")
Back to Journals » Cancer Management and Research » Volume 12
CCNY Accelerates Cylcin E Expression to Regulate the Proliferation of Laryngeal Carcinoma Cells via MEK/ERK Signaling Pathway
Authors Zhao X , Jiang M, Wang Z, Chen X , Wang H , Yue W , Cai C
Received 16 December 2019
Accepted for publication 27 May 2020
Published 23 June 2020 Volume 2020:12 Pages 4889—4898
DOI https://doi.org/10.2147/CMAR.S241620
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Antonella D'Anneo
Xiaoting Zhao,1,2 Mei Jiang,1,2 Ziyu Wang,2 Xiaohong Chen,3 Hongzhen Wang,4 Wentao Yue,1 Chao Cai5,6
1Central Laboratory, Beijing Obstetrics and Gynecology Hospital, Capital Medical University, Beijing, People’s Republic of China; 2Department of Cellular and Molecular Biology, Beijing Chest Hospital, Capital Medical University/Beijing Tuberculosis and Thoracic Tumor Research Institute, Tongzhou, Beijing, People’s Republic of China; 3Department of Otolaryngology, Head and Neck Surgery, Beijing Tongren Hospital, Capital Medical University, Beijing, People’s Republic of China; 4Department of Oncology, Rizhao City Hospital of Traditional Chinese Medicine, Rizhao, Shandong, People’s Republic of China; 5Beijing YouAn Hospital, Capital Medical University, Beijing, People’s Republic of China; 6Beijing Chest Hospital, Capital Medical University/Beijing Tuberculosis and Thoracic Tumor Research Institute, Tongzhou, Beijing, People’s Republic of China
Correspondence: Chao Cai; Wentao Yue Email [email protected]; [email protected]
Background: Laryngeal carcinoma is a common cancer among head and neck tumors, accounting for 0.5– 1% new cancer cases or deaths of all tumors throughout the body. Despite improvements in diagnostic and therapy, the prognosis of laryngeal carcinoma patients still remains poor. Thus, it is very important to identify the biomarkers involved in the molecular pathogenesis of laryngeal carcinoma. Cyclin Y (CCNY) is a conserved cell cycle regulator that acts as a growth factor in many cancers. The clinical significance of CCNY in laryngeal carcinoma remains unknown. The function of CCNY in laryngocarcinoma was studied in this paper.
Materials and Methods: CCNY knock-out cells were constructed by CRISPR/CAS9 technique. CCNY overexpression cells were also constructed based on CCNY knock-out cells. Cell growth ability was detected by MTS assay, high-content cell analysis, colony formation assays, and anchorage-independent growth assays. The protein levels in laryngocarcinoma cells were determined by Western blot. The role of CCNY in cell cycle progression was evaluated by flow cytometry.
Results: CCNY knock-out cells and CCNY up-regulation cell models were obtained successfully. Suppression of CCNY expression inhibited Hep2 cell growth. Cell growth was enhanced by the up-regulation of CCNY. The percentage of cells in G1 phase was altered when CCNY expression was down-regulated or up-regulated. The phosphorylation level of MEK and ERK as well as cyclin E protein level was also regulated by the expression level of CCNY.
Conclusion: In laryngocarcinoma cell line Hep2 cells, cell proliferation was controlled by CCNY. The expression of CCNY was involved in the cell cycle progress of Hep2 cells. It indicated that CCNY could promote cell growth by activating MEK/ERK/cyclin E signaling pathway.
Keywords: laryngocarcinoma, CCNY, cell cycle, ERK, cyclin E
Introduction
Laryngeal carcinoma is a common tumor among head and neck malignancy, accounting for 0.5–1% new cancer cases or deaths of all tumors throughout the body.1,2 The larynx is an important vocal and respiratory organ, how to preserve the function of the larynx after eradicating the tumor has always been the problem of surgical treatment. Radiotherapy (RT) can be used in some early laryngeal cancer, but most patients suffer from impaired function of the mucous and glandular of the pharynx, xerostomia (sensation of dryness in the mouth), dysphagia (difficulty in swallowing), and significantly decreased life quality.3,4 Meanwhile, radiotherapy may induce thyroid cancer and other head and neck cancers sometimes.3 Laser or part resection of throat may retain the laryngeal function of patients with early laryngeal cancer. But after the laser treatment or part surgeries of throat, the patient’s quality of pronunciation is not satisfactory.5 For advanced laryngeal cancer and recurrent laryngeal cancer, the retention rate of laryngeal function is very low at present.6 Biomarkers represent important tools that contribute to diagnosis, prognostic or prediction, respectively. It is very important to find a biomarker which could impact diagnosis or prognosis for laryngeal carcinoma.
Cancer is caused by uncontrolled cell division and abnormal cell proliferation due to variety reasons.7 Cell division is driven by cyclins, cyclin-dependent kinases (CDK) and other components of the core cell cycle machinery.8,9 Present studies indicate that almost all the cyclins play a key role in tumorigenesis and cancer progression.10 CCNY (Cyclin Y) is a highly conserved cell cycle protein of the cyclin superfamily of proteins which is firstly found in Drosophila.11,12 CCNY is generally expressed in human tissue cells (especially in brain and lung tissues) with a very low level, only highly expressed in testicular tissue cells.13 However, high level of CCNY protein is found in various human tumor tissues and cell lines such as colorectal cancer, breast cancer and brain glioma.13–15 In our previous study, CCNY was highly expressed in lung cancer tissues and lung cancer cells comparing to adjacent normal tissues from lung cancer patients or HEK293 cells.16 In lung cancer cells, ovarian cancer cells, hepatocellular carcinoma cells, glioma cells and breast cancer cells, cell growth was regulated by the expression of CCNY.14–19 It seems that CCNY plays important roles in cancer cell proliferation and CCNY might be an effective biomarker in tumors. The function of CCNY in laryngeal carcinoma cells was explored in this paper.
Materials and Methods
Reagents
RPMI 1640 and FBS (Fetal Bovine Serum) were purchased from Thermo Fisher (Waltham, MA); anti-CCNY, anti-GAPDH, anti-ERK1/2, anti-MEK, anti-pERK1/2, anti-cyclin E and anti-pMEK antibodies were obtained from Abcam (Cambridge, England, UK); Muse® Count & Viability Kit and Muse™ Cell Cycle Kit were from Millipore (Bedford, MA, USA); CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) was bought from Promega (Madison, WI, USA); Bisbenzimide Hoechst 33342 were purchased from Sigma (St. Louis, MO).
Cell Line
Laryngeal carcinoma Hep2 cell line was kept by Beijing Chest Hospital, Capital Medical University/Beijing Tuberculosis and Thoracic Tumor Research Institute from 1986. The study and use of Hep2 were reviewed and approved by the Research Ethics Committee in Beijing Chest Hospital, Capital Medical University as well as Beijing Obstetrics and Gynecology Hospital, Capital Medical University.
Construction of Recombinant Lentiviral Vectors and Infection into Cells
Recombinant lentiviral vectors were designed and packaged in HEK293T cells by GeneChem (Shanghai, China). The sgRNA sequence designed based on the human ccny mRNA (NM_181698) sequence was 5ʹ-TGAATGTGCCATCGTCACCC-3ʹ, 5ʹ-TGAACAGTGTCCGAACGAAC-3ʹ, 5ʹ-TAGATATCTGTCCGGCCAAC-3ʹ.
For cellular infection, cells were cultured at 6000 cells/well in 96-well culture plate and infected with recombinant lentivirus 24 hrs later. Stable clones were selected and identified by Western blot.
MTS Assay
Cell proliferation ability was assessed using the MTS assay. Briefly, 4000 cells/well were subcultured in 96-well culture plates. For the next 4 days, 20 μL MTS were added to each well and incubated at 37°C for 3 hrs each day. The absorbance was recorded at 490 nm by a microplate reader. The assay was repeated at least three times and the data are presented as the mean ± standard error of the mean.
Cell Proliferation Assay
Cells were subcultured in 96-well culture plates at 37°C. The next day, cells were washed three times with RPMI 1640 medium and nuclei were stained with Hoechst 33342 for 10 mins. After washing 3 times in RPMI 1640 medium, the total number of cells were counted by Cellomics Array Scan HCS Reader. The assay was repeated at least three times and the data are presented as the mean ± standard error of the mean.
Colony Formation Assay
Cells were seeded in a 6-well plate at 300 cells/well and cultured in RPMI 1640 medium with 10% FBS. The cells were cultured for 8 days. At last, the cells were washed twice with PBS, stained with 2% Gentian violet for 20 mins, and then washed with water. The number of colonies with >50 cells in each well was counted. The assay was repeated at least three times in triplicate.
Anchorage-Independent Growth Assay
A total of 1000 cells were resuspended in 1 mL of 0.6% soft-agar in 6-well plates coated with 1.2% soft-agar. 21 days later, the number of colonies was counted manually. All experiments were performed in triplicate and at least repeated three times.
Western Blot Analysis
Total proteins (10 μg/well) were separated using 12% SDS-PAGE and then wet-transferred onto nitrocellulose membranes. After blocked by 5% milk, membranes were incubated with primary antibodies overnight at 4°C. Then, the membranes were incubated with corresponding HRP-conjugated secondary antibodies for 2 hrs at room temperature. Protein levels were detected by using the ECL reagents.
Cell Cycle Analysis
Cell cycle was detected by Muse™ Cell Cycle Kit. Briefly, cells were washed by PBS and fixed by ice-cold 70% ethanol overnight at −20°C. Ethanol-fixed cells were stained by Muse™ Cell Cycle Reagent and analyzed on Muse™ Cell Analyzer.
Statistical Analysis
All values are expressed as the mean ± SEM. For comparison, the statistical differences of 2 groups, unpaired Student’s t-test was used by SPSS Statistics, version 19.0. P<0.05 (*) was considered different; P<0.01 (**) was considered significantly different.
Results
Cell Proliferation Ability Was Inhibited by the Suppression of CCNY Expression
In order to determine the function of CCNY in laryngocarcinoma, CRISPR/CAS9 technique was used to construct CCNY knock-out cells. As shown in Figure 1A, three different sgRNAs, sgCCNY-1, 2, 3, were used via infection of recombinant lentivirus. The CCNY level was almost disappeared when sgCCNY-3 was used. It indicated that the sgCCNY was the most effective sgRNA. In order to exclude off-target effect, three clones (CCNY cas9 clone-1, CCNY cas9 clone-2, CCNY cas9 clone-3) derived from cells with sgCCNY3 were used in the following research (Figure 1B).
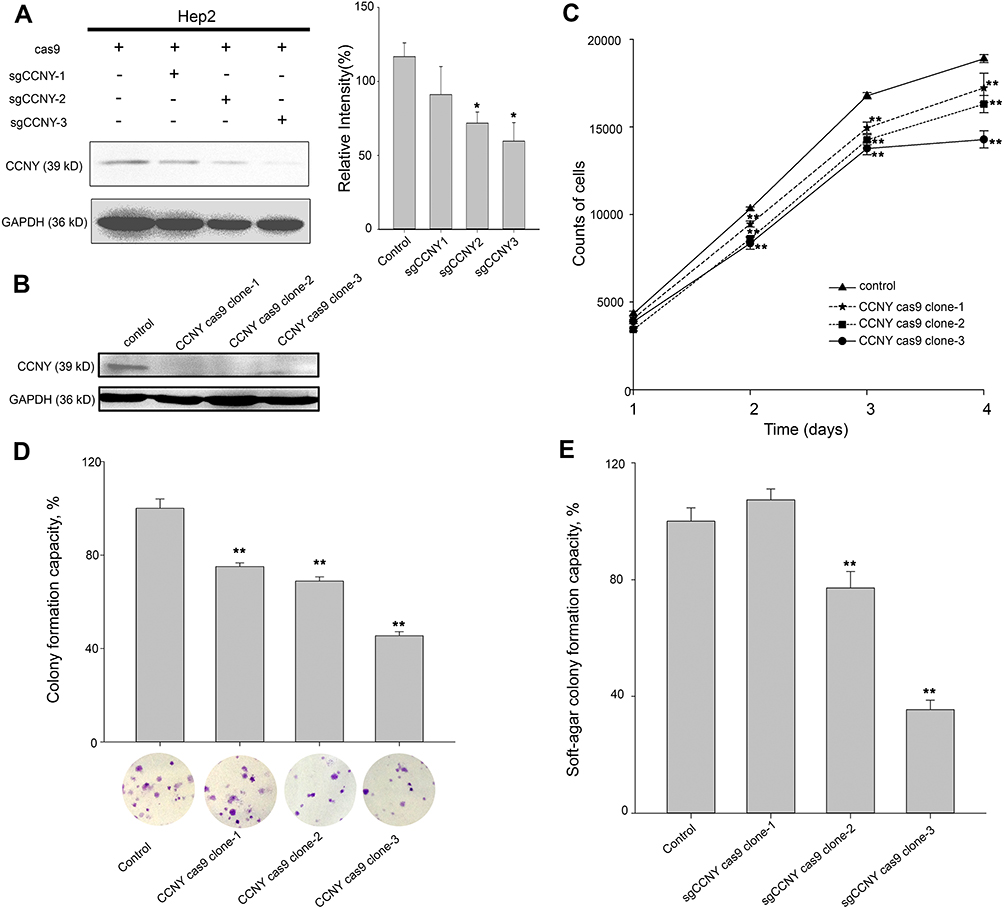 |
Figure 1 Cell growth ability was suppressed by the knock-out of CCNY. (A) CRISPR/CAS9 technique was used to construct the CCNY knockout cell strains. Three different sgCCNYs (sgCCNY-1, sgCCNY-2, sgCCNY-3) were used. The left panel, the expression level of CCNY was detected by Western blot, with GADPH used as the internal loading controls. The right panel, the level of CCNY was quantified by gray analysis. *P<0.05 (t-test). (B) Three CCNY knock-out cell clones (CCNY cas9 clone-1, CCNY cas9 clone-2, CCNY cas9 clone-3) derived from cells infected by recombinant lentivirus with sgCCNY-3 were identified by Western blot. (C) Cell growth curve was determined by cell counts. The total number of cells were counted by Cellomics ArrayScan HCS Reader. **P<0.01 (t-test). (D) Colony formation efficiency of cells. The data are expressed as the mean ± standard error. **P<0.01 (t-test). (E) Soft-agar colony formation ability of cells. The data are expressed as the mean ± standard error. **P<0.01 (t-test). |
Cell growth curve was drawn by cell counts. In Hep2 cells, cell proliferation was inhibited by the knock-out of CCNY (Figure 1C). Same results were obtained in colony formation assay and anchorage-independent growth assay (Figure 1D and E). The number of colonies were obviously decreased when the expression level of CCNY was suppressed.
Cell Growth Was Enhanced by the Up-Regulation of CCNY
As shown in Figure 2A, CCNY was re-expressed in CCNY knock-out Hep2 cells. MTS assay was used to detect the cell growth curve of Hep2 cells. As a result, cell growth ability was elevated when CCNY was re-expressed in Hep2 cells (Figure 2B). In colony formation assay, colonies were increased by the up-regulation of CCNY (Figure 2C). Similar results were got in the soft-agar formation assay (Figure 2D). It indicated that cell proliferation was regulated by the expression of CCNY.
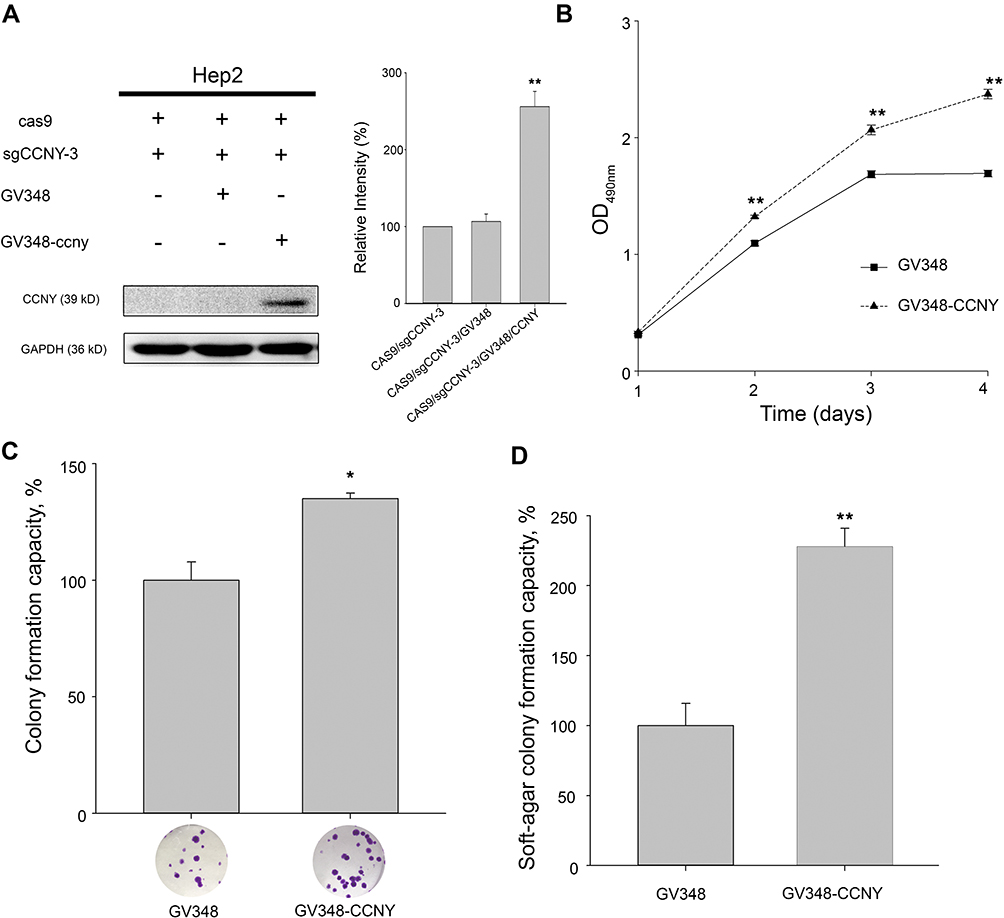 |
Figure 2 Cell proliferation was enhanced by the overexpression of CCNY. (A) CCNY was re-expressed in Hep2 cells by the infection of recombinant lentivirus. The left panel, the expression of CCNY was determined by WB. GAPDH is used as a loading control. The right panel, the level of CCNY was quantified by gray analysis. **P<0.01 (t-test). (B) Cells were subcultured in 96-well plates and an MTS assay was performed. Absorbance at 490 nm (y axis) was determined at every 24-hrs interval. The data are expressed as the mean ± standard error. **P<0.01 (t-test). (C) Colony formation efficiency of cells. The data are expressed as the mean ± standard error. *P<0.05 (t-test). (D) Soft-agar colony formation ability of cells. The data are expressed as the mean ± standard error. **P<0.01 (t-test). |
CCNY Promoted the Expression of Cyclin E via MAPK/ERK Signaling Pathway
In order to study the mechanism of CCNY, cell cycle analysis was carried out. As indicated in Figure 3, the percentage of cells in G0/G1 phase was increased in CCNY knock-out Hep2 cells. And the population of cells entering S phase was decreased by the suppression of CCNY expression. It illustrated that the down-regulation of CCNY leads to cell cycle G1/S arrest in Hep2 cells. Cell cycle distribution was also controlled by the up-regulation of CCNY in Hep2 cells. Cells entering S phase was increased obviously while cell population of cells in G0/G1 phase was obviously decreased due to the overexpression of CCNY in Hep2 cells (Figure 4). We thought that cell cycle progression could be regulated by the expression of CCNY.
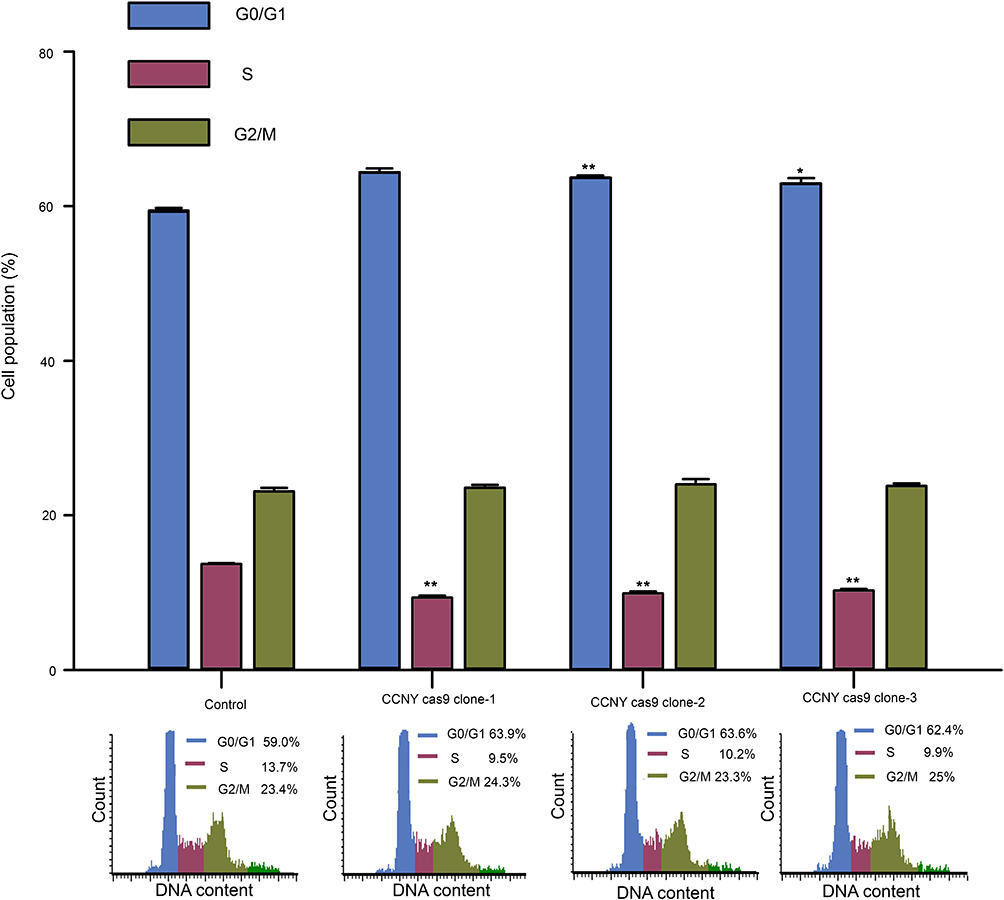 |
Figure 3 Cell cycle G1/S phase arrest was induced by the suppression of CCNY expression. Cell cycle distribution of cells was determined by flow cytometry. The histogram showed the statistical data expressed as the mean ± standard error. *P<0.05 (t-test), **P<0.01 (t-test). |
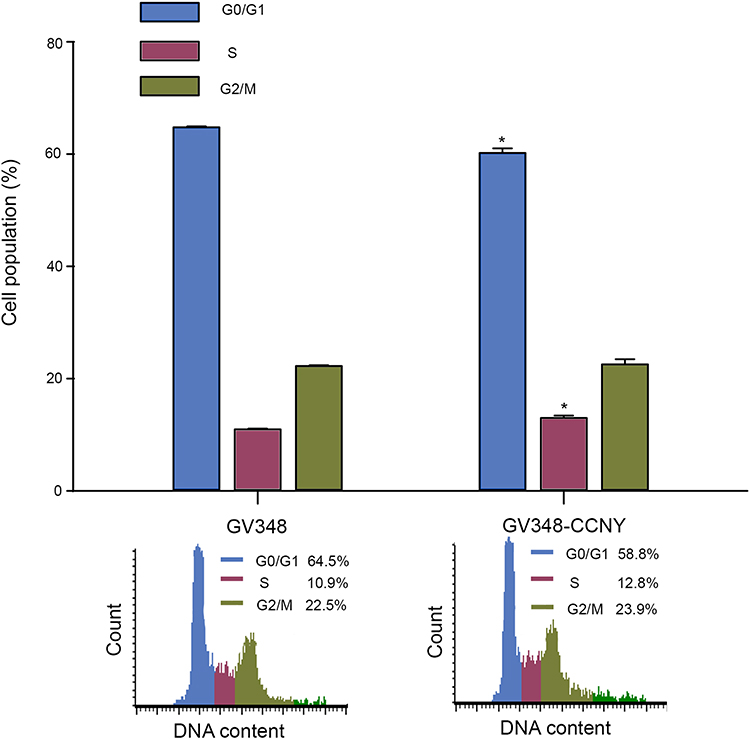 |
Figure 4 Cell cycle analysis. Cell cycle distribution of cells was determined by flow cytometry. The histogram showed the statistical data expressed as the mean ± standard error. *P<0.05 (t-test). |
The level of proteins regulating cell proliferation was also detected by Western blot. The phosphorylation level of MEK and ERK1/2 as well as cyclin E level was weakened when CCNY was down-regulated in Hep2 cells (Figure 5A). And phosphorylation level of MEK and ERK1/2 as well as cyclin E level was up-regulated when CCNY was re-expressed in Hep2 cells (Figure 5B). But the phosphorylation level of p38, Rb, Akt and β-catenin was not affected by the expression of CCNY (Figure 5C and D). It indicated that CCNY regulated cell cycle progression by promoting the expression of cyclin E via activating MAPK/ERK signaling pathway.
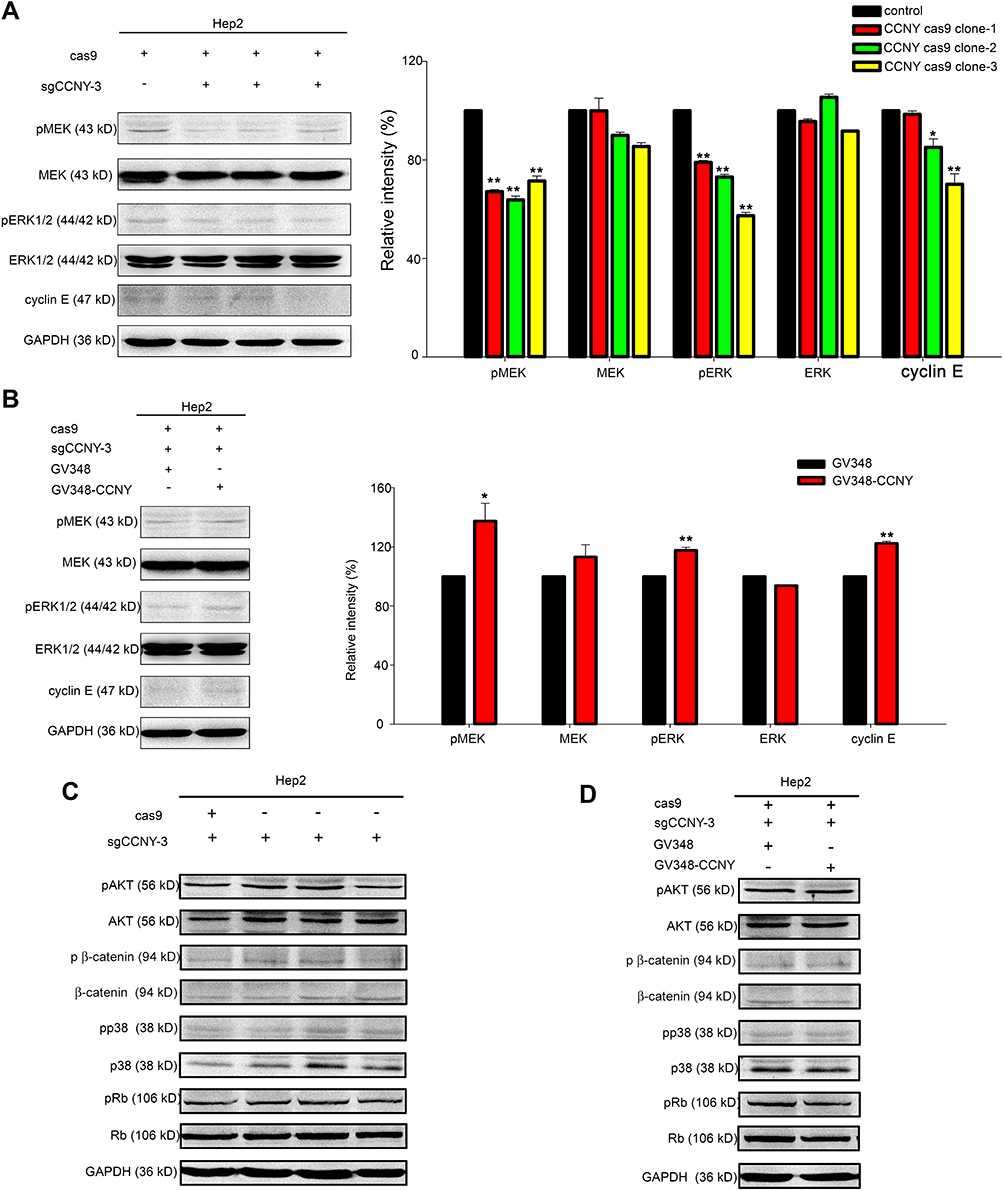 |
Figure 5 CCNY could induce cyclin E expression via MEK/ERK signaling pathway. (A and B) Western blot analysis of ERK1/2, MEK and their phosphorylated forms as well as cyclin E. GAPDH was used as internal loading controls. The levels of proteins were quantified by gray analysis as shown in the right panel. *P<0.05 (t-test), **P<0.01 (t-test). (C and D) The levels of pAKT, AKT, p β-catenin, β-catenin, pp38, p38, pRb, Rb were detected by Western blot. GAPDH was used as internal loading controls. |
Discussion
CCNY is a novel cyclin and plays very important roles in tumor cells. In this paper, we found that cell growth was obviously inhibited by the suppression of CCNY expression while cell proliferation ability was enhanced due to the up-regulation of CCNY expression in Hep2 cells, a kind of laryngeal carcinoma cells. In our results, cell cycle G1/S progression was controlled by CCNY. It indicated that CCNY regulated cell cycle progression by promoting the expression of cyclin E via activating MAPK/ERK signaling pathway.
In Hep2 cells, CCNY knock-out cells were constructed successfully by CRISPR/CAS9 technique. The function of CCNY was examined in 3 CCNY knock-out Hep2 cell clones to exclude the off-target effect. Cell growth was examined by cell counts, MTS, colony formation assay and soft-agar colony formation assay. Down-regulation of CCNY attenuated Hep2 cell growth, colony formation ability and soft-agar colony formation ability. The cell proliferation ability was recovered when CCNY was re-expressed in Hep2 cells. These results were similar with those obtained in many other tumor cells. In human breast cancer cells, RNAi-mediated down-regulation of CCNY resulted in a significant reduction in cell growth and colony formation ability.15 Overexpression of CCNY significantly increased the cell proliferation in hepatocellular carcinoma cells (HCC), and tumor growth was enhanced by the up-regulation of CCNY expression in the xenograft mouse model.19 There were similar results in lung cancer cells, breast cancer cells, glioma cells and so on.14,15,17 Above all, CCNY was involved in the cell growth regulation pathway in many cancers.
The main function of cyclin is the regulation of cell cycle progression. The cell cycle distribution of Hep2 cells was determined by flow cytometry. As a result, cell cycle G1/S progression was controlled by CCNY expression. G1/S arrest happened by the blocking of CCNY expression. CCNY expression could also promote G1/S progression in Hep2 cells. In lung cancer cells, cell cycle G1/S progression was blocked by CCNY sgRNA.16 Breast cancer cells were arrested in the G0/G1 phase by knockdown of CCNY.15 It indicated that CCNY regulated cell proliferation by promoting cell cycle G1/S progression.
CCNY, as a cyclin, needs to interact with a CDK to form a cyclin/CDK complex to regulate the cell cycle progression. It is said that the CDK interacting with CCNY is CDK14 (PFTK1) or CDK16 (PCTK1).20–22 Cell proliferation could be regulated by CCNY/PCTK1 complex.22,23 In HEK293 cells, the Wnt receptor LRP6 could be phosphorylated by CCNY/PFTK1 complex which could activate the canonical Wnt signaling pathway.24 It seemed that cell cycle G2/M progression was controlled by Wnt signaling pathway activated by CCNY/PFTK1.24,25 CCNY/PFTK1 complex could also activate Wnt signaling in HCC.25,26 In ovarian cancer A2780 cells, beta-catenin expression was increased in the nucleus when CCNY was overexpressed. It was thought that CCNY might promote cell proliferation via activating the Wnt signaling pathway.18 But we got different results in Hep2 cells, and cell cycle G1/S progression not G2/M was affected by the expression of CCNY. These results were similar with that obtained in lung cancers and breast cancer cells.15,16 The expression of β-catenin as well as its phosphorylated level was not affected by the level of CCNY. We thought that CCNY served to cell growth regulation independent of Wnt/β-catenin pathway. In order to explore the CCNY mechanism of promoting cell growth, the activity of several signaling pathways as well as the related proteins involved in cell proliferation regulation was determined. As a result, the phosphorylated levels of p38, Akt and Rb were not changed while CCNY was down-regulated or up-regulated. It indicated that MEK/ERK signaling pathway was regulated by the expression of CCNY in Hep2 cells. The MAPK signaling pathway plays a critical role in many important cellular processes. And MEK/ERK pathway is one of the MAPK signaling pathways and serve important role in cell proliferation, drug resistance and malignant transformation.27 Cell cycle progression was also controlled by MEK/ERK, and levels of cyclin D1, cyclin E, Cdk2, as well as Cdk4 were regulated by MEK/ERK pathway.28,29 Due to the important function of CDK2/cyclin E at the G1/S phase transition,30 we also detected the expression level of cyclin E. As expected, the level of cyclin E was changed by down-regulation or up-regulation of CCNY. We thought that CCNY promoting cell cycle progression by regulating the expression of cyclin E.
Conclusions
In this study, the function of CCNY was explored in Hep2 cells. It indicated cell growth was controlled by the expression of CCNY. Further research illustrated that the G1/S phase transition was regulated by CCNY. We thought that the MEK/ERK signaling pathway could be activated by CCNY expression. At last, we also found that CCNY was involved in the cyclin E expression regulation. We thought that CCNY promoting cell cycle progression by stimulating the expression of cyclin E via MEK/ERK signaling pathway. Further studies are recommended to determine the role of CCNY as a potential biomarker in laryngeal carcinoma.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
2. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338
3. Amemiya K, Shibuya H, Yoshimura R, et al. The risk of radiation-induced cancer in patients with squamous cell carcinoma of the head and neck and its results of treatment. Br J Radiol. 2005;78(935):1028–1033. doi:10.1259/bjr/86352309
4. Siddiqui F, Movsas B. Management of radiation toxicity in head and neck cancers. Semin Radiat Oncol. 2017;27(4):340–349. doi:10.1016/j.semradonc.2017.04.008
5. Xu W, Han D, Hou L, et al. Voice function following CO2 laser microsurgery for precancerous and early-stage glottic carcinoma. Acta Otolaryngol. 2007;127(6):637–641. doi:10.1080/00016480600987776
6. Dias FL, Lima RA, Kligerman J, et al. Therapeutic options in advanced laryngeal cancer: an overview. ORL J Otorhinolaryngol Relat Spec. 2005;67(6):311–318. doi:10.1159/000090040
7. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi:10.1016/S0092-8674(00)81683-9
8. Arata Y, Takagi H. Quantitative studies for cell-division cycle control. Front Physiol. 2019;10:1022. doi:10.3389/fphys.2019.01022
9. Liu LM, Kolodziejczyk W, Sicinski A. The cell cycle in stem cell proliferation, pluripotency and differentiation. Nat cell Biol, 2019;21(9):1060–1067.
10. Stamatakos M, Palla V, Karaiskos I, et al. Cell cyclins: triggering elements of cancer or not? World J Surg Oncol. 2010;8:111. doi:10.1186/1477-7819-8-111
11. Liu D, Finley RL
12. Liu D, Guest S, Finley RL
13. Ying-Tao Z, Yi-Ping G, Lu-Sheng S, et al. Proteomic analysis of differentially expressed proteins between metastatic and non-metastatic human colorectal carcinoma cell lines. Eur J Gastroenterol Hepatol. 2005;17(7):725–732. doi:10.1097/00042737-200507000-00006
14. Xu Y, Wang Z, Wang J, et al. Lentivirus-mediated knockdown of cyclin Y (CCNY) inhibits glioma cell proliferation. Oncol Res. 2010;18(8):359–364. doi:10.3727/096504010X12644422320582
15. Yan F, Wang X, Zhu M, et al. RNAi-mediated downregulation of cyclin Y to attenuate human breast cancer cell growth. Oncol Rep. 2016;36(5):2793–2799. doi:10.3892/or.2016.5126
16. Yue W, Zhao X, Zhang L, et al. Overexpression of cyclin Y in non-small cell lung cancer is associated with cancer cell proliferation. Sci China Life Sci. 2010;53(4):511–516. doi:10.1007/s11427-010-0090-8
17. Yue W, Zhao X, Zhang L, et al. Cell cycle protein cyclin Y is associated with human non-small-cell lung cancer proliferation and tumorigenesis. Clin Lung Cancer. 2011;12(1):43–50. doi:10.3816/CLC.2011.n.006
18. Liu H, Shi H, Fan Q, et al. Cyclin Y regulates the proliferation, migration, and invasion of ovarian cancer cells via Wnt signaling pathway. Tumour Biol. 2016;37(8):10161–10175. doi:10.1007/s13277-016-4818-3
19. Shi K, Ru Q, Zhang C, et al. Cyclin Y modulates the proliferation, invasion, and metastasis of hepatocellular carcinoma cells. Med Sci Monit. 2018;24:1642–1653. doi:10.12659/MSM.906075
20. Jiang M, Gao Y, Yang T, et al. Cyclin Y, a novel membrane-associated cyclin, interacts with PFTK1. FEBS Lett. 2009;583(13):2171–2178. doi:10.1016/j.febslet.2009.06.010
21. Li S, Song W, Jiang M, et al. Phosphorylation of cyclin Y by CDK14 induces its ubiquitination and degradation. FEBS Lett. 2014;588(11):1989–1996. doi:10.1016/j.febslet.2014.04.019
22. Hernandez-Ortega S, Sanchez-Botet A, Quandt E, et al. Phosphoregulation of the oncogenic protein regulator of cytokinesis 1 (PRC1) by the atypical CDK16/CCNY complex. Exp Mol Med. 2019;51(4):44. doi:10.1038/s12276-019-0242-2
23. Si Y, Liu J, Shen H, et al. Fisetin decreases TET1 activity and CCNY/CDK16 promoter 5hmC levels to inhibit the proliferation and invasion of renal cancer stem cell. J Cell Mol Med. 2019;23(2):1095–1105. doi:10.1111/jcmm.14010
24. Davidson G, Shen J, Huang YL, et al. Cell cycle control of wnt receptor activation. Dev Cell. 2009;17(6):788–799. doi:10.1016/j.devcel.2009.11.006
25. Wang X, Jia Y, Fei C, et al. Activation/Proliferation-associated Protein 2 (Caprin-2) Positively Regulates CDK14/Cyclin Y-mediated Lipoprotein Receptor-related Protein 5 and 6 (LRP5/6) Constitutive Phosphorylation. J Biol Chem. 2016;291(51):26427–26434. doi:10.1074/jbc.M116.744607
26. Sun T, Co NN, Wong N. PFTK1 interacts with cyclin Y to activate non-canonical Wnt signaling in hepatocellular carcinoma. Biochem Biophys Res Commun. 2014;449(1):163–168. doi:10.1016/j.bbrc.2014.05.002
27. Yaeger R, Corcoran RB. Targeting alterations in the RAF-MEK pathway. Cancer Discov. 2019;9(3):329–341. doi:10.1158/2159-8290.CD-18-1321
28. McCubrey JA, Steelman LS, Chappell WH, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773(8):1263–1284. doi:10.1016/j.bbamcr.2006.10.001
29. Woods D, Parry D, Cherwinski H, et al. Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol Cell Biol. 1997;17(9):5598–5611. doi:10.1128/MCB.17.9.5598
30. Ghule PN, Seward DJ, Fritz AJ, et al. Higher order genomic organization and regulatory compartmentalization for cell cycle control at the G1/S-phase transition. J Cell Physiol. 2018;233(10):6406–6413. doi:10.1002/jcp.26741
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.