligand-binding site-directed mAbs (Figure 4D).70 Fab fragments of mAb E18 similarly inhibited expansion of pathogenic T-cells as did the intact antibody,72 and an SCFV derived from another anti-CD28 mAb, clone PV-1, also suppressed pathogenic T-cell responses in mice and concomitantly increased the frequencies of Treg cells among CD4+ T-cells.73 In this study, the benefits of intact CTLA-4 signaling upon CD28 blockade were revealed as inhibiting CTLA-4 signaling also reverted the protective effect of the anti-CD28 SCFV.73
For clinical use, a pegylated anti-CD28 SCFV named FR104, which was derived from the anti-CD28 mAb clone CD28.3, is in development.74 This reagent has already been shown to prevent graft rejection in monkeys75 and to inhibit the induction of xenogenic graft-versus-host disease by human T-cells transplanted into NOD SCID mice.74 The pegylated anti-CD28 SCFV also depended on intact CTLA-4 signaling to be efficient.74,75
Taken together, selective blockade of CD28–ligand interaction has been shown to allow for CTLA-4-dependent suppression of pathologic immune responses in preclinical animal models. This lays the basis for further development of FR104 designed to inhibit CD28-mediated co-stimulation in patients.
Boosting CD28-mediated co-stimulation
CTLA-4 blockade
The function of CTLA-4 as a negative regulator of adaptive immune responses led to the hypothesis that CTLA-4 blockade might liberate CD80/86 for interaction with CD28 and promote increased T-cell activation and proliferation (Figure 4E). Thus, it was thought that blockade of this immune checkpoint might break immunological tolerance to malignant T-cells and lead to enhanced T-cell-mediated rejection of tumors. Indeed, seminal work by Leach et al demonstrated that the in vivo application of CTLA-4-specific antibodies not only resulted in the rejection of preestablished tumors but also enhanced antitumor immunity after secondary tumor challenge.76 The fact that the tumor environment frequently expresses low levels of CD80/86 or even lacks these co-stimulatory proteins (reviewed in Seliger et al77) indicated that additional mechanisms might account for the in vivo effects of anti-CTLA-4 antibodies (reviewed in Blank and Enk78). In this regard, several groups could demonstrate that anti-CTLA-4 antibodies mediate Fcγ-receptor-dependent intratumoral depletion of Treg cells (the only cell type with constitutive surface expression of CTLA-4) resulting in increased intratumoral effector/regulatory T-cell ratios (Figure 4E).79–81
Based on the preclinical results, two fully human mAbs with specificity for human CTLA-4 – tremelimumab82 and ipilimumab83 – have been generated and evaluated in patients with late-stage melanoma. While tremelimumab failed to significantly improve the survival rate of advanced melanoma patients,84 two ipilimumab Phase III trials showed significant improvement of overall survival.85 In 2013, ipilimumab was, therefore, approved by the European Commission as first-line treatment for adult patients with advanced melanoma. The difference in clinical efficacy of the two mAbs might be explained by the isotypes – IgG1 (ipilimumab) versus IgG2 (tremelimumab). Human IgG1 binds to Fcγ-receptors with higher affinity than human IgG2,86 which may allow a more efficient in vivo depletion of the CTLA-4-expressing Treg cells. In order to exploit possible synergies between blockade of CTLA-4 and programmed cell death protein-1 (PD-1) – a further checkpoint of T-cell activation – ipilimumab is currently tested in combination with nivolumab, a PD-1-blocking antibody.87
The power of CD28SAs
CD28SAs are a unique class of mAbs with a particularly strong signaling capacity,88 which is most likely related to their lateral binding mode to the CD28 homodimer,89,90 thereby allowing clustering by lattice formation (Figure 4F). CD28SAs apparently activate T-cells without the need for “signal one” provided by TCR-mediated antigen recognition. The complete loss of CD28SA responses in T-cell lines lacking TCR signaling,9,91 and in circulating human T-cells that are deprived of cell contact-dependent “tonic” TCR signals that depend on cellular interactions in the tissues,92 indicates, however, that the ability of CD28SAs to polyclonally activate all CD28-expressing T-cells constitutes an extreme case of co-stimulation, where very strong CD28 signals amplify very weak TCR-derived signals to a level sufficient for T-cell activation. Indeed, the site of this amplification has been mapped to the SLP76/VAV1/ITK signalosome (Figure 3).9 In line with this conclusion, Treg cells, which, in contrast to all conventional T-cells not undergoing an immune response, are constantly stimulated by recognition of cognate (self-)antigen and hence display a higher level of phosphorylation in their TCR signal transduction machinery,93 and are activated by lower concentrations of CD28SAs than conventional T-cells (Figure 4F).94 This preferential activation of Treg cells by CD28SAs has been successfully exploited in multiple rodent models of autoimmunity, inflammation, solid organ or hematopoietic stem cell transplantation (summarized in Tabares et al95), and most recently, tissue repair after myocardial infarction96 and stroke.97
However, when the human CD28SA TGN1412 was tested in a first-in-human (FIH) trial in 2006, the healthy volunteers experienced a life-threatening cytokine storm,98 which resulted from the activation of tissue-resident CD4+ effector memory cells.92,99 This effect had not been anticipated by primate toxicity testing because macaques downregulate CD28 on this small T-cell subset.99 Furthermore, human peripheral blood mononuclear cells do not respond to soluble TGN1412 (now renamed TAB08) because, as mentioned above, they lack the pre-activation of the TCR signaling machinery that depends on cellular interactions in tissues but is lost in the circulation.92,100 In the meantime, an assay was developed in which the response of circulating T-cells to CD28SAs is restored by resetting them to tissue-like conditions.92 In this system, it was shown that just like in rodents, application of low CD28SA concentrations results in the selective activation of Treg cells (Figure 4F).95 This finding was verified in vivo in a new Phase I study employing very low doses of the drug, which led to the selective release of the Treg signature cytokine IL-10 into the circulation,95 and further clinical trials in RA patients have been initiated with encouraging results.101
Concluding remarks
The central positioning of the CD28/CD80/86/CTLA-4 receptor–ligand system in the initiation and control of effector and Treg cell responses makes it an attractive target for therapeutic modulation of the immune system. Blockade of the shared ligands CD80/86 is now firmly established in RA management, with other applications still in the experimental phase. Selective blockade of CD28 co-stimulation without interfering with CTLA-4 ligation by using monovalent mAb derivatives is a promising alternative, which is in late preclinical development. Conversely, releasing only the “break” by blocking CTLA-4 has proven effective in boosting immune attack on cancer, with melanoma being at the most advanced stage in clinical use. Finally, the unique responsiveness of Treg cells to low-dose application of CD28SAs, which has proven effective in a vast array of immunopathologies in rodent models, has recovered from its setback during a catastrophic FIH trial, which now is recognized as due to unexpected overdosing. The next years will show whether as observed in those models, mobilization and recruitment of Treg cells will benefit patients with autoimmune and inflammatory disorders.
Acknowledgments
The authors like to thank the Deutsche Forschungsgemeinschaft (Research Unit 2123, SFB688, HU 295/12-1, BE 4080/2-1) and the Deutsche José Carreras Leukämie-Stiftung e.V. (R 13/25) for financial support.
Disclosure
TH is a consultant to TheraMAB LLC which develops clinical use of CD28SA. The other authors report no conflicts of interest in this work.
References
Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13(4):227–242. | |
Waterhouse P, Penninger JM, Timms E, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995; 270:985–988. | |
Sanderson K, Scotland R, Lee P, et al. Autoimmunity in a phase I trial of a fully human anti-cytotoxic T-lymphocyte antigen-4 monoclonal antibody with multiple melanoma peptides and Montanide ISA 51 for patients with resected stages III and IV melanoma. J Clin Oncol. 2005; 23(4):741–750. | |
Tai X, Van Laethem F, Sharpe AH, Singer A. Induction of autoimmune disease in CTLA-4-/- mice depends on a specific CD28 motif that is required for in vivo costimulation. Proc Natl Acad Sci U S A. 2007; 104(34):13756–13761. | |
Sanchez-Lockhart M, Kim M, Miller J. Cutting edge: a role for inside-out signaling in TCR regulation of CD28 ligand binding. J Immunol. 2011;187(11):5515–5519. | |
Sanchez-Lockhart M, Rojas AV, Fettis MM, et al. T cell receptor signaling can directly enhance the avidity of CD28 ligand binding. PLoS One. 2014;9(2):e89263. | |
Collins AV, Brodie DW, Gilbert RJ, et al. The interaction properties of costimulatory molecules revisited. Immunity. 2002;17(2):201–210. | |
Walker LS, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol. 2011;11(12):852–863. | |
Dennehy KM, Elias F, Na SY, Fischer KD, Hunig T, Luhder F. Mitogenic CD28 signals require the exchange factor Vav1 to enhance TCR signaling at the SLP-76-Vav-Itk signalosome. J Immunol. 2007; 178(3):1363–1371. | |
Boucher LM, Wiegmann K, Fütterer A, et al. CD28 signals through acidic sphingomyelinase. J Exp Med. 1995;181(6):2059–2068. | |
Mueller N, Avota E, Collenburg L, Grassme H, Schneider-Schaulies S. Neutral sphingomyelinase in physiological and measles virus induced T cell suppression. PLoS Pathog. 2014;10(12):e1004574. | |
O’Byrne D, Sansom D. Lack of costimulation by both sphingomyelinase and C2 ceramide in resting human T cells. Immunology. 2000;100(2):225–230. | |
Abboushi N, El-Hed A, El-Assaad W, et al. Ceramide inhibits IL-2 production by preventing protein kinase C-dependent NF-kappaB activation: possible role in protein kinase Ctheta regulation. J Immunol. 2004;173(5):3193–3200. | |
Kumari S, Curado S, Mayya V, Dustin ML. T cell antigen receptor activation and actin cytoskeleton remodeling. Biochim Biophys Acta. 2014;1838(2):546–556. | |
Saito T, Yokosuka T, Hashimoto-Tane A. Dynamic regulation of T cell activation and co-stimulation through TCR-microclusters. FEBS Lett. 2010;584(24):4865–4871. | |
Boomer JS, Green JM. An enigmatic tail of CD28 signaling. Cold Spring Harb Perspect Biol. 2010;2(8):a002436. | |
Muscolini M, Camperio C, Capuano C, et al. Phosphatidylinositol 4-phosphate 5-kinase alpha activation critically contributes to CD28-dependent signaling responses. J Immunol. 2013;190(10):5279–5286. | |
Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol. 2007;7(9):690–702. | |
Dennehy KM, Kerstan A, Bischof A, Park JH, Na SY, Hunig T. Mitogenic signals through CD28 activate the protein kinase Ctheta-NF-kappaB pathway in primary peripheral T cells. Int Immunol. 2003;15(5):655–663. | |
Egen JG, Allison JP. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity. 2002;16(1):23–35. | |
Rudd CE. The reverse stop-signal model for CTLA4 function. Nat Rev Immunol. 2008;8(2):153–160. | |
Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. 2009;229(1):12–26. | |
Bachmann MF, Kohler G, Ecabert B, Mak TW, Kopf M. Cutting edge: lymphoproliferative disease in the absence of CTLA-4 is not T cell autonomous. J Immunol. 1999;163(3):1128–1131. | |
Friedline RH, Brown DS, Nguyen H, et al. CD4+ regulatory T cells require CTLA-4 for the maintenance of systemic tolerance. J Exp Med. 2009;206(2):421–434. | |
Rudolph M, Hebel K, Miyamura Y, Maverakis E, Brunner- Weinzierl MC. Blockade of CTLA-4 decreases the generation of multifunctional memory CD4+ T cells in vivo. J Immunol. 2011;186(10):5580–5589. | |
Yao S, Zhu Y, Zhu G, et al. B7-h2 is a costimulatory ligand for CD28 in human. Immunity. 2011;34(5):729–740. | |
Diehn M, Alizadeh AA, Rando OJ, et al. Genomic expression programs and the integration of the CD28 costimulatory signal in T cell activation. Proc Natl Acad Sci U S A. 2002;99(18):11796–11801. | |
Riley JL, Mao M, Kobayashi S, et al. Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc Natl Acad Sci U S A. 2002;99(18):11790–11795. | |
Appleman LJ, Berezovskaya A, Grass I, Boussiotis VA. CD28 costimulation mediates T cell expansion via IL-2-independent and IL-2-dependent regulation of cell cycle progression. J Immunol. 2000;164(1):144–151. | |
Mohapatra S, Agrawal D, Pledger WJ. p27Kip1 regulates T cell proliferation. J Biol Chem. 2001;276(24):21976–21983. | |
Kovalev GI, Franklin DS, Coffield VM, Xiong Y, Su L. An important role of CDK inhibitor p18(INK4c) in modulating antigen receptor-mediated T cell proliferation. J Immunol. 2001;167(6):3285–3292. | |
Fraser JD, Irving BA, Crabtree GR, Weiss A. Regulation of interleukin-2 gene enhancer activity by the T cell accessory molecule CD28. Science. 1991;251(4991):313–316. | |
Granelli-Piperno A, Nolan P. Nuclear transcription factors that bind to elements of the IL-2 promoter. Induction requirements in primary human T cells. J Immunol. 1991;147(8):2734–2739. | |
Miller J, Baker C, Cook K, et al. Two pathways of costimulation through CD28. Immunol Res. 2009;45(2–3):159–172. | |
Shapiro VS, Truitt KE, Imboden JB, Weiss A. CD28 mediates transcriptional upregulation of the interleukin-2 (IL-2) promoter through a composite element containing the CD28RE and NF-IL-2B AP-1 sites. Mol Cell Biol. 1997;17(7):4051–4058. | |
Kim HP, Imbert J, Leonard WJ. Both integrated and differential regulation of components of the IL-2/IL-2 receptor system. Cytokine Growth Factor Rev. 2006;17(5):349–366. | |
Boise LH, Minn AJ, Noel PJ, et al. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 1995;3(1):87–98. | |
Khoshnan A, Tindell C, Laux I, Bae D, Bennett B, Nel AE. The NF-kappa B cascade is important in Bcl-xL expression and for the anti-apoptotic effects of the CD28 receptor in primary human CD4+ lymphocytes. J Immunol. 2000;165(4):1743–1754. | |
Kerstan A, Hunig T. Cutting edge: distinct TCR- and CD28-derived signals regulate CD95L, Bcl-xL, and the survival of primary T cells. J Immunol. 2004;172(3):1341–1345. | |
Frauwirth KA, Riley JL, Harris MH, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16(6):769–777. | |
Jacobs SR, Herman CE, Maciver NJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. 2008;180(7):4476–4486. | |
Rathmell JC, Vander Heiden MG, Harris MH, Frauwirth KA, Thompson CB. In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol Cell. 2000; 6(3):683–692. | |
Rulifson IC, Sperling AI, Fields PE, Fitch FW, Bluestone JA. CD28 costimulation promotes the production of Th2 cytokines. J Immunol. 1997;158(2):658–665. | |
Rodriguez-Palmero M, Hara T, Thumbs A, Hunig T. Triggering of T cell proliferation through CD28 induces GATA-3 and promotes T helper type 2 differentiation in vitro and in vivo. Eur J Immunol. 1999; 29(12):3914–3924. | |
Oosterwegel MA, Mandelbrot DA, Boyd SD, et al. The role of CTLA-4 in regulating Th2 differentiation. J Immunol. 1999;163(5):2634–2639. | |
Akbar AN, Henson SM. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat Rev Immunol. 2011;11(4):289–295. | |
Weng NP, Akbar AN, Goronzy J. CD28(-) T cells: their role in the age-associated decline of immune function. Trends Immunol. 2009;30(7):306–312. | |
Thewissen M, Somers V, Hellings N, Fraussen J, Damoiseaux J, Stinissen P. CD4+CD28null T cells in autoimmune disease: pathogenic features and decreased susceptibility to immunoregulation. J Immunol. 2007;179(10):6514–6523. | |
Martens PB, Goronzy JJ, Schaid D, Weyand CM. Expansion of unusual CD4+ T cells in severe rheumatoid arthritis. Arthritis Rheum. 1997;40(6):1106–1114. | |
Weng NP, Levine BL, June CH, Hodes RJ. Regulated expression of telomerase activity in human T lymphocyte development and activation. J Exp Med. 1996;183(6):2471–2479. | |
Parish ST, Wu JE, Effros RB. Sustained CD28 expression delays multiple features of replicative senescence in human CD8 T lymphocytes. J Clin Immunol. 2010;30(6):798–805. | |
Strioga M, Pasukoniene V, Characiejus D. CD8+ CD28- and CD8+ CD57+ T cells and their role in health and disease. Immunology. 2011; 134(1):17–32. | |
Sakaguchi S, Ono M, Setoguchi R, et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. | |
Soligo M, Camperio C, Caristi S, et al. CD28 costimulation regulates FOXP3 in a RelA/NF-kappaB-dependent mechanism. Eur J Immunol. 2011;41(2):503–513. | |
Tang Q, Henriksen KJ, Boden EK, et al. Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J Immunol. 2003;171(7):3348–3352. | |
Salomon B, Lenschow DJ, Rhee L, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12(4):431–440. | |
Gogishvili T, Luhder F, Goebbels S, Beer-Hammer S, Pfeffer K, Hunig T. Cell-intrinsic and -extrinsic control of Treg-cell homeostasis and function revealed by induced CD28 deletion. Eur J Immunol. 2013;43(1):188–193. | |
Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol. 2014;15(11):1070–1078. | |
Guo F, Iclozan C, Suh WK, Anasetti C, Yu XZ. CD28 controls differentiation of regulatory T cells from naive CD4 T cells. J Immunol. 2008;181(4):2285–2291. | |
Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174(3):561–569. | |
Linsley PS, Nadler SG. The clinical utility of inhibiting CD28-mediated costimulation. Immunol Rev. 2009;229(1):307–321. | |
Knoerzer DB, Karr RW, Schwartz BD, Mengle-Gaw LJ. Collagen-induced arthritis in the BB rat. Prevention of disease by treatment with CTLA-4-Ig. J Clin Invest. 1995;96(2):987–993. | |
Bluestone JA, St Clair EW, Turka LA. CTLA4Ig: bridging the basic immunology with clinical application. Immunity. 2006;24(3):233–238. | |
Larsen CP, Pearson TC, Adams AB, et al. Rational development of LEA29Y (belatacept), a high-affinity variant of CTLA4-Ig with potent immunosuppressive properties. Am J Transplant. 2005;5(3):443–453. | |
Wojciechowski D, Vincenti F. Belatacept in kidney transplantation. Curr Opin Organ Transplant. 2012;17(6):640–647. | |
Krummel MF, Sullivan TJ, Allison JP. Superantigen responses and co-stimulation: CD28 and CTLA-4 have opposing effects on T cell expansion in vitro and in vivo. Int Immunol. 1996;8(4):519–523. | |
Dengler TJ, Szabo G, Sido B, et al. Prolonged allograft survival but no tolerance induction by modulating CD28 antibody JJ319 after high-responder rat heart transplantation. Transplantation. 1999;67(3):392–398. | |
Yu XZ, Albert MH, Martin PJ, Anasetti C. CD28 ligation induces transplantation tolerance by IFN-gamma-dependent depletion of T cells that recognize alloantigens. J Clin Invest. 2004;113(11):1624–1630. | |
Hunig T. Manipulation of regulatory T-cell number and function with CD28-specific monoclonal antibodies. Adv Immunol. 2007;95:111–148. | |
Poirier N, Blancho G, Vanhove B. A more selective costimulatory blockade of the CD28-B7 pathway. Transpl Int. 2011;24(1):2–11. | |
Dennehy KM, Elias F, Zeder-Lutz G, et al. Cutting edge: monovalency of CD28 maintains the antigen dependence of T cell costimulatory responses. J Immunol. 2006;176(10):5725–5729. | |
Beyersdorf N, Ding X, Blank G, Dennehy KM, Kerkau T, Hunig T. Protection from graft-versus-host disease with a novel B7 binding site-specific mouse anti-mouse CD28 monoclonal antibody. Blood. 2008; 112(10):4328–4336. | |
Zhang T, Fresnay S, Welty E, et al. Selective CD28 blockade attenuates acute and chronic rejection of murine cardiac allografts in a CTLA-4-dependent manner. Am J Transplant. 2011;11(8):1599–1609. | |
Poirier N, Mary C, Dilek N, et al. Preclinical efficacy and immunological safety of FR104, an antagonist anti-CD28 monovalent fab’ antibody. Am J Transplant. 2012;12(10):2630–2640. | |
Poirier N, Azimzadeh AM, Zhang T, et al. Inducing CTLA-4-dependent immune regulation by selective CD28 blockade promotes regulatory T cells in organ transplantation. Sci Transl Med. 2010; 2(17):17ra10. | |
Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–1736. | |
Seliger B, Marincola FM, Ferrone S, Abken H. The complex role of B7 molecules in tumor immunology. Trends Mol Med. 2008;14(12):550–559. | |
Blank CU, Enk A. Therapeutic use of anti-CTLA-4 antibodies. Int Immunol. 2015;27(1):3–10. | |
Simpson TR, Li F, Montalvo-Ortiz W, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210(9):1695–1710. | |
Bulliard Y, Jolicoeur R, Windman M, et al. Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med. 2013;210(9):1685–1693. | |
Selby MJ, Engelhardt JJ, Quigley M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1(1):32–42. | |
Ribas A. Clinical development of the anti-CTLA-4 antibody tremelimumab. Semin Oncol. 2010;37(5):450–454. | |
Boasberg P, Hamid O, O’Day S. Ipilimumab: unleashing the power of the immune system through CTLA-4 blockade. Semin Oncol. 2010; 37(5):440–449. | |
Ribas A, Kefford R, Marshall MA, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. 2013;31(5):616–622. | |
Wolchok JD, Hodi FS, Weber JS, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci. 2013;1291:1–13. | |
Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood. 2012;119(24):5640–5649. | |
Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–133. | |
Bischof A, Hara T, Lin C-H, Beyers A, Hünig T. Autonomous induction of proliferation, JNK and NFkB activation in primary resting T-cells by mobilized CD28. Eur J Immunol. 2000;30:876–882. | |
Lühder F, Huang Y, Dennehy KM, et al. Topological requirements and signaling properties of T cell-activating, anti-CD28 antibody superagonists. J Exp Med. 2003;197(8):955–966. | |
Evans EJ, Esnouf RM, Manso-Sancho R, et al. Crystal structure of a soluble CD28-Fab complex. Nat Immunol. 2005;6(3):271–279. | |
Levin SE, Zhang C, Kadlecek TA, Shokat KM, Weiss A. Inhibition of ZAP-70 kinase activity via an analog-sensitive allele blocks T cell receptor and CD28 superagonist signaling. J Biol Chem. 2008;283(22):15419–15430. | |
Römer PS, Berr S, Avota E, et al. Preculture of PBMCs at high cell density increases sensitivity of T-cell responses, revealing cytokine release by CD28 superagonist TGN1412. Blood. 2011;118(26):6772–6782. | |
Andersson J, Stefanova I, Stephens GL, Shevach EM. CD4+CD25+ regulatory T cells are activated in vivo by recognition of self. Int Immunol. 2007;19(4):557–566. | |
Beyersdorf N, Gaupp S, Balbach K, et al. Selective targeting of regulatory T cells with CD28 superagonists allows effective therapy of experimental autoimmune encephalomyelitis. J Exp Med. 2005;202(3):445–455. | |
Tabares P, Berr S, Römer PS, et al. Human regulatory T cells are selectively activated by low-dose application of the CD28 superagonist TGN1412/TAB08. Eur J Immunol. 2014;44(4):1225–1236. | |
Weirather J, Hofmann UD, Beyersdorf N, et al. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. 2014;115(1):55–67. | |
Na SY, Mracsko E, Liesz A, Hunig T, Veltkamp R. Amplification of regulatory T cells using a CD28 superagonist reduces brain damage after ischemic stroke in mice. Stroke. 2015;46(1):212–220. | |
Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355(10):1018–1028. | |
Eastwood D, Findlay L, Poole S, et al. Monoclonal antibody TGN1412 trial failure explained by species differences in CD28 expression on CD4+ effector memory T-cells. Br J Pharmacol. 2010;161(3):512–526. | |
Stefanova I, Dorfman JR, Germain RN. Self-recognition promotes the foreign antigen sensitivity of naive T lymphocytes. Nature. 2002; 420(6914):429–434. | |
Tyrsin D. New Mechanism of Action for Treatment of Autoimmune Diseases; 2014 [cited 23, January, 2015]. Available from: http://viewer.zmags.com/publication/a12191bd#/a12191bd/14. | |
Seah SG, Carrington EM, Ng WC, et al. Unlike CD4+ T-cell help, CD28 costimulation is necessary for effective primary CD8+ T-cell influenza-specific immunity. Eur J Immunol. 2012;42(7):1744–1754. | |
Dolfi DV, Duttagupta PA, Boesteanu AC, et al. Dendritic cells and CD28 costimulation are required to sustain virus-specific CD8+ T cell responses during the effector phase in vivo. J Immunol. 2011;186(8):4599–4608. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.