")
Back to Journals » Infection and Drug Resistance » Volume 15
CCL25 Inhibition Alleviates Sepsis-Induced Acute Lung Injury and Inflammation
Authors Xia D , Wang S, Liu A, Li L, Zhou P, Xu S
Received 4 December 2021
Accepted for publication 20 May 2022
Published 25 June 2022 Volume 2022:15 Pages 3309—3321
DOI https://doi.org/10.2147/IDR.S352544
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Suresh Antony
Demeng Xia,1,* Sheng Wang,2,* Anwei Liu,1,3,* Lei Li,2 Panyu Zhou,2 Shuogui Xu2
1Luodian Clinical Drug Research Center, Shanghai Baoshan Luodian Hospital, Shanghai University, Shanghai, People’s Republic of China; 2Department of Emergency, Changhai Hospital, The Naval Medical University, Shanghai, People’s Republic of China; 3Department of Critical Care Medicine, Hospital of Southern Theatre Command of PLA, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shuogui Xu; Panyu Zhou, Department of Emergency, Changhai Hospital, Naval Medical University, Xiangyin Road, Shanghai, 200433, People’s Republic of China, Tel +8613176535161 ; +8619821317892, Email [email protected]; [email protected]
Purpose: Acute lung injury (ALI) is a common clinical syndrome with high mortality. The chemokine ligand 25 (CCL25) is involved in inflammation, leukocyte trafficking and immunoregulation. However, the role and mechanism of CCL25 in ALI are not fully understood yet. The aim of this study was to explore the relationship between acute lung injury and CCL25.
Patients and Methods: In this study, we first examined chemokine expression in sepsis patients and found that serum CCL25 expression levels were relatively high in sepsis patients compared to healthy individuals. Based on this, we designed in vitro and in vivo experiments to verify the validity of the theory. In vitro, we used lipopolysaccharide-stimulated human pulmonary microvascular endothelial cells (HPMECs). In vivo, we established male C57BL/6 mice cecal ligation puncture (CLP) model of sepsis.
Results: In vitro, we used lipopolysaccharide-stimulated human pulmonary microvascular endothelial cells (HPMECs) and found significantly higher expression of CCL25 by enzyme-linked immunosorbent assay. Inhibition of CCL25 resulted in a significant decrease in the expression of inflammatory cytokines in HPMECs. In addition, we found that CCL25 promoted increased endothelial permeability by reducing the expression of tight junction proteins and was associated with activation of the P38 MAPK pathway by measuring the transepithelial electrical resistance and fluorescence intensity of fluorescein isothiocyanate. Results from luciferase assays and chromatin immunoprecipitation assays showed that inhibition of NF-κB activity in HPMECs decreased CCL25 expression, but addition of recombinant CCL25 increased cell permeability and inflammatory cytokine expression. In vivo, we established male C57BL/6 mice cecal ligation puncture (CLP) model of sepsis. We found that inhibition of CCL25 significantly reduced inflammatory cytokine expression in a CLP-induced sepsis model, thereby alleviating lung tissue damage in mice.
Conclusion: Our study suggests that CCL25 contributed to the development of ALI by modulating the functions of microvascular endothelial cells.
Keywords: CCL25/CCR11, inflammatory cytokines, cell permeability, acute lung injury, P38, NF-κB
Introduction
Acute lung injury (ALI), characterized by pulmonary edema, severe hypoxemia, neutrophil infiltration and inflammatory process,1 is a common clinical pulmonary disorder.2 ALI has a high mortality rate, threatening millions of traumatic infectious patients despite various therapeutic strategies that have been made in the past few decades.3 The main pathological change of ALI is acute inflammatory responses with disruption of endothelial integrity, accumulation of inflammatory cells, interstitial edema and leakage of proteins into the alveolar space.4 Endothelial dysfunction and pulmonary microvascular hyper-permeability to fluids and proteins are the hallmarks of ALI. Lipopolysaccharide (LPS), a component of bacterial cell membrane, can significantly increase lung permeability, thereby reducing the expression of SIRT1 and tight junction proteins (TJs).5 It has also been widely used to establish mouse ALI model due to its ability to induce the recruitment of inflammatory cells and the production of inflammatory cytokines.6,7 Furthermore, sepsis, a systemic condition with several degrees of severity due to dysregulated activation of the hemostatic and innate immune systems, is a lethal syndrome and a main cause of ALI.8
In recent years, studies have shown that chemokines are closely related to the pathogenesis of septic lung injury and ALI.9,10 Chemokines belong to a series of small molecule cytokine superfamilies with similar structure and 20–70% amino acid homology.11 They can be divided into four families: alpha-chemokine (CXC subgroup), beta-chemokine (CC subgroup), gamma-chemokine (C subgroup) and delta-chemokine (CX3C subgroup).12 The chemokine (C–C motif) ligand 25 (CCL25), also known as thymus-expressed chemokine, was found in the thymus and the intestinal epithelium of humans and mice.13 Through controlling the recruitment of many kinds of immune cells, especially innate immune cells, CCL25 plays an essential role in regulating inflammation, leukocyte trafficking and immune differentiation.14 It has been proved that CCL25 is involved in the activation of Surfactant protein C-1 by toll-like receptor 4, which disrupts Treg/Th17 homeostasis and leads to lung injury after necrotizing enterocolitis.15 In addition, studies have shown that NF-κB plays a crucial role in the pathogenesis of sepsis-induced organ damage, typified by the Toll-like receptor 4-NF-κB pathway.16,17 However, neither the impact of CCL25 on the development of ALI nor whether CCL25 is involved in regulating the NF-κB pathway has been reported in detail.
In this study, we treated the human pulmonary microvascular endothelial cells (HPMECs) with LPS in vitro and established mouse sepsis model using cecal ligation and puncture (CLP) method in vivo to address the involvement of CCL25 in the pathogenesis of ALI, thereby providing theoretical basis for the diagnosis and treatment of ALI. A schematic of the mechanism of CCL25 in the pathogenesis of ALI is provided in Figure 1.
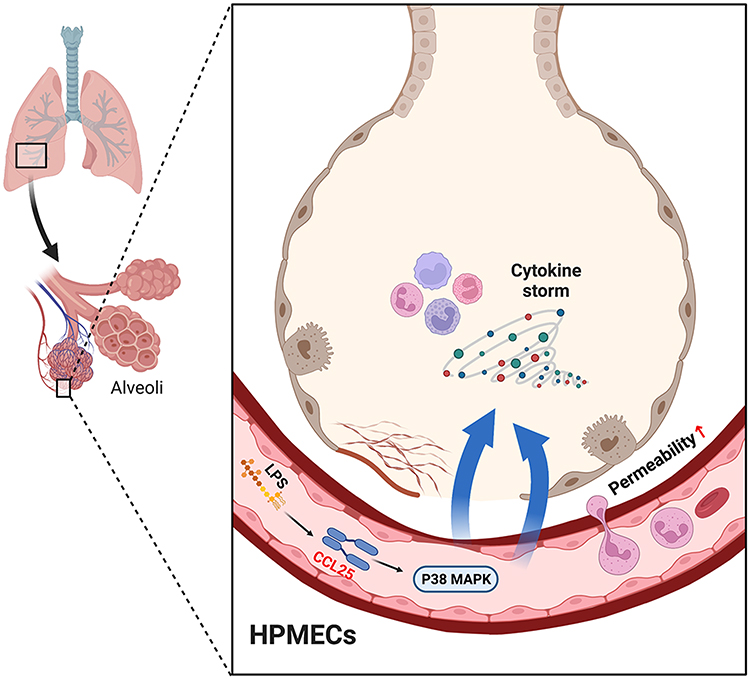 |
Figure 1 A schematic of the mechanism of CCL25 in the pathogenesis of ALI. |
Materials and Methods
Human Samples
All samples were selected from inpatients who were hospitalized between October 2017 and July 2019 in the intensive care unit (ICU). Sepsis, defined by the European Society of Intensive Care Medicine and the Society of Critical Care Medicine (Sepsis-3),18,19 refers to a life-threatening organ dysfunction caused by a dysregulated host response to infection. Patients with suspected infection and a total SOFA score ≥2 points within 48 h of ICU were eligible for inclusion in this study. The exclusion criteria were as follows: (1) patients aged <18 years; (2) parenteral nutrition within 48 h after ICU admission; (3) pregnant; (4) refuse to participate in the study; and (5) infections that were difficult to confirm by blood culture. The blood from 30 enrolled patients was sampled within 48 h after ICU admission (Table 1). In addition, 30 healthy control serum samples were collected from volunteers who had not been hospitalized or had a history of medication within the past 6 months, and were shown to be normal in physical examination at our hospital. This study was approved by the Ethics Committee of Shanghai Changhai Hospital, and informed consents were signed by the participants.
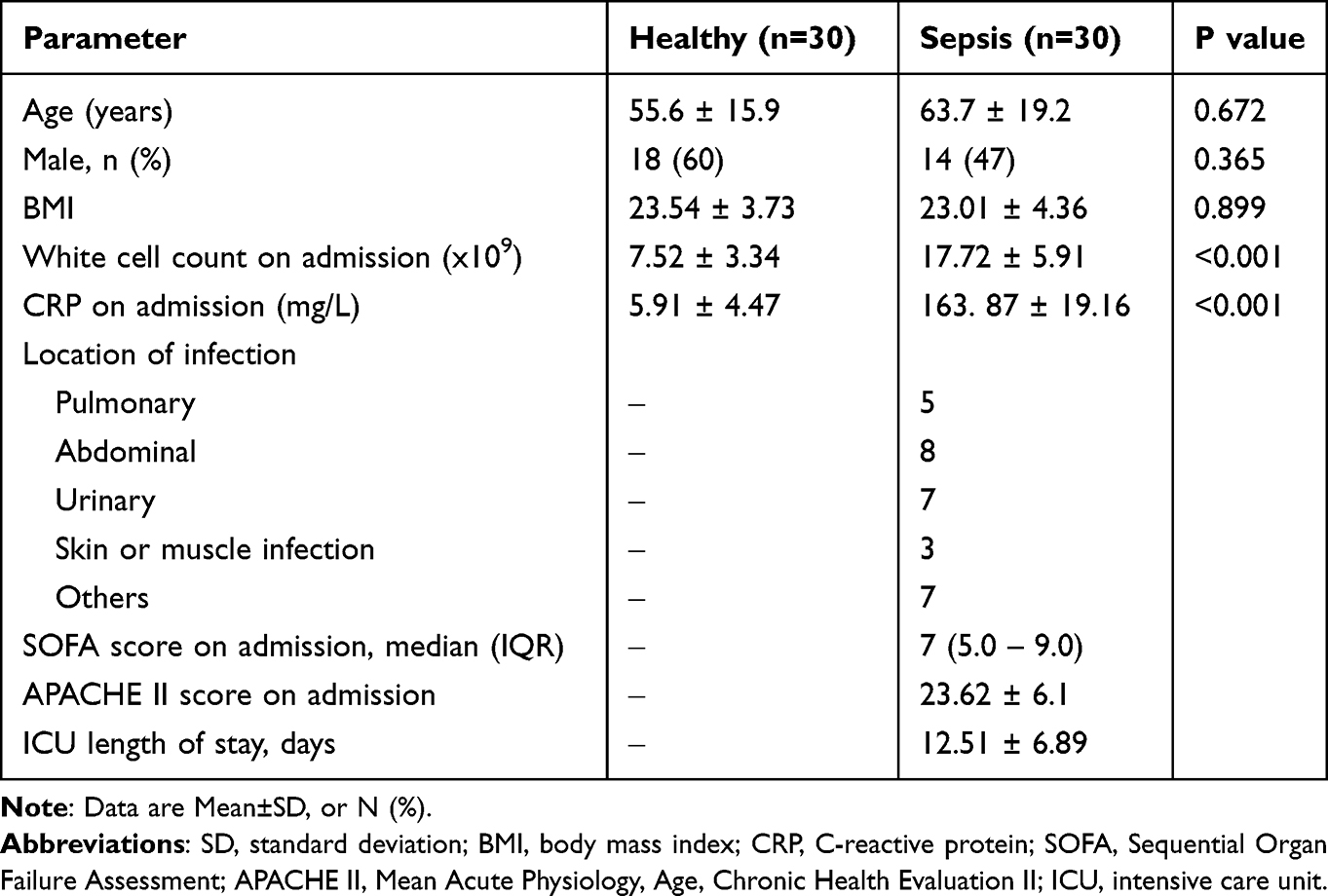 |
Table 1 General Characteristics of the Participants Involved in This Study |
Cell Culture
HPMECs were obtained from the American Type Culture Collection (Manassas, VA), and incubated in Dulbecco’s Modified Eagle Medium (Hyclone, Logan, Utah, USA) with 10% fetal bovine serum (Thermo Fisher Scientific, Inc.) and 1% penicillin/streptomycin (Solarbio) in an incubator containing 5% CO2 at 37°C. After that, the cells were incubated with LPS at 0, 25, 50, 100 and 200 ng/mL, respectively, for 24 h. Additionally, it was treated with anti-CCL25 and recombinant CCL25 (Abcam).
Enzyme-Linked Immunosorbent Assay (ELISA)
Expressions of CCL19, CCL21, CCL25, TNF-α, IL-1β and IL-6 in serum or HPMEC supernatants were measured by ELISA kits (Beijing Chenglin Biotechnology, Co. Ltd). In brief, capture mAb (50 μL) was added to the ELISA plate for incubation and washed 5 times with PBST. Then, the plate was treated with bovine serum albumin (50 μL) blocking solution (10 mg/mL). Serum (50 μL) and biotin-conjugated detector mAb (50 μL) were applied to the wells and the plate was incubated for 30 min at 37°C, and probed with 10 mg/mL of avidin-HRP. Tetramethylbenzidine substrate solution 50 μL (10 mg/mL) was added for color reaction of the Ag-Ab complex and absorbance was measured using the automated ELISA reader (Bio-Rad model 550, Irvine, CA) at 450 nm after final rinsing with PBST. The concentration was determined by comparing the optical density to a standard curve.
Quantitative Polymerase Chain Reaction (Q-PCR)
RNA isolation was performed by Trizol method and the concentration and the purity of RNA were measured spectrophotometrically. Reverse transcription was performed by the RevertAid First Strand cDNA Synthesis Kit (#K1622, Fermentas). Relative gene expression was calculated by ABI Prism 7300 SDS Software. The primers used are CCL25-forward, 5’ GCCTGCTGCGATATTCTAC 3’; CCL25-reverse, 5ʹGCTGATGGGATTGCTAAAC 3; β-actin-forward, 5’ AATGAGCGGTTCCGTTGC 3’; β-actin-reverse, 5’ TCTTCATGGTGCTGGGAG 3’. β-actin was included as the internal control. The mRNA levels were all quantified via SYBR Green PCR Kit (Thermo Fisher) on an ABI7300 system (Applied Biosystems) and normalized by β-actin using the 2−ΔΔCt method.20
Western Blot
RIPA buffer (BYL40825, JRDUN Biotechnogy, Shanghai, China) containing protease inhibitor and phosphatase inhibitors were added to HPMEC and lung tissues to fully lyse the cells and the tissues. The homogenates were centrifuged at 12,000 g for 10 min at 4°C and the BCA assay kit (PICPI23223, Thermo) was used to measure total protein concentration. Further, moderate proteins were subjected to 10% sodium dodecyl polyacrylamide gel electrophoresis and transferred to the PVDF membrane which was blocked with 5% skim milk (BYL40422, BD repackaging) at room temperature for 1 h and then incubated with the primary antibody which was diluted to the desired concentration for 2 h at room temperature, anti-CCL25 (1:10,000, Ab200343, Abcam), anti-occludin (1:50,000, Ab167161, Abcam), anti-zonula occludens-1 (ZO-1) (1:1000, #13663, CST), anti-P38 (1:1000, #8690, CST), anti-p-P38 (1:1000, #4511, CST), anti-NF-κB (1:1000, Ab16502, Abcam), anti-H3 (1:1000, 17,168-1-AP, Proteintech) and anti-β-actin (1:1000, Ab8227, Abcam). Subsequently, the membrane was washed and probed with an HRP-conjugated goat anti-rabbit secondary antibody (1:1000, A0208, Beyotime Biotechnology, China) for 1h at 37°C. The bands were visualized by the enhanced chemiluminescence (ECL) method.
Measurement of Transepithelial Electrical Resistance (TEER)
TEER measurement was used widely in monitoring cell growth and evaluating the cell–cell tight junction integrity.21 Resistance meter and electrode were calibrated. Then, the electrode was rinsed with sterilized electrolyte solution after its functional detection. Cells of each group were inoculated into the upper chamber of a 24-well Transwell plate (1×104 cells per well). Culture medium was added to 100 μL of upper chamber and 600 μL of lower chamber in a 5% CO2 incubator at room temperature. 24h later, the culture medium was changed, and cells were overgrown 48h later. The TEER of cells in each group was measured by a resistor. Meanwhile, a blank well was set up to determine the TEER. The resistance per unit area was calculated according to the following formula: TEER (resistance per unit area, Ω·cm2) = (R experiment – R blank) (resistance measurement, Ω) × effective membrane area (cm2).
FITC-Dextran Uptake
FITC was known to be a large fluorescent substance and fluorescence value could reflect permeability of mucosal epithelium.22 The logarithmic growth cells of each group were inoculated into the upper chamber of the 24-well Transwell plate (1×105 cells per well). A 100 μL and 600 μL of culture medium were added to the upper and lower chamber, respectively, in a 5% CO2 incubator at room temperature. The culture medium was changed per day. Cells were cultured to the full concentration. Starvation cells in the upper chamber of serum-free medium were replaced for 4h before the experiment and were given corresponding treatment in line with experimental groups. Following this, FITC-dextran (1 mg/mL) was added in an incubator with 5% CO2 at 37°C for 5 min. The basal culture medium was used as the base value, and the medium was supplemented for incubation for 24 h. The fluorescence intensity of FITC was detected by absorbing 200 μL from the substrate culture medium using a microplate analyzer (excitation wavelength: 490 nm, emission wavelength: 520 nm), and the permeability rate was calculated to the standard curve.
Animals
Male C57BL/6 mice aged 6–8 weeks (weighing 18–22 g) were obtained from the hospital. Animal experiments were performed according to the Guide for the Care and Use of Laboratory Animals. The experimental procedures were approved by the Ethics Committee of Shanghai Changhai Hospital. Thirty-six mice were randomly categorized into the control group, CLP model control group (IgG), CLP model group (Anti-CCL25, Abcam) with 12 in each group. Control group: with no treatment; IgG group: cecal ligation and puncture after aseptic laparotomy; Anti-CCL25 group: cecal ligation and puncture after aseptic laparotomy, and then treated with 5 mg/kg anti-CCL25 for 72 h. The mice were not fasted with water 12 h before the model was established.
CLP Sepsis Model
The operating table was sterilized by ultraviolet light for more than 30 minutes, and the surgical instruments were treated under high temperature and pressure sterilization. The mice were intraperitoneally anesthetized with pentobarbital sodium (1%, 40 mg/kg). After the anesthesia, the skin was prepared in the surgical area, disinfected with 75% alcohol, and the incision was opened about 2 cm along the midline of the abdomen to open the abdominal cavity. Then, the cecum was probed. After the 4–0 silk thread was ligated in the middle of the cecum, the 12-gauge needle was utilized for puncturing the cecum to the tip of the cecum twice, and the proper amount of intestinal tube was squeezed into the abdominal cavity, and the cecum was returned to the abdominal cavity. The abdominal incision was sutured layer by layer and routinely sterilized. Following this, the animals were returned to the cage, water and free activities were allowed. The environment and diet were the same as those of the normal control group. Samples were harvested 48h after CLP.23
Hematoxylin-Eosin Staining (HE Staining)
Mice were anesthetized with ether and their lungs were removed. The lung biopsy tissue was fixed at 4% paraformaldehyde, then dehydrated, transparent, waxed and embedded in paraffin. Lung tissues were cut into 5 μm for histological examination. After dewaxing and dehydration, tissue samples were stained with HE. Stained slides were analyzed by optical microscope under the same conditions. All groups were examined at 200x magnification. Briefly, the dewaxed and hydrated sections were transferred into water; then gradient ethanol dehydration: 100% ethanol for 2 min, 90% ethanol for 1 min, 80% ethanol for 1 min; washed with distilled water for 5 min; HE staining kits were used for staining for 3–5 min. Following this, rinsed with water for 5 min; 1% ethanol-hydrochloric acid differentiation for 30s; washed with water for 30s, and over-washed with distilled water for 5s; dyed with 0.5% eosin solution; ethanol dehydration: 90% ethanol for 1 min, 95% ethanol for 1 min, 100% ethanol for 1 min; Xylene was transparent and sealed with neutral resin.
Pathology of Lung Injury Score
After HE staining, the lung injury was scored according to the following pathology scoring criteria. 0 point: intact alveolar wall without thickening, no inflammatory infiltrates, no congestion. 1 point: slight diffuse inflammatory cell infiltration in the alveolar wall, no thickening of the alveolar wall. 2 points: marked and extensive inflammatory cell infiltrate with slight thickening of the alveolar wall (1–2 times). 3 points: severe inflammatory cell infiltrate with 2–3-fold thickening of the alveolar wall in individual areas. 4 points: severe inflammatory cell infiltrate with marked thickening of the alveolar wall and 25–50% solidification of the lung tissue. 5 points: severe inflammatory cell infiltrate with marked thickening of the alveolar wall and >50% solidification of the lung tissue.
Luciferase Analysis
HPMEC cells were seeded in a 6-well plate and transfected with 1.5 μg of promoter reporter plasmid (pGL3-Enhancer-p CCL25) and 20 ng of internal plasmid (pGL3-Enhancer). The pGL3-Enhancer-p CCL25 was designed and synthesized by JRDUN Biotechnology (Shanghai) Co., Ltd. Luciferase activity was determined using Dual-Luciferase Reporter Assay kit (Promega).24
Chromatin Immunoprecipitation (ChIP) Assay
HPMEC cells25 were cultured in 10-cm dishes and then cross-linked using 1% formaldehyde, followed by lysing in SDS lysis buffer. The sonicated cell lysates were diluted with ChIP buffer. A 10-μL sample was removed as a 2% input sample. The remaining 500 μL of diluted chromatin was incubated with anti-NF-κB (ab32536, Abcam) or anti-IgG overnight at 4°C. After elution of chromatin and reversal of cross-links, extracted DNA samples were finally subjected to PCR analysis to amplify the potential NF-κB binding sites on the CCL25 promoter.
Statistical Analysis
Data were expressed as mean ± SD. Statistical comparisons were made by one-way ANOVA using GraphPad Prism 7.0 software. Use Tukey’s multiple comparisons test to correct for multiple comparisons. P<0.05 was considered statistically significant. All experiments were repeated for at least three times.
Results
Serum Levels of CCL19, CCL21 and CCL25 in Sepsis Patients
Serum samples from 30 sepsis patients and 30 healthy individuals were collected for ELISA analyzation. As shown in Figure 2, the serum levels of CCL19, CCL21 and CCL25 in sepsis patients were significantly higher compared with those in healthy controls, with CCL25 had the most prominent upregulation (P<0.001) and supported by extensive literature. Therefore, CCL25 was selected for the following experiments.
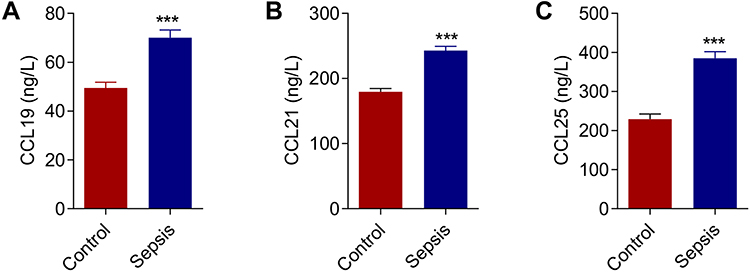 |
Figure 2 Expression levels of chemokines in sepsis patients. (A) Expression levels of chemokine CCL19 in sepsis patients. (B) Expression levels of chemokine CCL21 in sepsis patients. (C) Expression levels of chemokine CCL25 in sepsis patients. P<0.05 indicated significant difference, ***P<0.001. The experiments were repeated three times. |
Up-Regulation of CCL25 Expression in HPMEC Upon LPS Treatment
HPMECs were treated with 0, 25, 50, 100 and 200 ng/mL LPS. Q-PCR and Western blotting assays showed a significant increase in protein expression and relative mRNA expression of CCL25, and the upregulation was more pronounced with higher LPS concentrations (Figure 3A) (P<0.05). Furthermore, we found that the protein expression and relative expression of mRNA of CCL25 were more significantly upregulated after 48h of LPS induction (Figure 3B), suggesting that CCL25 may be involved in LPS-induced ALI. The relative expression of ccl25mnra is shown in Figure 3C and D.
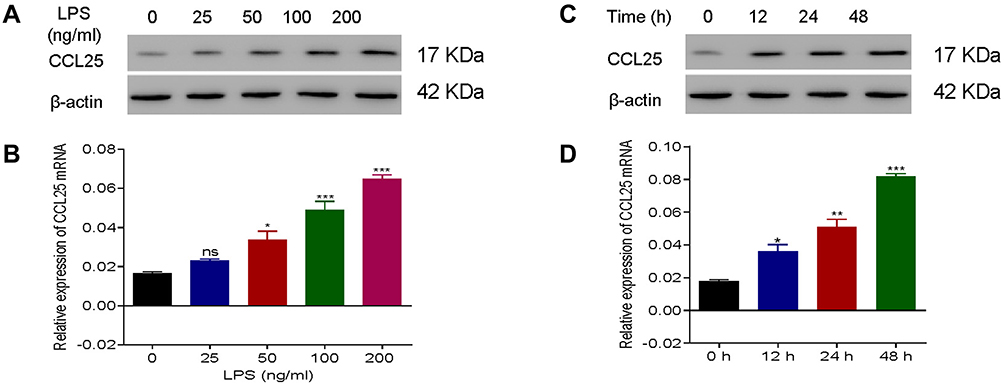 |
Figure 3 Up-regulation of CCL25 expression in HPMEC upon LPS treatment. (A) Protein expression and relative mRNA expression of CCL25 after treatment of HPMEC with 0, 25, 50, 100 and 200 ng/mL LPS, respectively, compared with 0 ng/mL LPS group. (B) Protein expression and mRNA relative expression of CCL25 at 0h, 12h, 24h, 48h after treatment of HPMEC with 100 ng/mL LPS. (C and D) The relative expression of CCL25 mRNA. *P<0.05, **P<0.01, ***P<0.001, compared with the 0 h group. The experiments were repeated three times. |
Anti-CCL25 Reduced Inflammatory Cytokine Expression and Endothelial Permeability in LPS-Treated HPMECs
To explore the effect of CCL25on inflammatory cytokine expression and endothelial permeability, HPMECs were treated with 100 ng/mL LPS in the presence of 3 different concentrations of anti-CCL25 (0, 100 and 200 ng/mL). Compared to the LPS+0 ng/mL anti-CCL25 group, TEER values and occludin and ZO-1 expressions were significantly increased in anti-CCL25 group (P<0.01) (Figure 4A and F), whereas the fluorescence intensity of FITC was significantly decreased, suggesting that CCL25 neutralization prevented LPS-induced increase of endothelial permeability (P<0.01) (Figure 4B). ELISA results revealed that the expression of pro-inflammatory cytokines TNF-α and IL-1β was significantly reduced in HPMECs after the addition of anti-CCL25, and the higher the concentration of anti-CCL25, the more significant the inhibition (P<0.01) (Figure 4C–E). Further, we found that anti-CCL25 reduced the phosphorylation activity of P38 in LPS-induced HPMECs (Figure 4F). There was no significant difference in total P38 level between different groups. The results suggested that CCL25 neutralization protected HPMECs from LPS-induced downregulation of tight-junction proteins, and the consequent increase of endothelial permeability.
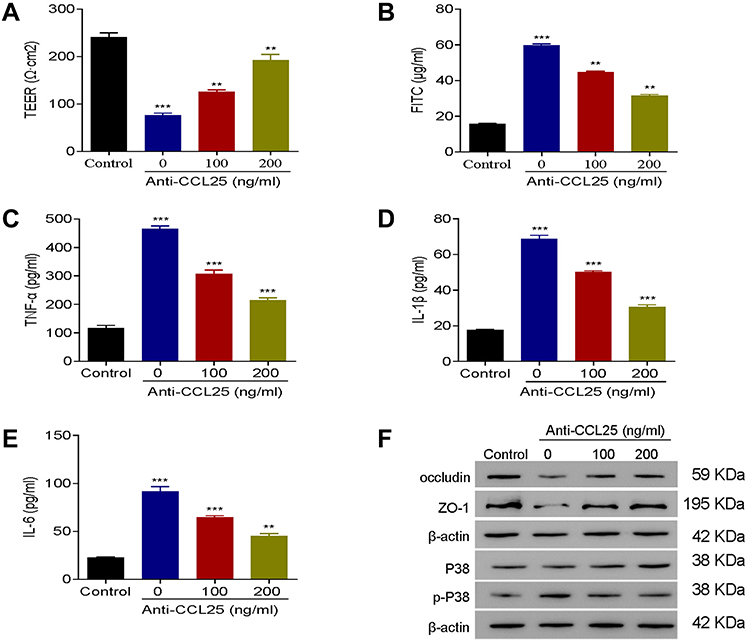 |
Figure 4 Anti-CCL25 reduced inflammatory cytokine expressions and endothelial permeability in LPS-treated HPMECs. 0, 100 and 200 ng/mL Anti-CCL25 were added to 100 ng/mL LPS-induced HPMECs, respectively. (A) TEER assay. (B) FITC fluorescence intensity assay. (C) ELISA detect to the expression of TNF-α. (D) ELISA to detect the expression of IL-1β. (E) ELISA to detect the expression of IL-6. (F) Western blotting to detect the protein expression of P38, p-P38, occludin and ZO-1. P<0.05 indicated significant difference, ***P<0.001, compared with control group; **P<0.01, ***P<0.001, compared with LPS+ 0 ng/mL Anti-CCL25 group. The experiments were repeated three times. |
Recombinant CCL25 Increased Endothelial Permeability in HPMECs
To explore the effect of recombinant CCL25 on cell permeability, TEER value and fluorescence intensity of FITC were detected. The results exhibited that the TEER values were down-regulated by recombinant CCL25 dose-dependently (P<0.01) (Figure 5A), while the fluorescence intensity of FITC was elevated with the increase of the concentration of recombinant CCL25 (P<0.05) (Figure 5B).
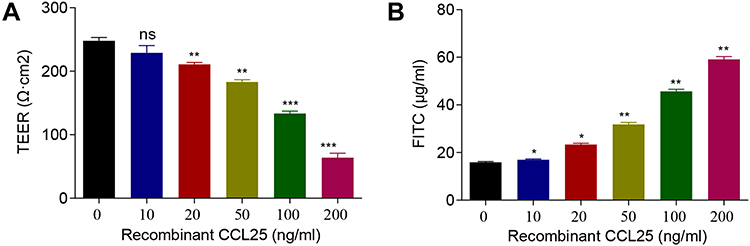 |
Figure 5 Recombinant protein CCL25 increased endothelial permeability and CCL25 expression in HPMECs. HPMEC were treated with 0, 25, 50, 100 and 200 ng/mL recombinant CCL25. (A) TEER assay. (B) FITC fluorescence intensity assay. P<0.05 indicated significant difference, *P<0.05, **P<0.01, ***P<0.001. The experiments were repeated three times. |
P38 Inhibition Reduced Inflammatory Cytokines Expression and Endothelial Permeability in CCL25-Treated HPMECs
To further analyze the relationship between CCL25 and P38 signaling, we treated HPMEC with CCL25 and divided the cells into a control group, a vector group and an SB203580 group. In the SB203580 group, 20 μM SB203580, a P38 inhibitor, was added. The results showed that the TEER values, as well as occludin and ZO-1 expressions in SB203580-treated group were significantly higher compared with the vehicle group (P<0.05; P<0.001) (Figure 6A and F). The fluorescence intensity of FITC, and the expressions of TNF-α, IL-1β, IL-6 and p-P38 were significantly down-regulated (P<0.05), yet the P38 expression was not significantly different between groups (P>0.05) (Figure 6B–F). These results indicate that CCL25 up-regulates the expression of inflammatory cytokines and endothelial permeability through promoting P38 phosphorylation.
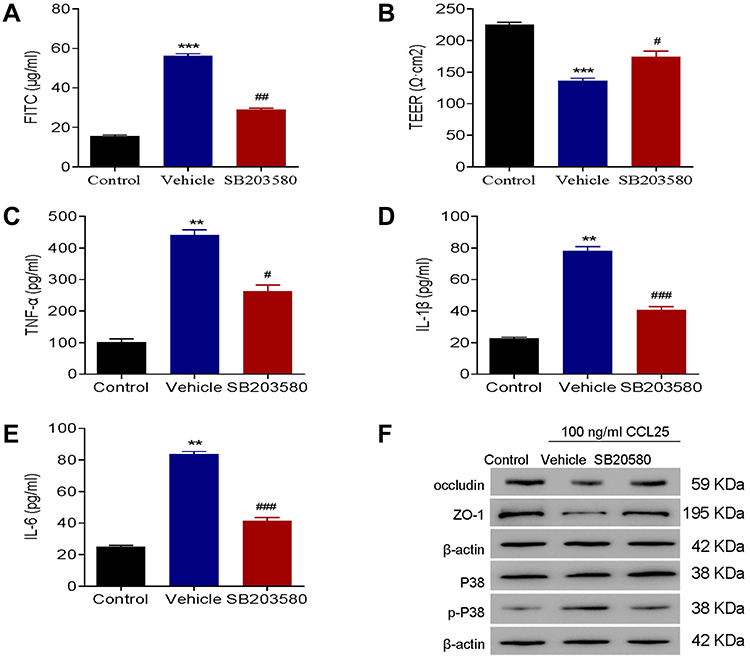 |
Figure 6 P38 inhibition reduced inflammatory cytokines expressions and endothelial permeability in CCL25-treated HPMEC. (A) FITC fluorescence intensity assay. (B) TEER assay. (C) ELISA detect to the expression of TNF-α. (D) ELISA to detect the expression of IL-1β. (E) ELISA to detect the expression of IL-6. (F) Western blotting to detect the protein expression of P38, p-P38, occludin and ZO-1. P<0.05 indicated significant difference. **P<0.01, ***P<0.001, compared with control group; #P<0.05, ##P<0.01, ###P<0.001, compared with vehicle group. The experiments were repeated three times. |
NF-κB Inhibitor Suppressed Expression of CCL25
NF-κB also plays an important role in cell inflammation. To explore the effects of NF-κB on CCL25 expression in ALI, HPMECs were treated with LPS 100 ng/mL and PDTC10 μmol/L, a NF-κB inhibitor. As shown in Figure 7A–C, CCL25 expression was significantly increased and NF-κB p65 translocated to the nucleus after LPS stimulation compared to controls, and PDTC significantly reversed this change. Luciferase assays and ChIP showed that PDTC prevented NFkB from binding to the CCL25 promoter (Figure 7D and E), suggesting that NF-κB was directly bound to the CCL25 promoter and activated its expression in HPMECs.
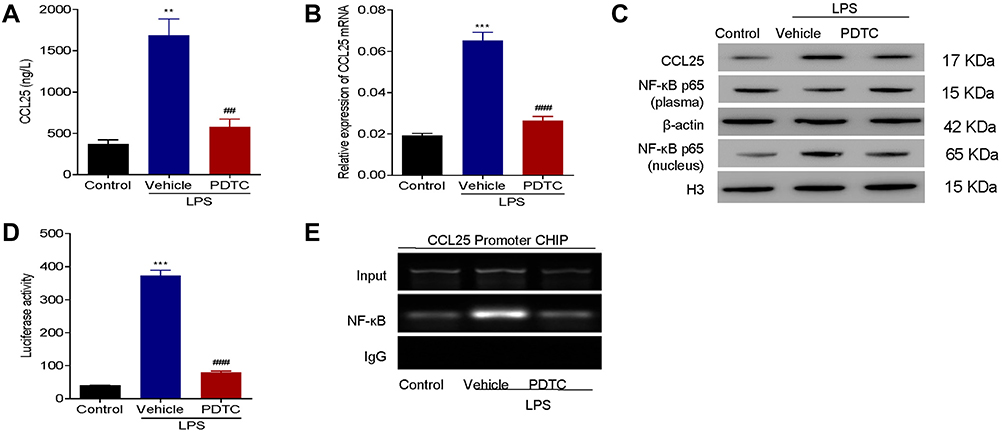 |
Figure 7 NF-κB inhibitor suppressed expression of CCL25. (A) The expression of CCL25. (B) Relative expression of CCL25 mRNA. (C) Western blotting to detect the protein expression of CCL25, NF-κB (plasma) and NF-κB (nucleus). (D) Luciferase assays. (E) ChIP analysis. The input was used as an internal positive control. P<0.05 indicated significant difference. **P<0.01, ***P<0.001, compared with control group; ##P<0.01, ###P<0.001, compared with vehicle group. The experiments were repeated three times. |
The Combination of NF-κB Inhibitor with Recombinant CCL25 Increased Cell Permeability and Inflammatory Cytokine Expression
To further confirm the efficacy of NF-κB on the expression of CCL25, HPMECs were treated with 100 ng/mL of recombinant CCL25 in the presence of PDTC 10 μmol/L. We observed a marked enhancement in TEER values and the expression of occludin and ZO-1, which were partially reversed after CCL25 treatment (Figure 8A and G).
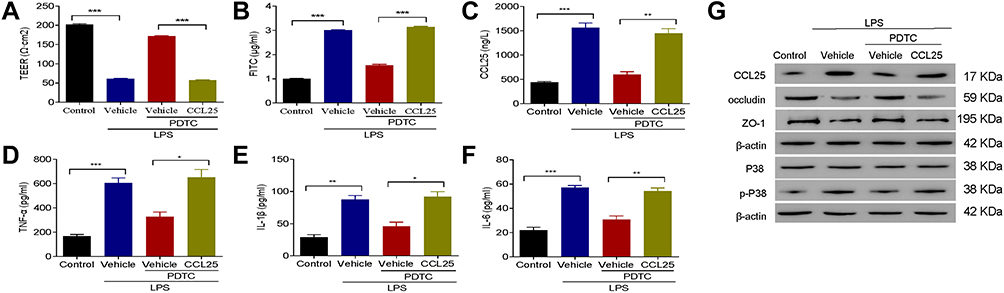 |
Figure 8 The combination of NF-κB inhibitor with recombinant CCL25 increased cell permeability and inflammatory cytokine expression. (A) TEER assay. (B) FITC fluorescence intensity assay. (C) ELISA detect to the expression of CCL25. (D) ELISA detect to the expression of TNF-α. (E) ELISA to detect the expression of IL-1β. (F) ELISA to detect the expression of IL-6. (G) Western blotting to detect the protein expression of CCL25, P38, p-P38, occludin and ZO-1. P<0.05 indicated significant difference, *P<0.05, **P<0.01, ***P<0.001. The experiments were repeated three times. |
After the addition of recombinant CCL25, the fluorescence intensity of FITC and the expression of CCL25, TNF-α, IL-1β and IL-6 were significantly up-regulated, and P38 was significantly activated in HPMECs (Figure 8B–G). These results indicated that PDTC down-regulated the expression of inflammatory cytokines and endothelial permeability, whereas CCL25 reversed the aforementioned effect induced by PDTC.
Anti-CCL25 Reduced the Levels of Inflammatory Cytokines and Improves the Survival of Sepsis Mice
We then established mouse sepsis model using CLP method. ELISA results showed that the IL-1β, IL-6 and TNF-α expression levels were significantly reduced in anti-CCL25 group (P<0.05) (Figure 9A–C). As shown in Figure 9D, p-P38 protein expression was reduced in the anti-CCL25 group and protein expression of occludin and ZO-1 was upregulated compared to the IgG group. Besides, compared with the control group, CLP induced obvious interstitial edema with massive infiltration of the inflammatory cells into the interstitium, and seriously damaged the pulmonary architecture. These pathological changes were alleviated by the addition of anti-CCL25, suggesting that CCL25 blockage reduced lung injury in mice with ALI (Figure 9E and F). Further, the survival rate in anti-CCL25 group was higher than that in IgG group (Figure 9G).
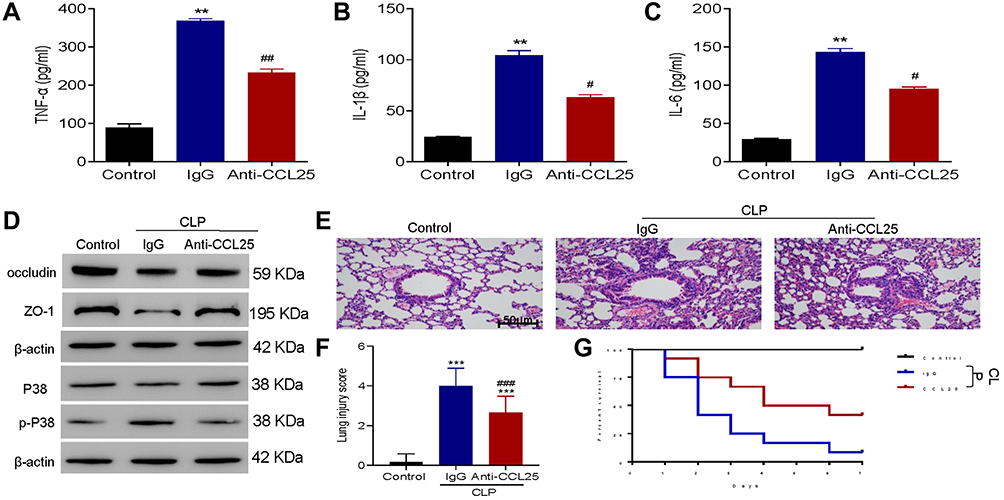 |
Figure 9 Anti-CCL25 reduced the levels of inflammatory cytokines and improved the survival of sepsis mice. Totally 36 mice were divided into control group, CLP model control group (IgG), CLP model group (Anti-CCL25), with 12 in each group. (A) ELISA detect to the expression of TNF-α. **P<0.01 compared with control group; ##P<0.01 compared with IgG group. (B) ELISA to detect the expression of IL-1β. **P<0.01 compared with control group; #P<0.05 compared with IgG group. (C) ELISA to detect the expression of IL-6. **P<0.01 compared with control group; #P<0.05 compared with IgG group. (D) Western blotting to detect the protein expression of P38, p-P38, occludin and ZO-1. (E) HE staining of lung injury, magnification: x200. (F) Pathological score of lung injury. ***P<0.001, compared with control group; ###P<0.001, compared with IgG group. (G) Survival rate. P<0.05 indicated significant difference. The experiments were repeated three times. |
Discussion
The present study revealed that CCL25 caused the increase of inflammatory cytokines expressions and cell permeability through increasing the phosphorylation of P38, and hence contributing ALI pathology. Our results indicated that inhibition of CCL25 and P38 by NF-κB inhibitors upregulated the expression of endothelial barrier tight-junction protein, repaired pulmonary mucosa barrier and lowered the levels of inflammatory cytokines. The survival rate of sepsis mice was improved after CCL25 inhibition. The results suggest that CCL25 might be a therapeutic target for ALI.
It is well known that CCL25 plays an important role in a variety of diseases. It has been proved that CCL25 mediated the migration and activation of hepatic stellate cells, and CCR9 deficiency could attenuate liver damage in mice.26 Downregulation of CCL25 could prevent impaired of β-cell function for the treatment of type 2 diabetes.27 Bdeir et al have showed that platelet-derived chemokines (CXCL7 and CXCL4) can promote the pathogenesis of ALI by activating neutrophil chemotaxis and vascular permeability.9 Blockade of chemokine receptors has a protective effect on PMVEC in septic ALI.28 Our study found for the first time that the expression levels of CCL25 was increased after LPS treatment in HPMEC, indicating that CCL25 might be essential in the pathogenesis of ALI in vitro and in vivo.
In ALI, various inflammatory cytokines such as TNF-α, IL-6 and IL-1β can induce activation of pulmonary endothelial cells, migration of white blood cells, granulocyte granulation and capillary leakage, and the accumulation of edema fluid further hinders the perfusion and oxygen exchange of alveolar cells.29–32 Therefore, we detected upregulated expressions of TNF-α, IL-6 and IL-1β in LPS-treated HPMEC and sepsis mice mode, which were downregulated by CCL25 neutralization. After activation of P38,33 the transcription factor AP-1 could be activated to enhance the transcription and expression of TNF-α and IL-1β.34 Studies have shown that knockout of chemokines (such as CXCL16) or inhibition of p38 signaling could reduce LPS permeability and LPS-induced lung injury.35,36 Consistent with this finding, our results showed that when P38 phosphorylation was inhibited, the fluorescence intensity of FITC, inflammatory cytokine expressions including TNF-α, IL-6 and IL-1β were reduced in HPMEC after the CCL25, suggesting that CCL25 may up-regulate the expression of inflammatory cytokines and cell permeability through promoting P38 phosphorylation. In addition, the CCR9-CCL25 chemokine axis has an important role in shaping the tumor microenvironment directly and through immune cells and is involved in attracting immune cells in tumors.37 Studies have shown that hepatic endothelial CCL25 mediates the recruitment of CCR9+ intestinal homing lymphocytes to the liver in primary sclerosing cholangitis.38 Trivedi et al reported that immune-mediated liver injury complicated by inflammatory bowel disease is caused by CCL25-mediated recruitment of mucosal lymphocytes by CCR9 activation.39 In conjunction with our study, CCL25 is involved in regulating the expression of inflammatory factors, suggesting that CCL25 may also have a potential role in the recruitment of inflammatory cells in ALI. However, this needs to be verified in future studies. Moreover, tight junction proteins including ZO-1 and occludin were crucial for maintaining the mechanical barrier and controlling the permeability of mucosal epithelium.40 Moreover, alveolar endothelial barrier function was influenced due to the down-regulation of ZO-1 expression.41 Similarly, we found that LPS-induced downregulation of these tight-junction protein was also rescued by CCL25 neutralization.
The expression of various chemokines is strictly regulated, many of which are induced by inflammatory responses and transcription factor NF-κB.42 An NF-κB-based pathway regulates the expression of inflammatory, cell adhesion molecules and chemokines such as CCL2 and CXCL8.43,44 The NF-κB pathway also plays an important role in the effect of P38 MAPK on LPS-induced production of pro-inflammatory factors and expression of chemokines.36 Consistently, we found that NF-κB had binding sites on the CCL25 promoter and regulated the expression of CCL25. Therefore, these findings strongly support the positioning of NF-κB as a primary modulator for the treatment of ALI, whose inhibition can reduce the harmful effects of CCL25 and may lead to a significant down-regulation of inflammatory and cell permeability events throughout ALI. Of course, we still need to conduct further experiments to verify the link between CCL25 and NF-kB pathway in the follow-up study.
Conclusions
In conclusion, our data indicated that elevated CCL25 expression resulted in decreased levels of endothelial barrier tight-junction protein and increased levels of inflammatory cytokines in HPMEC and sepsis mice model. Moreover, CCL25 promoted ALI through phosphorylation of P38, while NF-κB regulated CCL25. Although further studies are needed to validate this conclusion, our study demonstrates that CCL25 may be served as a therapeutic target for ALI.
Abbreviations
ALI, acute lung injury; CCL25, chemokine ligand 25; HPMECs, human pulmonary microvascular endothelial cells; CLP, cecal ligation puncture; LPS, lipopolysaccharide; ECL, enhanced chemiluminescence; BMI, body mass index; CRP, C-reactive protein; SOFA, Sequential Organ Failure Assessment; APACHE II, Mean Acute Physiology, Age, Chronic Health Evaluation II; ICU, intensive care unit.
Ethics Statement
The experimental procedures were approved by the Ethics Committee of Shanghai Changhai Hospital. The in vivo study was conducted according to the NIH guidelines for the care and use of laboratory animals (NIH Publication No. 85-23 Rev. 1985) and was approved by the Research Center for Laboratory Animals of Naval Medical University of China [STCSM2017-3-7]. The study was conducted in accordance with the Declaration of Helsinki.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Wu Q, Sun G, Yuan X, et al. Tubeimoside-1 attenuates LPS-induced inflammation in RAW 264.7 macrophages and mouse models. Immunopharmacol Immunotoxicol. 2013;35(4):514–523. doi:10.3109/08923973.2013.810643
2. Aziz M, Ode Y, Zhou M, et al. B-1a cells protect mice from sepsis-induced acute lung injury. Mol Med. 2018;24(1):26. doi:10.1186/s10020-018-0029-2
3. Zhang TZ, Yang SH, Yao JF, Juan DU, Yan TH. Sangxingtang inhibits the inflammation of LPS-induced acute lung injury in mice by down-regulating the MAPK/NF-κB pathway. Chin J Nat Med. 2015;13(12):889–895. doi:10.1016/S1875-5364(15)30094-7
4. Letsiou E, Wang H, Belvitch P, Dudek S, Sammani S. ID: 109: parkin mediates endothelial pro-inflammatory responses in acute lung injury. J Investig Med. 2016;64(4):
5. Fu C, Hao S, Xu X, et al. Activation of SIRT1 ameliorates LPS-induced lung injury in mice via decreasing endothelial tight junction permeability. Acta Pharmacol Sin. 2019;40(5):630–641. doi:10.1038/s41401-018-0045-3
6. Andrzejczak D, Woldan-Tambor A, Zawilska JB. The effects of tiagabine on lipopolysaccharide (LPS)-induced proinflammatory cytokine release from primary rat microglial cell cultures. Pharmacol Rep. 2016;127:352–357.
7. Fragoso IT, Ribeiro EL, Gomes FODS, et al. Diethylcarbamazine attenuates LPS-induced acute lung injury in mice by apoptosis of inflammatory cells. Pharmacol Rep Pr. 2016;69(1):81. doi:10.1016/j.pharep.2016.09.021
8. Zhao H, Zhao M, Wang Y, Li F, Zhang Z. Glycyrrhizic acid prevents sepsis-induced acute lung injury and mortality in rats. J Histochem Cytochem. 2016;64(2):125. doi:10.1369/0022155415610168
9. Bdeir K, Gollomp K, Stasiak M, et al. Platelet-specific chemokines contribute to the pathogenesis of acute lung injury. Am J Respir Cell Mol Biol. 2017;56(2):261–270. doi:10.1165/rcmb.2015-0245OC
10. Hwaiz R, Rahman M, Zhang E, Thorlacius H. Platelet secretion of CXCL4 is Rac1-dependent and regulates neutrophil infiltration and tissue damage in septic lung damage. Br J Pharmacol. 2015;172(22):5347–5359. doi:10.1111/bph.13325
11. Betts MJ, Guigó R, Agarwal P, Russell RB. Exon structure conservation despite low sequence similarity: a relic of dramatic events in evolution? EMBO J. 2014;20(19):5354–5360. doi:10.1093/emboj/20.19.5354
12. Mellado M, Rodríguez-Frade JM, Vila-Coro AJ, et al. Chemokine receptor homo- or heterodimerization activates distinct signaling pathways. EMBO J. 2014;20(10):2497–2507. doi:10.1093/emboj/20.10.2497
13. Lu IN, Chiang BL, Lou KL, et al. Cloning, expression and characterization of CCL21 and CCL25 chemokines in zebrafish. Dev Comp Immunol. 2012;38(2):203–214. doi:10.1016/j.dci.2012.07.003
14. Gupta P, Sharma PK, Mir H, et al. CCR9/CCL25 expression in non-small cell lung cancer correlates with aggressive disease and mediates key steps of metastasis. Oncotarget. 2014;5(20):10170–10179. doi:10.18632/oncotarget.2526
15. Jia H, Sodhi CP, Yamaguchi Y, et al. Toll like receptor 4 mediated lymphocyte imbalance induces nec-induced lung injury. Shock. 2019;52(2):215–223. doi:10.1097/SHK.0000000000001255
16. Wang YM, Ji R, Chen WW, et al. Paclitaxel alleviated sepsis-induced acute lung injury by activating MUC1 and suppressing TLR-4/NF-kappaB pathway. Drug Des Devel Ther. 2019;13:3391–3404. doi:10.2147/DDDT.S222296
17. Kawai T, Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol Med. 2007;13(11):460–469. doi:10.1016/j.molmed.2007.09.002
18. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
19. Shankar-Hari M, Harrison DA, Rubenfeld GD, Rowan K. Epidemiology of sepsis and septic shock in critical care units: comparison between sepsis-2 and sepsis-3 populations using a national critical care database. Br J Anaesth. 2017;119(4):626–636. doi:10.1093/bja/aex234
20. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402. doi:10.1006/meth.2001.1262
21. Kaiser M, Pereira S, Pohl L, et al. Chitosan encapsulation modulates the effect of capsaicin on the tight junctions of MDCK cells. Sci Rep. 2015;5:10048. doi:10.1038/srep10048
22. Karaki SI, Ishikawa J, Tomizawa Y, Kuwahara A. Effects of ε ‐viniferin, a dehydrodimer of resveratrol, on transepithelial active ion transport and ion permeability in the rat small and large intestinal mucosa. Physiol Rep. 2016;4(9):
23. Hwang JS, Kim KH, Park J, et al. Glucosamine improves survival in a mouse model of sepsis and attenuates sepsis-induced lung injury and inflammation. J Biol Chem. 2019;294(2):608–622. doi:10.1074/jbc.RA118.004638
24. Xie W, Lu Q, Wang K, et al. miR-34b-5p inhibition attenuates lung inflammation and apoptosis in an LPS-induced acute lung injury mouse model by targeting progranulin. J Cell Physiol. 2018;233(9):6615–6631. doi:10.1002/jcp.26274
25. Das PM, Ramachandran K, vanWert J, Singal R. Chromatin immunoprecipitation assay. Biotechniques. 2004;37(6):961–969. doi:10.2144/04376RV01
26. Chu PS, Nakamoto N, Ebinuma H, et al. C-C motif chemokine receptor 9 positive macrophages activate hepatic stellate cells and promote liver fibrosis in mice. Hepatology. 2013;58(1):337–350. doi:10.1002/hep.26351
27. Atanes P, Lee V, Huang GC, Persaud SJ. The role of the CCL25-CCR9 axis in beta-cell function: potential for therapeutic intervention in type 2 diabetes. Metabolism. 2020;113:154394. doi:10.1016/j.metabol.2020.154394
28. Zhu X, Zou Y, Wang B, et al. Blockade of CXC chemokine receptor 3 on endothelial cells protects against sepsis-induced acute lung injury. J Surg Res. 2016;204(2):288–296. doi:10.1016/j.jss.2016.04.067
29. Ji S, Wen Y, Lai Q, Li M, Zhang P. [Hypoxia combined with TNF-α induces apoptosis of cultured human pulmonary microvascular endothelial cells via activation of the STAT3 rather than ERK1/2 signaling pathway]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2016;32(7):896. Chinese.
30. Gennai S, Monsel A, Hao Q, Park J, Matthay MA, Lee JW. Microvesicles derived from human mesenchymal stem cells restore alveolar fluid clearance in human lungs rejected for transplantation. Am J Transplant. 2015;15(9):2404–2412. doi:10.1111/ajt.13271
31. de Carlo TE, Chin AT, Joseph T, et al. Distinguishing diabetic macular edema from capillary nonperfusion using optical coherence tomography angiography. Ophthalmic Surg Lasers Imaging Retina. 2016;47(2):108–114. doi:10.3928/23258160-20160126-02
32. Alomar SY, Gentili A, Zaibi MS, Kępczyńska MA, Trayhurn P. IL-1β (interleukin-1β) stimulates the production and release of multiple cytokines and chemokines by human preadipocytes. Arch Int Physiol. 2016;122(3):6. doi:10.3109/13813455.2016.1156706
33. Song HL, Zhang X, Wang WZ, et al. Neuroprotective mechanisms of rutin for spinal cord injury through anti-oxidation and anti-inflammation and inhibition of p38 mitogen activated protein kinase pathway. Neural Regen Res. 2018;13(1):128–134. doi:10.4103/1673-5374.217349
34. Kim KW, Im J, Jeon JH, Lee HG, Yun CH, Han SH. Staphylococcus aureus induces IL-1β expression through the activation of MAP kinases and AP-1, CRE and NF-κB transcription factors in the bovine mammary gland epithelial cells. Comp Immunol Microbiol Infect Dis. 2011;34(4):347–354. doi:10.1016/j.cimid.2011.04.004
35. Wang W, Weng J, Yu L, Huang Q, Jiang Y, Guo X. Role of TLR4-p38 MAPK-Hsp27 signal pathway in LPS-induced pulmonary epithelial hyperpermeability. BMC Pulm Med. 2018;18(1):178. doi:10.1186/s12890-018-0735-0
36. Tu GW, Ju MJ, Zheng YJ, et al. CXCL16/CXCR6 is involved in LPS-induced acute lung injury via P38 signalling. J Cell Mol Med. 2019;23(8):5380–5389. doi:10.1111/jcmm.14419
37. Mir H, Singh S. CCL25 signaling in the tumor microenvironment. Adv Exp Med Biol. 2021;1302:99–111.
38. Eksteen B, Grant AJ, Miles A, et al. Hepatic endothelial CCL25 mediates the recruitment of CCR9+ gut-homing lymphocytes to the liver in primary sclerosing cholangitis. J Exp Med. 2004;200(11):1511–1517. doi:10.1084/jem.20041035
39. Trivedi PJ, Bruns T, Ward S, et al. Intestinal CCL25 expression is increased in colitis and correlates with inflammatory activity. J Autoimmun. 2016;68:98–104. doi:10.1016/j.jaut.2016.01.001
40. Feng SR, Chen ZX, Cen JN, Shen HJ, Wang YY, Yao L. [Critical roles of matrix metalloproteinases secreted by leukemic cells in the pathogenesis of central nervous system leukemia]. Zhonghua Xue Ye Xue Za Zhi. 2016;37(12):1070. Chinese. doi:10.3760/cma.j.issn.0253-2727.2016.12.012
41. Yang J, Wan Y, Liu H, Bi J, Lu Y. C2-ceramide influences alveolar epithelial barrier function by down-regulating Zo-1, occludin and claudin-4 expression. Toxicol Meth. 2017;27(4):293–297. doi:10.1080/15376516.2017.1278812
42. Korbecki J, Kojder K, Barczak K, et al. Hypoxia alters the expression of CC chemokines and CC chemokine receptors in a Tumor-A literature review. Int J Mol Sci. 2020;21(16):5647. doi:10.3390/ijms21165647
43. Katanov C, Lerrer S, Liubomirski Y, et al. Regulation of the inflammatory profile of stromal cells in human breast cancer: prominent roles for TNF-alpha and the NF-kappaB pathway. Stem Cell Res Ther. 2015;6:87. doi:10.1186/s13287-015-0080-7
44. Yang RC, Chang CC, Sheen JM, Wu HT, Pang JH, Huang ST. Davallia bilabiata inhibits TNF-alpha-induced adhesion molecules and chemokines by suppressing IKK/NF-kappa B pathway in vascular endothelial cells. Am J Chin Med. 2014;42(6):1411–1429. doi:10.1142/S0192415X1450089X
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.