")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 15
Catatonia in Down syndrome: systematic approach to diagnosis, treatment and outcome assessment based on a case series of seven patients
Authors Miles JH , Takahashi N, Muckerman J , Nowell KP , Ithman M
Received 13 April 2019
Accepted for publication 31 July 2019
Published 20 September 2019 Volume 2019:15 Pages 2723—2741
DOI https://doi.org/10.2147/NDT.S210613
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Judith H Miles,1,2 Nicole Takahashi,2 Julie Muckerman,2 Kerri P Nowell,2,3 Muaid Ithman4
1Department of Child Health, University of Missouri Healthcare, Columbia, MO, USA; 2Thompson Center for Autism and Neurodevelopmental Disorders, University of Missouri, Columbia, MO, USA; 3Department of Health Psychology, University of Missouri Healthcare, Columbia, MO, USA; 4Department of Psychiatry, University of Missouri Health Care, Columbia, MO, USA
Correspondence: Judith H Miles
Thompson Center for Autism and Neurodevelopmental Disorders, 205 Portland Street, Columbia, MO 65211, USA
Tel +1 573 884 6838
Fax +1 573 884 1151
Email [email protected]
Objective: The goal is to expand our knowledge of catatonia occurring in adolescents and young adults with Down syndrome (DS) by describing the first prospective, consecutive, well-characterized cohort of seven young people with DS diagnosed with catatonia and treated between 2013 and 2018, and to assess each patient’s treatment responses. Longitudinal assessment of each patient’s response to treatment is intended to provide clinicians and psychiatrists a firm foundation from which assess treatment efficacy.
Study design: Young adults with Down syndrome were consecutively enrolled in the study as they were diagnosed with catatonia. A comprehensive data set included medical, laboratory, developmental, demographic, family, social and genetic data, including query into disorders for which individuals with DS are at risk. Catatonia was diagnosed based on an unequivocal history of regression, positive Bush-Francis Catatonia Rating Scale and positive response to intravenous lorazepam. Patients’ longitudinal progress was monitored using the Catatonia Impact Scale (CIS) developed for this purpose.
Results: Seven consecutive DS patients, who presented with unequivocal regression were diagnosed with catatonia and treated for 2.7–6 years using standard-of-care therapies; primarily GABA agonist, lorazepam, electroconvulsive therapy (ECT) and glutamate antagonists (dextromethorphan/quinidine, memantine, minocycline). Responses to each treatment modality were assessed at clinic visits and through weekly electronic CIS reports.
Conclusion: Seven young adults with DS were diagnosed with catatonia; all responded to Lorazepam and/or ECT therapy with good to very good results. Though ECT most dramatically returned patients to baseline, symptoms often returned requiring additional ECT. Dextromethorphan/quinidine, not used until mid-2017, appeared to reduce the reoccurrence of symptoms following ECT. Though all seven patients improved significantly, each continues to require some form of treatment to maintain a good level of functioning. Findings of a significant number of autoimmune disorders and laboratory markers of immune activation in this population may guide new diagnostic and treatment opportunities.
Keywords: lorazepam, electroconvulsive therapy, dextromethorphan/quinidine, benzodiazepines, Trisomy 21, Bush-Francis Catatonia Rating Scale
Introduction
Down syndrome (DS), occurs in around one in 800 live births, yielding a prevalence of 200,000–250,000 children and adults in the United States.1–3 A free-standing extra chromosome 21 (Trisomy 21) occurs in 95% of cases. Approximately 2% have a mosaic karyotype with a mixture of normal and trisomy 21 cells. In 3% the trisomy results from a chromosome translocation. Children with DS are born most often to families with no previous DS history.4
Recently the DS medical community has come together around the fact that adolescents and young adults with DS are at risk of significant functional regression as adolescents or young adults.5 When medical conditions, common to DS, such as seizures, infections, celiac, metabolic and cardiac diseases have been excluded, catatonia has emerged as the common diagnosis.6–11 Though the three previous reports of catatonia occurring in DS indicate it is eminently treatable, prospective studies have been limited by the number of patients and the length of medical follow-up.6–8 This has led to unease by physicians and families in the choice of treatments and even some concern about the legitimacy of the catatonia diagnosis in DS, autism and other neurodevelopmental disorders.
The purpose of this publication is to expand knowledge of catatonia occurring in Down syndrome by describing a prospective, consecutive, well-characterized cohort of seven young people with DS who were diagnosed with catatonia and treated at University of Missouri Health Care between 2013 and 2018, including demographics, medical status, laboratory and neurologic evaluations, responses to standard-of-care therapeutic modalities and preliminary outcomes. We hope this will demystify the diagnosis of catatonia in DS, facilitate the diagnostic process, inform treatment decisions and provide a basis for treatment expectations. Finally, we hope to dispel any doubt that catatonia in this population is the same disorder as catatonia encountered in patients with other medical, neurodevelopmental and psychiatric disorders.
We present evidence that DS patients who have developed catatonia are relatively easy to diagnose utilizing the Bush-Francis Catatonia Rating Scale (BFCRS) and respond to acknowledged catatonia treatments.9,12–22 Use of this information by a medical team experienced in care of patients with genetic disorders and psychiatry allowed us to successfully treat seven young adults.
Materials and methods
Families were either self or physician referred to the Thompson Center for evaluation of regression. Most had found our previous paper online, through a friend or DS organization.6 The seven families live in Missouri. Out of state families were assisted in locating appropriate care closer to home. Each patient was evaluated following a protocol, developed for patient #1 and updated as new information has become available. Diagnosis of the catatonia syndrome was based on an incontrovertible history of regression, identification of clinical catatonia symptoms and a diagnostic trial of lorazepam.
The BFCRS, published in 1996, is considered the gold standard diagnostic measure, based on good inter-rater reliability and concordance, and a track record of use.17,12,15 It is easy to administer, especially when unfamiliar German descriptors are converted to English. The 23 items each query a specific catatonia symptom including changes in motor activity (reduced movement, freezing or maintaining a posture, alternating episodes of hyperactivity), unusual movements (stereotypies, grimacing, blinking, motor or vocal tics, posturing, ambitendency, automatic obedience), changes in speech (mutism, reduced meaningful speech, echolalia), changes in food or fluid intake (reduced appetite, slow food intake), negativism (refusal to engage in previously enjoyed activities) and a variety of autonomic abnormalities including temperature, blood pressure, respiratory rate and sweating. The Screening score is generated by summing the number of positive responses to questions 1–14. The Severity score sums the severity levels of all 23 symptoms.17 The BFCRS is considered positive when three or more of the 14 screening symptoms are present, though the highest diagnostic confidence is achieved with a screening score of four or more symptoms.12
Families completed a 23-page history form capturing pregnancy, health, neurologic, regression, medication, sleep, autonomic nervous system, behavioral, diet, fever, GI, sensory and family history. Medical and neurologic examinations were augmented by medical tests selected to identify known causes of catatonia, medical problems common to DS, and additional causes of regression that may mimic catatonia. The standard laboratory testing protocol is provided in Supplementary materials, Protocol 1.
Once catatonia was clinically diagnosed and medical evaluations had excluded most infectious, autoimmune, metabolic and neurologic causes, a diagnosis of idiopathic catatonia was stipulated and treatment initiated. Most patients had previously been prescribed a variety of antidepressants, psychiatric, anti-epileptic and sleep medications which had not ameliorated their symptoms; all had been discontinued prior to this study. Treatment was begun with lorazepam, the acknowledged initial treatment for catatonia.6,9,20–25 Based on response, the lorazepam dose was gradually increased until 2 mg increases failed to provide improvement or the dose reached approximately 20 mg/day in divided doses. A number of other medications, previously reported as efficacious for some patients with catatonia including N-acetyl cysteine (NAC), minocycline, and dextromethorphan + quinidine (Nuedexta@) were used as clinically indicated.26–30 Failure to achieve a close to baseline recovery prompted recommendation for bilateral electroconvulsive therapy (ECT). All except the first patient continued to receive catatonia medications during the course of ECT. Resurgence of symptoms often prompted repeated courses of ECT, or maintenance treatments.
Patient follow-up consisted of monthly clinic visits and weekly symptom reports completed by families using the electronic, email distributed, Catatonia Impact Scale (CIS) (Supplementary materials; Protocol 2). The CIS adhered to the format of the fully validated Autism Impact Measure (AIM), developed by Kanne and Mazurek.31,32 and queried symptoms enumerated in the BFCRS, DSM-5 and current medical literature. Thirty questions provided a longitudinal assessment of both symptom frequency and impact on functioning, using a 5-point Likert scale. Description of the CIS is in preparation.33
University of Missouri Institutional Review Board approval (Project # 1210315) was obtained to collect clinical data with storage in the Thompson Center Database, which can be analyzed and published in an effort to increase knowledge of catatonia in DS. All parents/legal guardians provided written, informed consent regarding data collection, and storage on University of Missouri servers, for presentation at medical and parents’ meetings and publication of anonymous results. In addition, all parents/legal reviewed their Case Reports and provided written informed consent for the case details to be published. This study was conducted in accordance with the Declaration of Helsinki. Weekly collection of data using the Catatonia Impact Screen (CIS) from families via email provides longitudinal records of each patient’s response to therapies. Deidentified data can be made available to the research community.
Case reports
Cases are presented in the order they were diagnosed (numbered 1–7), such that the length of time each patient has been treated is less with succeeding cases. Age is at presentation for diagnosis.
Case: 1: BK – 18 yo male (1st visit 3/13; lorazepam initiation 6/13; follow-up 6 yrs)
BK is a Caucasian male with no previous psychiatric problems, aged 18 years at presentation. His only medical problem was alopecia areata treated since age 12 years. Despite moderate-range intellectual disability (ID) based on school testing prior to development of catatonia, he demonstrated higher-than-expected adaptive functioning, including playing the piano, reading and working part-time. He pitched Special Olympics Softball and excelled in team soccer. Family history was positive for adult-onset hypothyroidism, Raynaud’s and anxiety. There were no identifiable initiating stressors. Symptoms emerged at age 18 years, 4 months, with progressive psychomotor slowing, episodic immobility, freezing, reduced speech and eye-blink rate, loss of hedonic capacity, grimacing and holding his tongue out, repeated shrugging and turning of the shoulders, slow eating, and stereotyped movements of the fingers. He became negativistic. There were no sleep problems and minimal snoring. Neurological examination revealed overall motor slowing with general but mild body stiffness, anisocoria that was considered within normal range by neuro-ophthalmology and a change from right to left-handedness for eating. Neurologic studies were unremarkable, including brain MRI and routine and 24 hr EEGs. Extensive neurologic and medical evaluations, as described in Methods, to identify conditions that may cause or mimic catatonia failed to provide an alternative diagnosis. The pretreatment BFCRS Screening score was 8 and the Severity score 25. Based on these findings, his response to a diagnostic trial of lorazepam and lack of evidence to suggest another underlying medical or neuropsychiatric disorder, BK was diagnosed with unspecified catatonia.35 Lorazepam was titrated to a maximum dose of 20 mg/day over an 11-month period. Parental report, based on their subjective symptom levels endorsed a 75% return to baseline function. Other medications were tried including minocycline, which was discontinued within a week due to dizziness, and memantine, used for a short period, appeared to exacerbate catatonia symptoms. Due to failure to achieve baseline functions, bifrontal ECT treatments were initiated after 15 months resulting in an immediate and robust response, with more spontaneous speech and fluidity of movement. He maintained this response on no other catatonia specific treatments for approximately 2 years. Gradual reemergence of symptoms including loss of spontaneous speech, motor slowing and freezing, and decreased interest in activities led to reinstitution of medical treatment. Low dose lorazepam appeared to stabilize symptoms; he has returned to playing the piano, working part time and exercising. Parents did not see a benefit of NAC or dextromethorphan/quinidine and decided against repeat ECT, though he is not completely back to baseline. He is on 3.5 mg of lorazepam per day, which parents increase to 4.5 mg if symptoms increase. Current BFCRS Screening scale is 1 and Severity scale 2.5.
Case 2: MS – 19 yo female (1st visit 8/13; lorazepam initiation 10/13; follow up 5.2 yrs)
MS was diagnosed with Trisomy 21 Down syndrome shortly after birth. Patent Foramen Ovale closed spontaneously. Early milestones were delayed with walking at 22 months and toilet training at 8 years. She was described as an easygoing baby with good eye contact, who reached to be picked up, and was cuddly with immediate family. During the second year, she plateaued and regressed in many skills. She lost her previously obtained 10 words, became aloof with poor social initiation, decreased eye contact, fascination with lights, repetitive finger movements, and inspection of objects with a sideways glance. At 4 ½ years of age she was diagnosed with autism and a 16p13.11-p12.3 duplication. Duplications in this region have occasionally been reported in subjects with autism, neuropsychiatric and neurocognitive disorders including one child with catatonia and Autism.36–38 Because of the commonality of small duplications in neurodevelopmental disorders, its significance is unclear.
Despite ear infections requiring multiple pressure equalizing tubes, adenoidectomy at age 4 years, strabismus surgery was at age 6 years, celiac disease diagnosed at age 7 years and Hashimoto’s thyroiditis at age 15 years, MS is considered medically healthy by her parents. She wears glasses for myopia and astigmatism. In elementary school, she was described as engaged and willing to participate in activities. She was included in the regular classroom with an aide, read at a first-grade level, told jokes and was identified as having moderate to severe ID. The Cattell-Binet given by the school at age 11 years provided a FSIQ of 29. When, not actively engaged she preferred to sit on the floor and repetitively shake her dolls.
Parents think regression started at age 13 years with waxing and waning symptoms over the next 7 years. In middle school, when placed in a special education class, she began a new behavior whereby she had her dolls scream, “I hate you”, “Get out of my face”, in a voice that mimicked some aggressive classmates. Having a one-on-one aide decreased the behavior but when support was discontinued she became more distressed. At age 19 (March 2013) MS developed abrupt, severe regression coincident with an incident at school when a fellow student was restrained. She became withdrawn, off in her own world. She stared without appearing to notice things, sat all day with her head down repeating phrases and slapping her legs, and become combative when given directions. She lost self-help skills such as toileting, buckling her seatbelt and self-feeding. She began using her left hand for some tasks. Sleep was impaired, she refused previously enjoyed activities, engaged in repetitive rocking, echolalia and exhibited facial grimaces. Medications prescribed by their local psychiatrist included aripiprazole, risperidone, an SSRI and clonidine. With risperidone, she developed a temperature of 104 and incontinence, which resolved with medication discontinuation. Incipient neuroleptic malignant syndrome was suspected. In August 2013, a diagnosis of catatonia was made based on history, examination, positive BFCRS Screening (12) and Severity (29) scores and a positive response to 2 mg IV lorazepam. Extensive neurologic and medical evaluations failed to provide an alternative diagnosis. Brain MRI revealed mild hypoplasia of the cerebellar vermis and the EEGs consistently showed background slowing.
Oral lorazepam was gradually increased to 9 mg/day with intermittent but unsustained periods of improvement. Trials of minocycline, N-acetyl-cysteine, and folinic acid did not provide appreciable improvement. Memantine was discontinued when the family noted increased agitation. ECT administered February through April 2014 reduced symptoms. Its discontinuation led to gradual recurrence. A second 12 treatment series was followed by maintenance ECT which decreased symptoms during the treatment week which waned by the next treatment. Because of the failure to more completely control the symptoms intermittent ECT was discontinued. Reintroduction of lorazepam was recommended but rejected by her family, fearing MS would revert to her severe impairment level at diagnosis. Her symptom level remained stable with a scattering of good and bad days. Dextromethorphan/quinidine, a glutamate antagonist, was started in November 2016 at 1 tablet/day, increased to bid dosing in January 2017. Her parents reported she seemed happier, was sitting up straight and looking others in the face, had lost body and facial stiffness, was talking in short phrases without prompting, willing to participate in activities with her therapists, and sleeping well most nights, without enuresis. In July 2017, parents noted a gradual return of symptoms with irritability, poorer sleep and louder vocalizations. Reinstituting ECT was discussed. Family elected to continue the dextromethorphan/quinidine. Over the next two years she has cycled between episodes of reduced and increased symptoms. Repeat ECT is being considered. Parents note she does best when a therapist or family member helps her maintain interactions and activities.
Case 3: BC – 33 yo female, (1st visit 1/14; lorazepam initiation 6/14; follow up 5 yrs)
BC was diagnosed with Down syndrome shortly after birth. She did well as an infant and remained healthy throughout childhood. Mother recalls she was social, talkative and engaged. At age 25 yo, her father died unexpectedly, after which she began to regress with a decline in speech, activity level and social interactions. Her family was led to believe this was a typical course for DS. At age 33 yrs, she was referred to the Thompson Center and was diagnosed with unspecified catatonia, complicating DS, based on ten Catatonia symptoms. She also developed periods of incontinence, loss of daily living skills like showering, and developed compulsive moving of objects from room to room. Subsequent to lorazepam administration, she seemed more engaged and interactive and started to read again. Despite increasing lorazepam gradually to 15 mg/day, improvement was not sustained. Trials of minocycline, memantine and N-acetyl cysteine did not stimulate improvement. Sertraline caused significant side effects (sleep, dizziness, anorexia) and was discontinued when pharmacogenomic testing revealed reduced CYP2C19 enzyme function, which decreases the rate of clearance of sertraline thereby causing side effects. Lorazepam was continued at 15 mg/day in 3 doses. BC received 17 ECT treatments from 8/8/16 through 10/12/16, resulting in significant improvement. Over the next 6 months, symptoms gradually reoccurred. Treatment was augmented with dextromethorphan/quinidine two tablets/day and a second course of 16 ECT treatments. She responded well and her mother estimated that she was back to 85% of baseline functioning. Dextromethorphan/quinidine 20/10 was continued; remission lasted approximately 8 months. In August 2018 she received a third course of 14 ECT treatments. She continues on monthly maintenance ECT, lorazepam and dextromethorphan/quinidine. She is considered almost at baseline with BFCRS scores of 2 and 3 (1-17-19).
Case 4: PL – 25 yo female (1st visit 6/15; lorazepam initiation 7/15; follow up 3.5 yrs)
PL was diagnosed with Trisomy 21 DS shortly after birth. She did well as an infant; a small ASD closed spontaneously. She remained healthy throughout childhood. Mother reports she was a social, talkative, interactive with an excellent sense of humor. She read at a 5th grade level, was independent with activities of daily living, and got along well with classmates. At age 19 years, PL was hospitalized with an idiopathic left transverse and sigmoid sinus thrombosis with tentorial hemorrhage. Hypercoagulability evaluation was normal. She was treated with anticoagulants for 6 months and is maintained on 81 mg aspirin daily. Recent MRI (6/17) indicated recanalization of the left cerebral sinuses. At age 20 years she was diagnosed with hypothyroidism, easily controlled with Synthroid.
Parents noticed in about ninth grade, she was moving slower and was less interested in activities. By her junior year she seemed slower but still planned to work and get married. Following High School there was marked regression. She became inactive, sitting abnormally still and staring for long periods. She was slow going up and down stairs, paused/froze when reaching for things, and took at least an hour to eat a meal. By the time she was evaluated she had lost most spoken language, did not respond to most questions, used repetitive phrases such as “Steve got mad today”, and had developed facial grimacing with odd facial expressions, stereotypic hand movements and a rigid tailor’s sitting posture with a straight back. She would dwell on bad events, made poor eye contact and was resistant to suggestions and instructions. She angered easily and had crying episodes. Affect was flat and she had lost interest in family and friends. She needed help dressing. She was withdrawn from the adult workshop as she could no longer participate in piece work. Comprehensive medical and psychiatric evaluations led to diagnoses of celiac disease and moderate obstructive sleep apnea, both were treated but did not ameliorate catatonia symptoms.
PL was diagnosed with catatonia, complicating DS, with a BFCRS Screening score of 9 and Severity score of 21 and started on lorazepam 2 mg/day. Initially she seemed more engaged and interactive and started to read again. Lorazepam doses up to 8 mg were observed to improve symptoms quickly but by the end of two hours, the effect would wear off. For about a year, the family maintained behavioral equilibrium with 2 mg doses every two hours. Trials of minocycline and N-acetyl cysteine did not improve symptoms. She cycled good and bad days. Dextromethorphan/quinidine, 1 tablet a day, improved symptoms with better engagement, talking, telling stories and enjoying outings. When improvement stalled, a course of 19 ECT sessions was started in March 2017 and dextromethorphan/quinidine was increased to twice a day. Symptoms improved including resolution of previously unexplained facial flushing. In September she moved into a residential Independent Living Home, attends a day program and is doing well. She was considered almost at baseline with BFCRS Screening and Severity scores of 3 and 2.5 (11-1-18). Recent reduction of lorazepam to 6 mg/day led to significant symptoms, which resolved quickly as lorazepam was restored to 13 mg/day.
Case 5: JT, 26 yo female, (1st visit 11/15; lorazepam initiation 12/15; follow up 3.2 yrs)
Diagnosed with Trisomy 21 DS shortly after birth, JT was considered healthy with spontaneous closures of a PDA at 6 months and ASD at 8 years. Hypothyroidism was diagnosed at age 8 years. She wore glasses for myopia and esotropia. The main health concern was early obesity, which was complicated by hidradenitis treated with long-term minocycline. Milestones were described as typical for Down syndrome with walking independently at 30 months. There were no episodes of regression below the age of 3 years, and no overt autistic symptoms, though she had a number of stereotypic behaviors, attachments to specific objects, and resistance to changes in routines. JT did well in special education classes where she preferred writing to talking which was in 3–4-word phrases. She read at a 3rd grade level and loved to knit, using a knitting form. As a child, JT was described as friendly and eager to please. She took meticulous care of her room, not wanting others to dust, as they put things in the wrong place. She attended to all her self-care and made her own breakfast. She liked feminine dresses, jewelry and often wore a tiara. As an adult, she worked in a daily workshop. In September 2015, JT missed a few days at work with non-specific complaints and crying jags but otherwise seemed fine. In early October, she developed an explosive diarrheal illness, refused to eat and retreated to bed. In the Emergency Room, a head CT, abdominal ultrasound, EKG and screening blood work were normal; gastroenteritis was considered likely. Following discharge, she continued to seem ill. She shook her hands in what was described as a tic-like behavior, had arm posturing, refused to eat or drink, and became mute and unresponsive to questions. She got stuck trying to move, had freezing episodes, and trouble climbing up and down stairs. She spent the majority of her time lying in bed staring at the ceiling, or sitting in a chair and staring straight ahead. She also developed repetitive tongue clicking, facial grimaces and repetitive finger movements as well as remaining in a rigid position with resistance to moving. She developed enuresis and required assistance with self-care skills in which she had previously been independent.
When evaluated in Regression Clinic she had been immobile for 6 weeks. Medical evaluations led to new diagnoses of celiac disease and autoimmune liver disease. The BFCRS Screening score was 9 and Severity score 26. She responded to a test dose of 2 mg of IV lorazepam becoming more active, looking at things and getting up to walk around. She was diagnosed with unspecified catatonia and started on lorazepam 2 mg/day. Her family had significant problems giving her daily medication due to irregular sleep patterns. Her dose was gradually increased to 14 mg/day, dextromethorphan/quinidine was added to the lorazepam resulting a significant improvement in CIS scores. She was talking more, amenable to getting dressed, and more social. By Christmas 2018, family perceived her to be at her baseline or possibly better, presumably reflecting concurrent diagnosis and treatment of previously unrecognized celiac disease.
She remained well, continuing on dextromethorphan/quinidine BID and lorazepam 12 mg/day. In September, JS developed abdominal pain and diarrhea and was diagnosed with acute B cell precursor lymphoblastic leukemia. She died following her third round of chemotherapy with liver failure and septic shock at age 29 years.
Case 6: CN, 19 yo female (1st visit 3/16; lorazepam initiation 5/16; follow up 2.8 years)
CN was diagnosed with mosaic Trisomy 21 DS shortly after birth. An arterial-ventricular canal defect was successfully repaired at 6 months of age. Except for mild exotropia, for which she had muscle revision, she remained healthy. Early development was mildly delayed with independent walking at 18 months. Parents reported her foremost impediment had been severe aphasia and apraxia for which she was in continuous speech therapy; prior to the regression she spoke in four-word sentences. She read at the 2nd grade level. Though there was never consideration of autism, her mother described her as shy, with poor eye contact and limited imaginative play. She had no echolalia or motor stereotypies, loved parties and had friendships in school. At 16–17 years of age, CN’s parents noted she was moving slower, had lost much of her speech and answered questions with just one word. She was evaluated by Child Neurology and treated for anxiety, poor sleep, depression and possible psychosis based on increasing self-talk. Medical treatments included fluoxetine, sertraline, escitalopram, clonidine, zolpidem and trazodone without improvement. She was self-referred to the University of Missouri in March 2016. Symptoms included slow walking, mutism with some whispering, weight loss, sitting still for long periods, and getting stuck/freezing when trying to move, pick up a fork or open a door. She remained in rigid positions, resisting efforts by others to change her position. She had lost interest in Special Olympics basketball and swinging which, she previously enjoyed. She lost self-help skills including bathing, shaving her legs and making her bed. Her behavior and mood were described as flat with staring and standing without moving.
At diagnosis of unspecified catatonia complicating DS, BFCRS Screening score was 9; Severity score was 18. Lorazepam, started at 2 mg/day, was gradually increased to 15 mg/day. Initially she seemed more engaged but symptoms continued; minocycline and N-acetyl cystine were added without improvement and were discontinued. She was started on dextromethorphan/quinidine in November 2016, resulting in improved interactions, which gradually faded. ECT was started in April 2017 with excellent response, which also dissipated. She remained stable but did not resume talking and was not as engaged in everyday activities. A short course of 4 ECT treatments returned CIS levels to near normal; stopping ECT allowed symptoms to return. In April 2018, a longer course of ECT returned symptoms to near normal, but required maintenance ECT to maintain. Currently she is treated with lorazepam 10 mg/day, dextromethorphan/quinidine 40 mg/day and maintenance ECT, in clusters of three sessions every 3 weeks. Parents report she is pretty much back to baseline, enjoying her day program, listening to music, doing art work and word searches.
Case 7: BI, 26 yo female, (First visit 6/16; lorazepam 7/16; Follow up 2.5 yrs)
BI was diagnosed with Down syndrome shortly after birth. Early motor development was within normal limits, sitting independently at 8 months and walking at 24 months. There were no developmental losses during childhood. She did well in school, read at a 4th grade level, spelled well and engaged in basic conversations. She sang in the choir, made friends, took Spanish in High School and participated in Special Olympics. She independently walked to school and to the store to buy groceries with her debit card. She graduated from High School in both regular and special classes. At 19 years, parents noted she stopped going to school events she had previously enjoyed. After graduation at age 21, she moved into a residential day service program. By 22 years, she stopped communicating verbally and became non-compliant with instructions. By the fall, she would just lie in bed or on the couch. When first seen in June 2016, she had no language, did not respond to questions or instruction and sat motionless for long periods. Her physical movements were slow, she held her head down, and exhibited repetitive movements including shoulder shrugging, facial grimacing, and repetitive hand movements. Her mood was depressed and she became combative and angry when pressed. She had lost activities of daily living and when taken to the shower would just stand.
BI was considered healthy. A possible seizure was observed with shaking of arms and legs in October 2015. She was hospitalized for 4 days with normal EEGs and basic labs. Brain MRI revealed focal encephalomalacia within the right caudate head reflective of a remote ischemic or inflammatory insult. Neurology concluded she did not have a seizure disorder and mother felt it was stress induced. Recent review of that admission by Psychiatry was deemed consistent with Catatonia
BI was diagnosed with unspecified catatonia in July 2016. She was begun on lorazepam 2 mg/day. Initial response to low dose lorazepam was minimal, partially due to medication refusal. As lorazepam dose increased to 14 mg/day and dextromethorphan/quinidine was added, she showed significant improvement with some talking, improved sleeping and eating, cooperating with her mother in showering, dressing and leaving the house. ECT started in March 2017 provided immediate improvement with some rebound symptoms which gradually diminished over the next 6 months on dextromethorphan/quinidine and lorazepam. When last seen in 2017, BFCRS Screening =5, Severity =4. Mother considered her about 80% baseline. Recent phone visit indicates increased symptoms; ECT is being considered.
Results
Each catatonia diagnosis was based on a history of significant regression and symptoms enumerated by the BFCRS. At diagnosis, all patients scored in the catatonia diagnostic range with Screening scores ranging from 8–12 with mean of 9 (Table 1) and Severity scores from 16 to 29 with mean of 20. The nine most common symptoms were mutism, immobility, getting stuck during motor activities, involuntary movements, perseveration or obsessive symptoms, negativism, and social withdrawal. Loss of independence in daily living skills and sleep disturbances were also common. Though not considered part of catatonia, emotional lability with episodes crying, sobbing, laughing or giggling was reported by all families. The diagnosis of catatonia was corroborated by detecting at least three core symptoms specified by the more diagnostically conservative DSM-5 criteria.35,39 Positive responses to customary medical treatments of catatonia known to affect the GABA/Glutamate neurotransmitter systems confirmed the diagnoses.
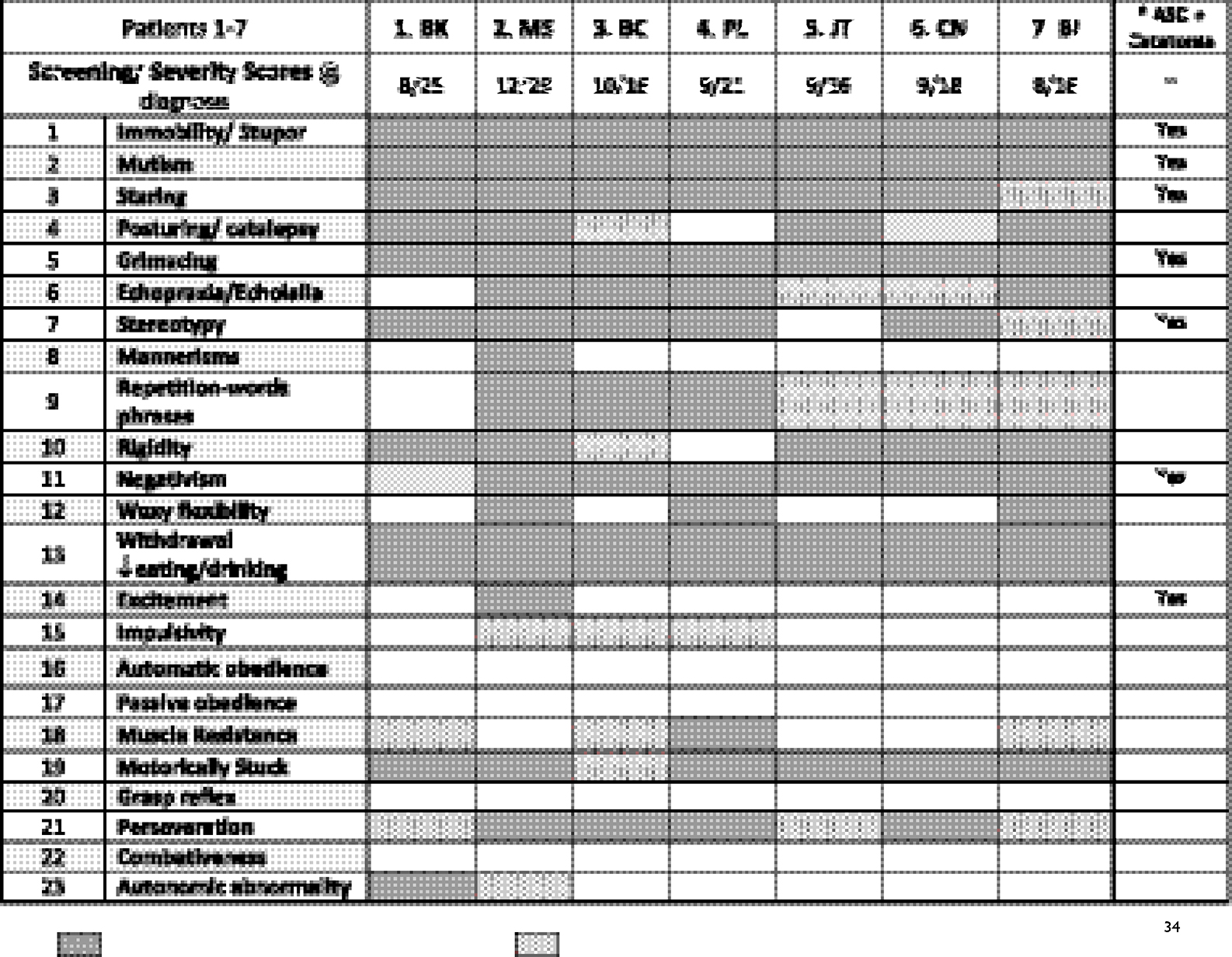 |
Table 1 Symptoms of catatonia in DS using the Bush-Francis Catatonia Rating Scale (BFCRS) |
Each family provided comprehensive medical, genetic and social histories and patients received thorough medical and neurologic examinations (Table 2). Previous genetic testing results confirmed complete trisomy 21 in 6 subjects and a mosaic karyotype in the other. One patient also carried a 16p13.11–12.3 duplication. Ages at diagnosis ranged from 19 to 33 years, with delays from symptom onset to diagnosis ranging from 3 months to 10 years. Six of the seven families described gradual onset with fluctuation in symptoms as their child became steadily more symptomatic. Three families described one or more early episodes resembling mild catatonia that appeared to resolve. The shortest intervals to diagnosis were one and nine months. Five families reported significant life stress prior to symptom onset including death and serious illness of fathers, divorce, anxiety caused by classmates and leaving high school. In most respects, these seven individuals resembled other young adults with DS. The only demographic outlier was the male to female ratio of 1:6 which differed from the commonly reported ratio of about 1.2:1. Medical histories were also consistent with Down syndrome. Premorbid adaptive functioning ranged from low to moderately high.
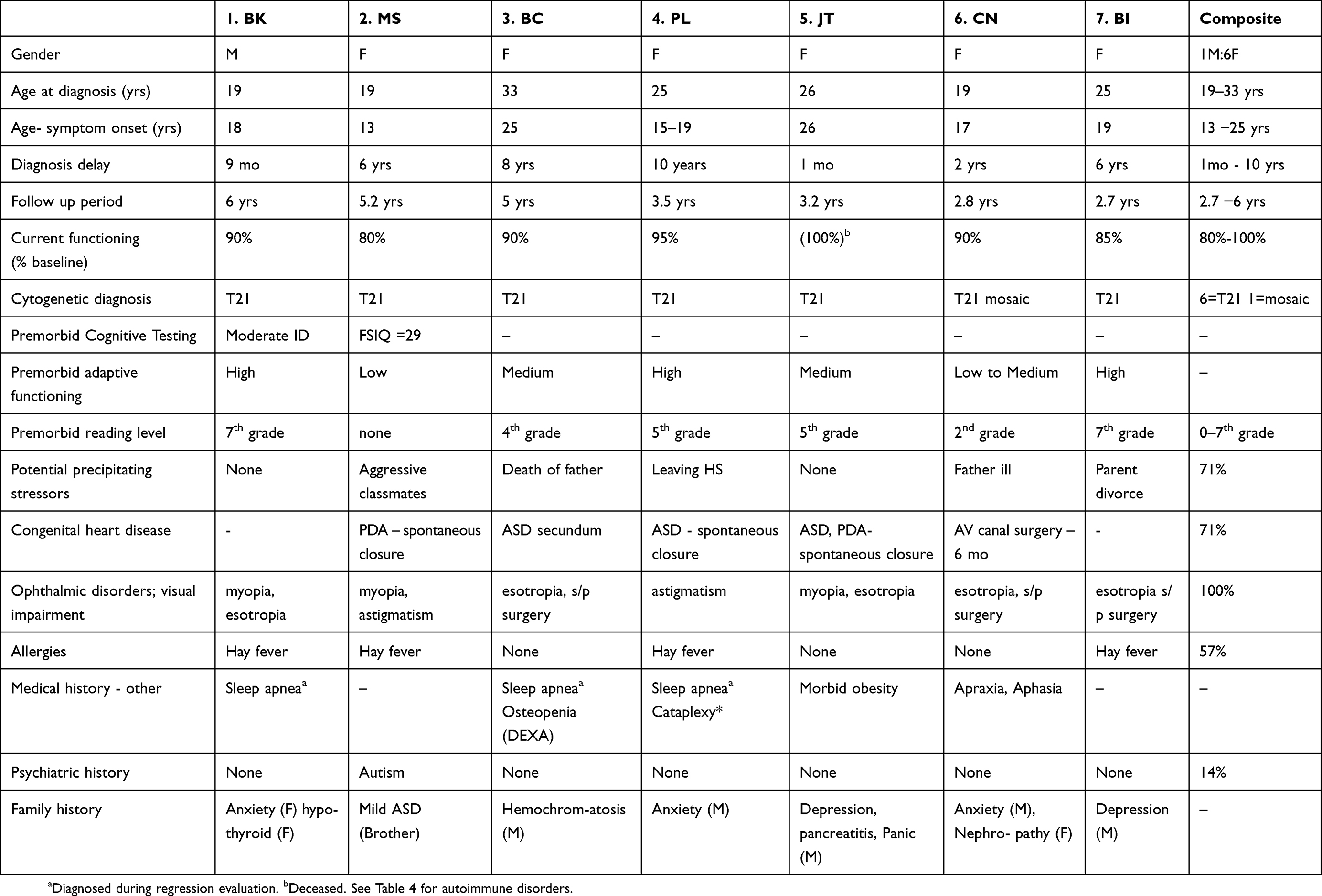 |
Table 2 Demographic, genetic, medical and psychiatric features in seven cases of DS and catatonia |
Neurologic symptoms and testing are tabulated in Table 3. None of the patients had evidence of seizures, encephalitis or other brain infections. Patient MS (#3), who was the most intellectually impaired, had background slowing on the EEG and mild vermis hypoplasia on brain MRI. Six of the seven patients had cerebral spinal fluid (CSF) studies including cell counts, glucose, pH, lactate and protein. Studies specifically aimed at causes of catatonia, included the CSF Paraneoplastic autoantibody Evaluation, which included antibodies to NMDA (N-methyl-D-aspartate)-type glutamate receptors, Glutamic acid decarboxylase (GAD), and others.40 CSF was also tested for Methyltetrahydrofolate, neurotransmitter metabolites (5HIAA, HVA, 30MD) and Neopterin (a marker for CNS immune system stimulation); all were within normal ranges.41 Plasma Paraneoplastic Autoantibody Evaluations obtained for all 7 patients were normal. Six of the seven patients were Vitamin D deficient or insufficient; oral Vitamin D supplementation led to normalization of levels. Three patients underwent overnight Polysomnography due to snoring, day time sleepiness or restless sleep. All had positive studies; two were treated and remain on CPAP; the milder patient declined CPAP.
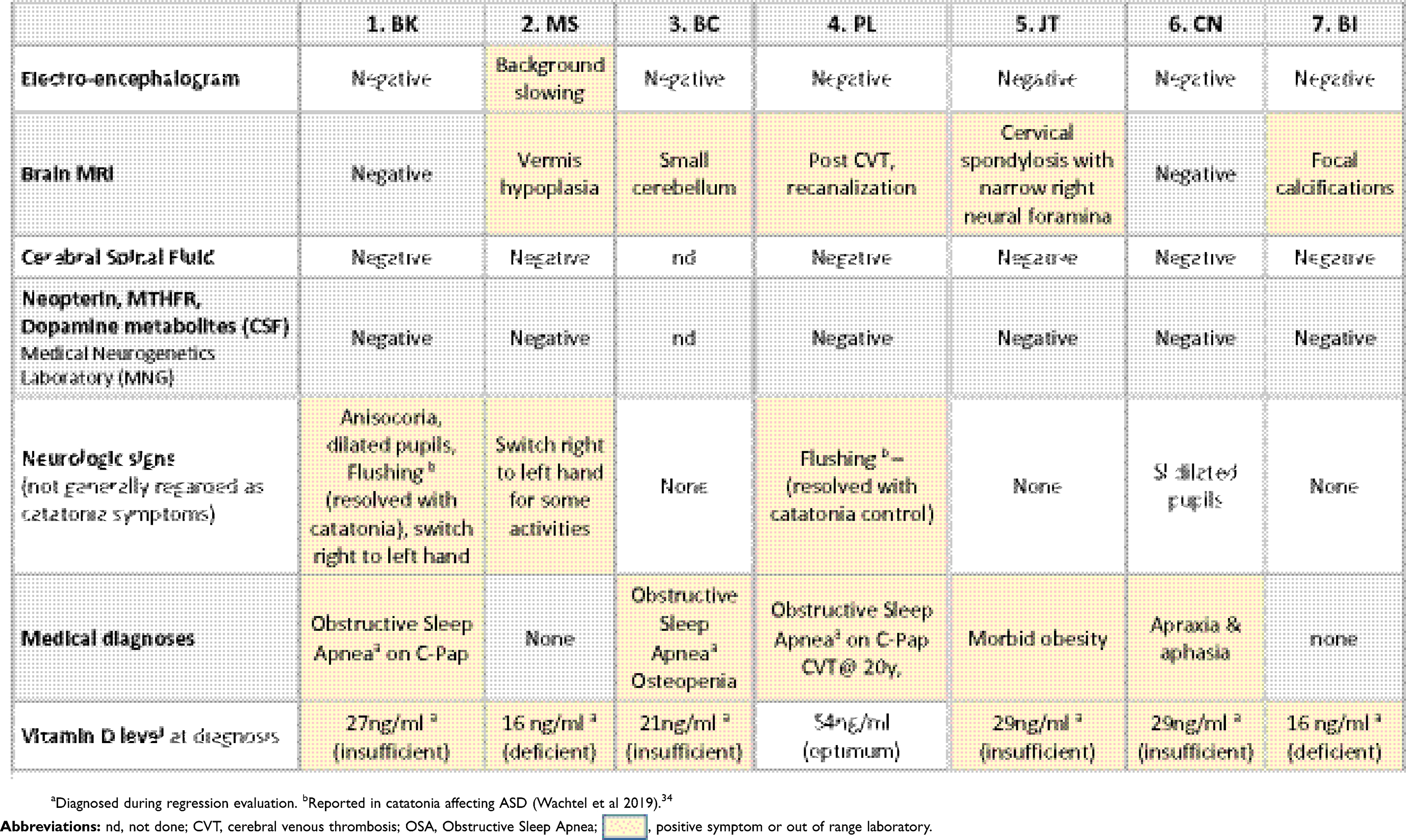 |
Table 3 Medical and neurologic features and test results in seven case of DS and catatonia |
Autoimmune disorders were diagnosed or confirmed in four patients, including three with celiac disease (42%), two of whom were diagnosed during the catatonia evaluation, three with hypothyroidism (43%), one immune hepatic disease (14%) and one alopecia areata (14%). (Table 4). Four patients had two or more autoimmune disorders. Only the four with recognized autoimmune disorders had elevated FANAs or ESRs and the three without diagnosed autoimmune disorders had no autoimmune markers other than an elevated ASO titer in one and elevated DNase antibody in the other. Paraneoplastic autoimmune panels including serum (7 patients) and CSF (4 patients) revealed no infectious or neoplastic markers, including GAD65, NMDA receptor and GABA receptor antibodies.40 On the other hand, the Cunningham panel which detects neural autoimmune antibodies associated with neuropsychiatric disorders including PANS, PANDAS and Sydenham’s chorea revealed high titers to one or more neural proteins in six of the seven patients.42–44 The CaM Kinase II (Calcium-dependent Calmodulin Protein Kinase II) activation, which is a measure of neural immune stimulation was elevated in 3 cases.
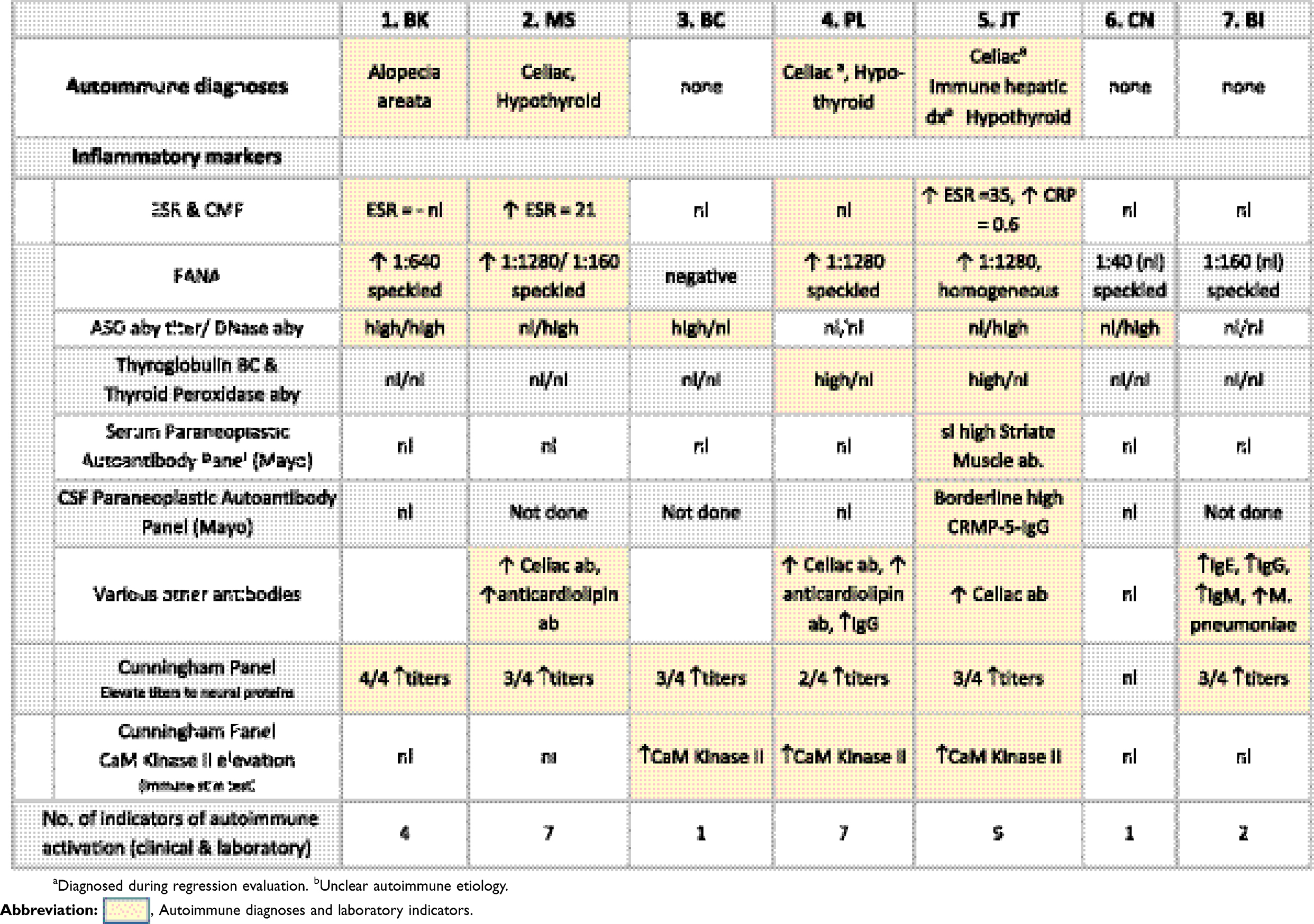 |
Table 4 Autoimmune diagnoses and testing in seven case of DS and catatonia |
Response to treatments
Each patient displayed a favorable reaction to the 2 mg IV test dose of lorazepam given at the first clinic visit (Table 5). Common observations were faster walking, and looking around at people and the clinic environment rather than looking down. For example, BC (#3), who had regressed 10 years earlier, sat in clinic without moving or acknowledging us. About 20 mins after receiving lorazepam, she began turning her head and looking around. She noticed a book on sea animals. With some prompting she read the descriptions under the pictures. She had not done that in years. In all cases parents noticed small improvements in symptoms ranging from faster walking or eating, looking around during the ride home, relaxed posture and less negativity lasting up to 24 hrs. Following medical evaluations, patients started 2 mg oral lorazepam each morning. As the doses were gradually increased, the amount of improvement varied. The first two patients, BK (#1) and MS (#2) started treatment before the CIS tracking measure was developed, hampering our ability to track specific symptoms. This was especially problematic for MS (#2) whose combination of DS, autism and catatonia made it problematic to accurately evaluate her response to low lorazepam doses, which led to relatively early introduction to ECT. All six patients who clearly responded to increasing doses of lorazepam, continued to have some symptoms including facial movements, shoulder shrugging, staring and resisting previously enjoyed activities. All appeared to reach a plateau where increasing lorazepam by 2 mg did not improve symptoms. At that time ECT was recommended as the next treatment. Unfortunately, Missouri’s requirement for court approval for individuals with diminished capacity to consent, often delayed ECT up to 6 months.
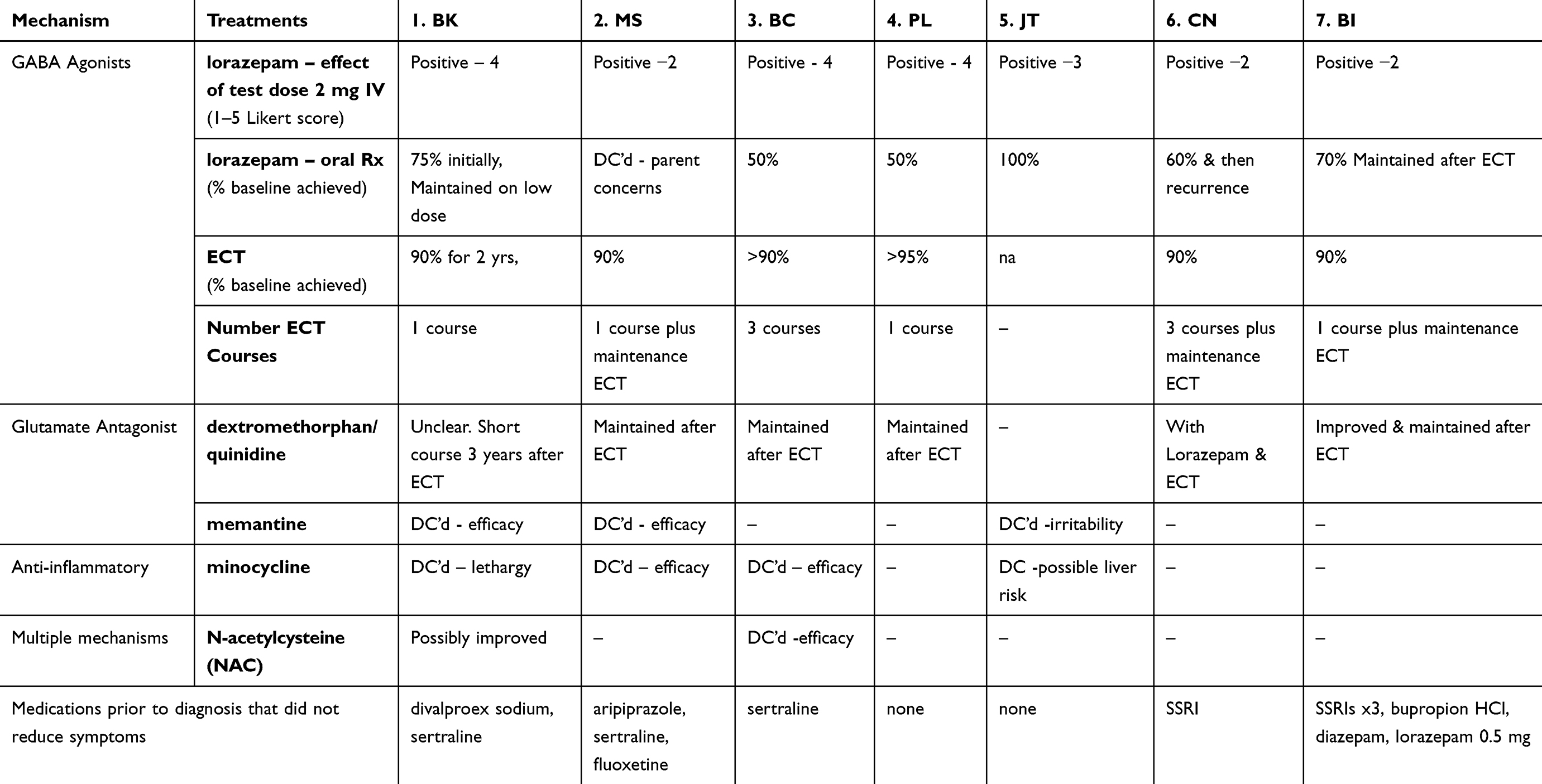 |
Table 5 Catatonia treatments and results in seven case of DS and catatonia |
Three glutamate antagonists, memantine, N-acetyl cysteine (NAC) and minocycline, were considered due to their anti-inflammatory properties and reports of some efficacy, and tried for limited periods. None provided sustained improvement and were eventually discontinued. In November 2014, positive outcomes with dextromethorphan/quinidine, another glutamate antagonist, were reported in adult psychiatric patients with catatonia.29 Subsequently, six of our seven patients were treated with dextromethorphan/quinidine starting with the 20 mg dextromethorphan a day, increasing to 40 mg after one week. Four appeared to have symptom improvement maintained for a number of months. In two cases, dextromethorphan/quinidine appeared to sustain improvement derived from ECT. As noted by others, response to glutamate antagonists was notably slower than response to lorazepam.45 Six patients received from one to three courses of ECT, two remained on maintenance. One young woman had 3 ECT courses, and continues on maintenance ECT, which her family reports maintains close to baseline functioning. The Catatonia Impact Scale (CIS) proved to be sensitive to small symptom changes providing a response trajectory which helped formulate timelier treatment decisions (Figure 1).33
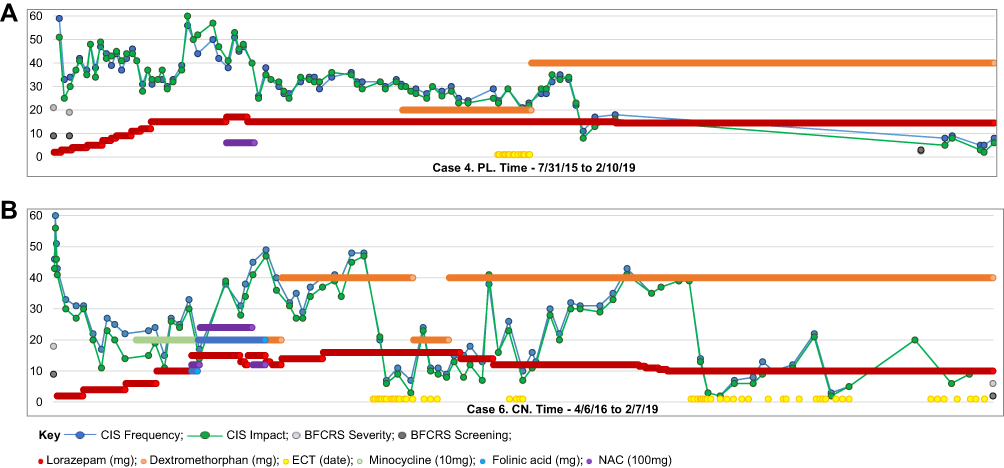 |
Figure 1 Longitudinal progress measured by parent reports using the Catatonia Impact Scale (CIS) and BFCRS. (A) Case 4. PL. From 7/31/15 to 2/10/19. (B) Case 2. CN. From 4/6/16 to 2/7/19. |
Discussion
For generations, catatonia symptoms were interpreted as signs of other named diagnoses, including schizophrenia, autism, encephalitis, cataplexy and even hysteria. As Shorter pointed out
recent revisions in the classification of psychiatric disease has afforded the opportunity to rescue catatonia from the swamp of competing diagnoses and establishing it as an independent illness entity in adults and children.49
Initially, this rested on recognition that catatonia was not a subtype of Schizophrenia.9,10,46 And more recently, that catatonia was not a symptom of autism as had been suggested.47,48 Catatonia is now accepted, by most psychiatrists, as a discrete syndrome, stemming from a variety of psychiatric, neurologic or medical causes.9,11,14,23,38,46,50–53 Adolescents and young adults with neurodevelopmental disabilities are merely the most recent group recognized at risk to develop catatonia.6,10,14,38,50,54 This overdue recognition should not come as a surprise, since until the 1980s, physicians did not believe individuals with DS were susceptible to the same psychiatric disorders or would respond to the same treatments as their typical peers.55–57
The first signs of regression in our patients were noted between 13 and 26 years, similar to published experience for Down syndrome and Autism.48,58,59 Most families described a gradual downhill course over a few months to 6 years. Time from onset of symptoms to diagnosis generally reflected the time it took families to persuade physicians and/or schools that the change was not behavioral or just part of DS. Only JT (#5) presented abruptly over a 2-week period. Our experience was similar to previous observations that 75% of young people with developmental disorders had more insidious onsets compared with abrupt onsets in 77% of patients with medical disorders.14 Stressful events or situations temporally coinciding with functional regression have been postulated as triggers or causes.60 In our patients, significant emotional trauma ranging from death or significant illness of a parent, divorce, a stressful class room and leaving a supportive high school environment were described. Moreover, all but one family reported that highly stressful situations continued to exacerbate catatonia symptoms. On the other hand, each of the seven young people, especially as they were responding to treatment, improved more rapidly when they were able to interact with friends, therapists and families. Removal from social situations appeared to exacerbate withdrawal.
Once the possibility of catatonia is acknowledged, the diagnostic process for individuals with Down syndrome is identical to that for patients with primary psychiatric diagnoses. The first step is to substantiate a history of unambiguous regression from baseline functioning. This may be more difficult for the clinician to appreciate in a young person with developmental and/or behavioral symptoms as opposed to an otherwise functional adult who is more easily identified as unwell even by those who had not known him before. The catatonia diagnosis is established by identifying specific clinical symptoms on the BFCRS.17,12,13,15 Patients with catatonia complicating psychiatric or medical diagnoses present with similar catatonia symptom profiles.12,14,61 As iterated by Francis (2010), “catatonia caused by psychiatric disorders and that due to medical conditions or toxicity shows similar structure and severity”.12 Waxy flexibility, excitement, automatic and passive obedience, grasp reflex and combativeness which were infrequently observed in our patients have also been reported less commonly in psychiatric patients.12,24 We found that night and day time enuresis which often accompanied severe catatonia improved as the catatonia was treated, implying a shared origin. Sleep problems with insomnia, early wakening and restlessness were also commonly reported and again resolved or greatly improved with resolution of the most severe catatonic symptoms, without other medical intervention. Verification of a catatonia diagnosis is based on the resolution of symptoms in response to standard first line treatments. This was clear in our patients, all of whom responded positively to a test dose of lorazepam (2 mg IV) and ECT. Prior treatments with antidepressants, antipsychotics, anxiolytics or sleep medications had not resolved catatonia symptoms.
Benzodiazepines and electroconvulsive therapy (ECT) remain the standard, first-line therapies for catatonia in both children and adults regardless of underlying psychiatric or medical disorders and are also the most clinically studied.16,20,21,62,63 The preponderance of reports in DS, Autism and other neurodevelopmental disorders affirms their efficacy in these populations.6–9,11,14,53,63 A recent Cochrane report of 31 articles revealed lorazepam response rates of 66–100% in Western countries; ECT response rates varied 59–100%.22 Though, excellent response to ECT is generally acknowledged, including endorsement from the American Academy of Child and Adolescent Psychiatry, rigorous evidence including random controlled trials and meta-analyses have not been performed.62–65 In addition, it is suggested a synergistic effect may be obtained by providing lorazepam and ECT simultaneously.66–68
Catatonia also responds to glutamate antagonists, which work through NMDA receptors, to raise GABA relative to glutamate. A number N-methyl-D-aspartate (NMDA) antagonists, including memantine, amantadine, N-acetyl cysteine (NAC), minocycline, topiramate and dextromethorphan/quinidine have each had proponents.26–29 In our patients, NMDA antagonists were used to supplement lorazepam when increasing GABA agonist dosages failed to elicit optimal improvement. In this limited clinical experience dextromethorphan/quinidine appeared the most effective controlling symptom rebound following ECT.
An unanticipated distinction between catatonia in our Down syndrome cohort and catatonia reported in patients with psychiatric and medical disorders is the chronicity of symptoms and dependence on sustained treatment to maintain recovery (Table 5). Each of the patients, followed from 2.7 to 6 years, eventually responded to therapy and is currently functioning close to their baseline. However, each remains on medications (Lorazepam - 3.5–16 mg/day, and/or dextromethorphan/quinidine 40/20 mg/day) and/or maintenance ECT. Response to ECT was clearly superior to medications (Figure 1). Reasons for the protracted nature of catatonia in DS is unclear. Consoli et al reported 75% of patients with developmental disorders had a chronic course, compared to only 17% of adolescents with a medical cause and 7% with underlying psychiatric disorders.14 A recent description of 22 patients with Autism and catatonia found that ECT provided marked benefit in all patients, though often required ongoing maintenance treatments.34 One plausible explanation for the catatonia’s chronicity in young people with neurodevelopmental disorders may be delayed diagnoses, which might render treatment more difficult. Based on this experience, Pelzer and colleagues recommend that patients with long term symptoms of catatonia be treated expediently with ECT, a recommendation our experience supports.22 At this time, we feel comfortable advising families that whereas we can’t promise recovery will be rapid, experience supports optimism that with standard of care treatments including lorazepam, ECT and possibly dextromethorphan/quinidine a return to baseline or close to baseline functioning is expected.
An additional objective of this study was to ascertain any clinical or laboratory characteristics that might foretell a risk of catatonia (Table 2, 3, 4). For the most part, our patients displayed typical DS features, including cytogenetic variation; six with isolated Trisomy 21 and one mosaic Trisomy 21. One (14%) was diagnosed with Autism, consistent with the 13–18% concordance commonly reported.69–72 That our patient MS (#2), who had the most severe intellectual disability and problematic behavior is consistent with previous reports that children with DS plus ASD tend to have more severe cognitive impairment, language deficits, stereotyped behavior, over activity, impulsivity and self-injurious behavior73–77 Likewise, medical diagnoses in our patients were compared to DS norms (Tables 2 and 3). Congenital heart diseases (71% vs 25–45% expected), ophthalmologic disorders (100% vs 40% expected), speech delays (100% vs almost 100% expected), autism spectrum disorders (14% vs 12–18%), obstructive sleep apnea (57% vs 30–66% expected) and osteoporosis (29% vs >50% expected) were the most common.78–82 Only, the incidence of congenital heart defects (71%) was appreciably higher than expected (25–45%). Of the five cardiac diagnoses, three resolved spontaneously and none of the five children had on-going cardiac problems requiring medications, activity limitations or close cardiac follow-up, making it unlikely that congenital heart defects provoked catatonia. Each family considered their child medically vigorous with expected growth parameters and general good health. Based on this small sample, we found no evidence that young people with DS who developed catatonia were less healthy than their typical DS peers. IQ scores were not consistently available for our patients, as parents had often declined school testing; once catatonia developed valid assessments were impossible. In lieu of psychometric data, we used each person’s highest reading level, which ranged from none to 7th grade, and found reading levels roughly corresponded to functional levels as described by parents and in school records.83 Functional abilities ranged from low to high functioning and roughly mirrored or surpassed cognitive functioning reported in DS adults (moderate-severe impairment in about 70%, mild-moderate impairment in 20% and 8% in the mild range).84
On the other hand, the DS+CAT patients appear to have more of some autoimmune disorders (Table 4). Hypothyroidism (Hashimoto’s thyroiditis and/or Graves’ disease) occurred in 57% vs 35% reported in DS alone, celiac disease was found in 43% vs 5–10% in DS and Alopecia areata in 14% vs 4–11% in DS.85–91 This is noteworthy in light of the elevated risk for catatonia in typical patients with autoimmune disorders including Celiac disease, Graves’s disease, Diabetes Type 1, Alopecia Areata, Lupus, Cerebral Folate deficiency and Paraneoplastic Autoimmune Encephalitis.,52,53,92,93 raising the question of a pathophysiologic relationship between DS and catatonia.
Laboratory corroboration of autoimmune activation occurred almost exclusively in our patients with autoimmune medical diagnoses. JT (#5), with four autoimmune disorders, had the most salient results including a speckled pattern FANA, elevated ESR and CRP plus borderline serum and CSF Paraneoplastic Autoantibody Panels. Since, autoimmune laboratory data have not been collected from healthy, age matched DS populations it is not possible to speculate whether laboratory evidence of autoimmune activation might predict or correlate with development of catatonia. Though none of our patients fit clinical criteria for PANS (Pediatric Acute Neurologic Syndrome), anecdotal reports of catatonia being misdiagnosed as PANS prompted us to obtain the Cunningham Panel, which is used in diagnostic evaluations for PANS, measuring levels of circulating antibodies directed against brain antigens, and measuring their ability to increase the activity of the CaMKII enzyme that upregulates neurotransmitters in the brain.42–44 Of the seven DS+Cat patients tested, six exhibited between 2 and 4 elevated anti-neuronal protein titers and three exhibited a positive neural immune stimulation assay. This suggests inclusion of the Cunningham Panel may help identify whether in DS, regression is related to autoimmunity.
An unexpected observation was the high (6:1) female to male ratio in our DS+Cat population, which mirrored the 6.5:1 ratio from three previous DS+Cat reports.6–8 This differs from sex ratios reported in unselected DS populations. One review of fifty-five publications found consistent 1.31:1 male to female ratios in Trisomy 21.94 Similar ratios are reported in the US, Europe and China.95,96 This information, plus the increased propensity for females to develop autoimmune disorders in the general population raises the question whether females with DS are at a higher risk for catatonia.
Young people with neurodevelopmental disorders present an additional diagnostic challenge for many clinicians. To the inexperienced, the individual symptoms of catatonia may be misconstrued as part of their neurodevelopmental disorder. Furthermore, for the 15–20% of individuals with Down syndrome also diagnosed with autism, their array of behavioral symptoms, some of which overlap with symptoms of catatonia, can steer clinicians away from the catatonia diagnosis. These impediments will only resolve through comprehensive descriptions in the medical literature.
Conclusion
A comprehensive evaluation of catatonia in young adults with Down syndrome confirmed that the diagnosis of catatonia in DS is straightforward when based on a clear history of functional regression, symptoms specified by the BFCRS and positive responses to lorazepam and/or electroconvulsive therapy. Furthermore, comparing catatonia in DS patients to catatonia described in Autism, in various psychiatric and medical disorders we observed no quantifiable differences in either the catatonia diagnostic process or catatonia symptoms. That is not to say that the underlying neurodevelopmental disorder may not influence the severity or tenor of the symptoms or the ease in reaching remission. For these reasons, further evaluation of catatonia in homogeneous patient groups is recommended.
Though our treatment protocols remained the same throughout the five years, some modifications were made based on increasing clinical experience. Significant changes were 1) lorazepam was continued during ECT after patient BK (#1); 2) lorazepam doses were increased more rapidly; 3) ancillary treatment modalities, including NAC, minocycline, and memantine were tried less often and for shorter periods before discontinuing; 4) dextromethorphan/quinidine was started earlier in the course and continued for longer periods. These clinical judgements developed in the course of patient care and can’t be construed as clinical trials. In addition, each young person presented with previously undetected health problems, with symptoms apparently overshadowed by the catatonia. Concurrent treatment of these health conditions undoubtedly optimized health and functioning, and also allowed us to track responses to the catatonia specific treatments, independently of symptoms related to specific medical disorders.
Finally, following these seven patients with DS and catatonia for three and a half to six years, provided a number of fresh observations including a high prevalence of autoimmune disorders (57%), laboratory evidence of immune activation and an enhanced female gender ratio (6 F:1 M), each of which raise the prospect of immunological influences. Future investigations, following these clues, may lead to a clearer understanding of catatonia triggers in DS.
Acknowledgments
The authors thank the Leda Sears Foundation, the Department of Child Health, and the Thompson Center for the grant and in-kind support of this work. Especially we thank the young people and their parents who participated fully in this endeavor including reviewing and refining the Catatonia Impact Scale.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Presson AP, Partyka G, Jensen KM, et al. Current estimate of down syndrome population prevalence in the United States. J Pediatr. 2013;163(4):1163–1168. doi:10.1016/j.jpeds.2013.06.055
2. de Graaf G, Buckley F, Skotko BG. Estimation of the number of people with down syndrome in the United States. Gen Med. 2017;19(4):439–447.
3. St. Louis AM, Kim K, Browne ML, et al. for the national birth defects prevention network. Prevalence trends of selected major birth defects: a multi-state population-based retrospective study, United States, 1999 to 2007. Birth Defects Res. 2017;109:1442–1450. doi:10.1002/bdr2.1113
4. Hunter A. Down syndrome. In: Cassidy SB, Allanson JE, Hoboken HJ, editors. Management of Genetic Syndromes.
5. Down Syndrome Medical Interest Group-USA (DSMIG-USA). Available from: http://www.dsmig-usa.org/.
6. Ghaziuddin N, Nassiri A, Miles JH. Catatonia in down syndrome; a treatable cause of regression. Neuropsychiatr Dis Treat. 2015;2(11):941–942. doi:10.2147/NDT.S77307
7. Jap SN, Ghaziuddin N. Catatonia among adolescents with down syndrome: a review and 2 case reports. J Ect. 2011;27(4):334–337. doi:10.1097/YCT.0b013e31821d37c6
8. Torr J, D’Abrera JC. Maintenance electroconvulsive therapy for depression with catatonia in a young woman with down syndrome. J Ect. 2014;30(4):332–336. doi:10.1097/YCT.0000000000000116
9. Dhossche DM, Wachtel LE. Catatonia is hidden in plain sight among different pediatric disorders: a review article. Pediatr Neurol. 2010;43(5):307–315. doi:10.1016/j.pediatrneurol.2010.07.001
10. Fink M. Rediscovering catatonia: the biography of a treatable syndrome. Acta Psychiatr Scand. 2013;127:1–47. doi:10.1111/acps.2012.127.issue-s441
11. Hauptman AJ, Benjamin S. The differential diagnosis and treatment of catatonia in children and adolescents. Harvard Rev Psychiat. 2016;24(6):378–395. doi:10.1097/HRP.0000000000000114
12. Francis A. Catatonia: diagnosis, classification and treatment. Curr Psychiatry Rep. 2010;12:180–185. doi:10.1007/s11920-010-0113-y
13. Sienaert P, Rooseleer J, De Fruyt J. Measuring catatonia: a systematic review of rating scales. J Affect Disord. 2011;135(1–3):1–9. doi:10.1016/j.jad.2011.02.012
14. Consoli A, Raffin M, Laurent C, et al. Medical and developmental risk factors of catatonia in children and adolescents: a prospective case-control study. Schizophr Res. 2012;137:151–158. doi:10.1016/j.schres.2012.02.012
15. Sakar S, Sakey S, Mathan K, Bharadwaj B, Kattimani S, Kajkumar R. Assessing catatonia using four different instruments: inter-rater reliability and prevalence in inpatient clinical population. Asian J Psychiatr. 2016;23:27–31. doi:10.1016/j.ajp.2016.07.003
16. Luchini F, Medda P, Mariani MG, Mauri M, Toni C, Perugi G. Electroconvulsive therapy in catatonic patients: efficacy and predictors of response. World J Psychiatry. 2015;22:182–192. doi:10.5498/wjp.v5.i2.182
17. Bush G, Fink M, Petrides G, Dowling F, Francis A. Catatonia I. Rating scale and standardized examination. Acta Psychiatr Scand. 1996;93(2):129–136. doi:10.1111/acp.1996.93.issue-2
18. Ghaziuddin N, Walter G, eds. Electroconvulsive Therapy in Children and Adolescents. New York: Oxford University Press; 2013.
19. Ghi D, Ghaziuddin N. Use of electroconvulsive therapy in the treatment of catatonia. Future Neurol. 2014;9(5):533–540. doi:10.2217/fnl.14.49
20. Sienaert P, Dhossche DM, Vancampfort D, De Hert M, Gazdag G. A clinical review of the treatment of catatonia. Front Psychiatry. 2014;5:181–190. doi:10.3389/fpsyt.2014.00181
21. Dhossche DM. Decalogue of catatonia in autism spectrum disorder. Front Psychiatry. 2014;5:1–4. doi:10.3389/fpsyt.2014.00001
22. Pelzer A, Heijden F, Boer E. Systematic review of catatonia treatment. Neuropsychiatric Dis Treat. 2018;14:317–326. doi:10.2147/NDT.S147897
23. Walther S, Strik W. Catatonia. CNS Spectr. 2016;21:341–348. doi:10.1017/S1092852916000274
24. Rosebush PI, Mazurek MF. Catatonia and its treatment. Schizophr Bull. 2010;36(2):239–242. doi:10.1093/schbul/sbp141
25. Francis A, Fink M, Appiani F, et al. Catatonia in diagnostic and statistical manual of mental disorders, fifth edition. J Ect. 2010;26(4):246–247. doi:10.1097/YCT.0b013e3181fe28bd
26. Beach SR, Gomex-Bernal F, Huffman JC, Fricchione GL. Alternative treatment strategies for catatonia: a systematic review. Gen Hosp Psychiatry. 2017;48:1–19. doi:10.1016/j.genhosppsych.2017.06.011
27. Carroll BT, Goforth HW, Thomas H, et al. Review of adjunctive glutamate antagonist therapy in the treatment of catatonic syndromes. J Neuropsychiatry Clin Neurosci. 2007;19(4):406–412. doi:10.1176/jnp.2007.19.4.406
28. Theibert HPM, Carroll BT. NMDA antagonists in the treatment of catatonia: a review of case studies from the last 10 years. Gen Hosp Psychiatry. 1918;51:132–133. doi:10.1016/j.genhosppsych.2017.10.010
29. Caplan JP Use of dextromethorphan/quinidine as a glutamate modulator in catatonia.
30. Turner JL, Mitchell JL, Carroll BT, Denysenko L. Dextromethorphan/quinidine withdrawal-emergent catatonia. Ann Clin Psychiatry. 2016;1:e8–e9.
31. Kanne SM, Mazurek MO, Sikora D, et al. The Autism Impact Measure (AIM): initial development of a new tool for treatment outcome measurement. J Autism Dev Disord. 2014;44(1):168–179. doi:10.1007/s10803-013-1862-3
32. Mazurek MO, Carlson C, Baker-Ericzén M, Butter E, Norris M, Kanne S. Construct validity of the Autism Impact Measure (AIM). J Autism Dev Disord. 2018. doi:10.1007/s10803-018-3462-8
33. Nowell KP, Muckerman J, Graves B, Takahashi N, Miles JH. A longitudinal measure of patient outcomes and treatment responses. In preparation, 2019.
34. Wachtel LE. Treatment of catatonia in autism spectrum disorders. Acta Psychiatr Scand. 2019;130(1):46–55. doi:10.1111/acps.12980
35. American Psychiatric Association (APA). Diagnostic and Statistical Manual of Mental Disorders: DSM-5.
36. Woodbury-Smith M, Paterson AD, O’Connor I, et al. A genome-wide linkage study of autism spectrum disorder and the broad autism phenotype in extended pedigrees. J Neurodevelop Disord. 2018;10:20–29. doi:10.1186/s11689-018-9238-9
37. Girirajan S, Dennis MY, Baker C, et al. Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am J Hum Gen. 2013;92:221–237. doi:10.1016/j.ajhg.2012.12.016
38. Raffin M, Consoli A, Giannitelli M, et al. Catatonia in children and adolescents: a high rate of genetic conditions. J. Am Acad Child Adolesc Psychiatry. 2018;57(7):518–525. doi:10.1016/j.jaac.2018.03.020
39. Tandon R, Heckers S, Bustillo J, et al. Catatonia in DSM-5. Schizophr Res. 2013;150:26–30. doi:10.1016/j.schres.2013.04.034
40. Mayo Clinic Laboratories. Available from: http://www.mayocliniclabs.com/customer-service.
41. Medical Neurogenetics Laboratories Atlanta GA USA. Available from: http://mnglabs.com.
42. Moleculara Laboratory, Oklahoma City OK USA. Available from: http://moleculara.com.
43. Chang K, Frankovich J, Cooperstock M, et al. Clinical evaluation of youth with Pediatric Acute-Onset Neuropsychiatric Syndrome (PANS): recommendations from the 2013 PANS consensus conference. J Child Adolesc Psychopharmacol. 2015;25(1):3–13. doi:10.1089/cap.2014.0084
44. Cox CJ, Zuccolo AJ, Edwards EV, et al. Antineuronal antibodies in a heterogeneous group of youth and young adults with tics and obsessive-compulsive disorder. J Child Adolescent Psychopharmacol. 2015;25(1):1–10. doi:10.1089/cap.2014.0048
45. Daniels J. Catatonia: clinical aspects and neurobiological correlates. J Neuropsychiatry Clin Neurosci. 2009;21:371. doi:10.1176/jnp.2009.21.4.371
46. Fink M, Shorter E, Taylor MA. Catatonia is not schizophrenia: kraepelin’s error and the need to recognize catatonia as an independent syndrome in medical nomenclature. Schizophr Bull. 2010;36:314–320. doi:10.1093/schbul/sbp059
47. Wing L, Shah A. Catatonia in autistic spectrum disorders. Br J Psychiatry. 2000;176:357–362. doi:10.1192/bjp.176.4.357
48. Wing L, Shah A. A systematic examination of catatonia-like clinical pictures in autism spectrum disorders. In: Dhossche DM, Wing L, Ohta M, Neumärker K-J, editors. Catatonia in Autism Spectrum Disorders. Amsterdam: Elsevier; 2006:22–39.
49. Shorter E. Making childhood catatonia visible, separate from competing diagnoses. Acta Psychiatr Scand. 2012;125(1):3–10. doi:10.1111/j.1600-0447.2011.01788.x
50. Ghaziuddin N, Dhossche D, Marcotte K. Retrospective chart review of catatonia in child and adolescent psychiatric patients. Acta Psychiatr Scand. 2012;125:33–38. doi:10.1111/j.1600-0447.2011.01778.x
51. Rustad JK, Landsman S, Ivkovic A, Finn CT, Stern TA. Catatonia: an approach to diagnosis and treatments. Prim Care Companion CNS Disord. 2018;20(1):17f02202. doi:10.4088/PCC.17f02202
52. Oldham MA. The probability that catatonia in the hospital has a medical cause and the relative proportions of its causes: a systematic review. Psychosomatics. 2018;59:333–340. doi:10.1016/j.psym.2018.04.001
53. Smith JH, Smith VD, Philbrick KL, Kumar N. Catatonia due to a general medical or psychiatric condition. J Neuropsychiatry Clin Neurosci. 2012;24:198–207. doi:10.1176/appi.neuropsych.11060120
54. Dhossche D, Bouman N. Catatonia in children and adolescents. J Am Acad Child Adolesc Psychiatry. 1997;36(7):870–871. doi:10.1097/00004583-199707000-00007
55. Myers BA, Pueschel SM. Psychiatric disorders in persons with down syndrome. J Nerv Ment Dis. 1991;79(10):609–613. doi:10.1097/00005053-199110000-00004
56. Mantry D, Cooper SA, Smiley E, et al. The prevalence and incidence of mental ill-health in adults with down syndrome. J Intellect Disabil Res. 2008;52:141–155. doi:10.1111/j.1365-2788.2007.00985.x
57. Visootsak J, Sherman S. Neuropsychiatric and behavioral aspects of trisomy 21. Curr Psychiatry Rep. 2007;9(2):135–140.
58. DeJong H, Bunton P, Hare DJ. A systematic review of interventions used to treat catatonic symptoms in people with autistic spectrum disorders. J Autism Dev Disord. 2014;44:2127–2136. doi:10.1007/s10803-014-2085-y
59. Kakooza-Mwesige A, Wachtel LE, Dhossche D. Catatonia in autism: implications across the life span. Eur Child Adolesc Psychiatry. 2008;17:327–335. doi:10.1007/s00787-008-0676-x
60. Devenny D, Matthews A. Regression: atypical loss of attained functioning in children and adolescents with down syndrome. Internat Rev Res Devel Disab. 2011;41:233–264.
61. Carroll BT, Kennedy JC, Goforth HW. Catatonic signs in medical and psychiatric catatonias. CNS Spectr. 2000;5(7):66–69.
62. Fink M, Kellner CH, McCall WV. Optimizing ECT technique in treating catatonia. J Ect. 2016;32:149. doi:10.1097/YCT.0000000000000345
63. Unal A, Altindag A, Demir B, Aksoy I. The use of lorazepam and electroconvulsive therapy in the treatment of catatonia: treatment characteristics and outcomes in 60 patients. J Ect. 2017;33(4):290–293. doi:10.1097/YCT.0000000000000433
64. Ghaziuddin N, Kutcher S, Knapp P, et al. Practice parameter for use of electroconvulsive therapy with adolescents. J Am Acad Child Adolesc Psychiatry. 2004;43:1512–1539. doi:10.1097/01.chi.0000142280.87429.68
65. Leroy A, Naudet F, Vaiva G, Francis A, Thomas P, Amad A. Is electroconvulsive therapy an evidence-based treatment for catatonia? A systematic review and meta-analysis. Eur Arch Psychiatry Clin Neurosci. 2018;268(7):675–687. doi:10.1007/s00406-017-0819-5
66. Dhossche DM, Shah A, Wing L. Blueprints for the assessment, treatment and future study of catatonia in autism spectrum disorders. Int Rev Neurobiol. 2006;72:267–284. doi:10.1016/S0074-7742(05)72016-X
67. Fink M, Taylor MA, Ghaziuddin N. Catatonia in autistic spectrum disorders: a medical treatment algorithm. Int Rev Neurobiol. 2006;72:233–244. doi:10.1016/S0074-7742(05)72014-6
68. Petrides G, Divadeenam K, Bush G, Francis A. Synergism of lorazepam and electroconvulsive therapy in the treatment of catatonia. Biol Psychiatry. 1997;1:375–381. doi:10.1016/S0006-3223(96)00378-2
69. Warner G, Howlin P, Salomone E, Moss J, Charman T. Profiles of children with down syndrome who meet screening criteria for autism spectrum disorder (ASD): a comparison with children diagnosed with ASD attending specialist schools. J Intel Disabil Res. 2017;61:75–82. doi:10.1111/jir.12344
70. Capone GT, Grados MA, Kaufmann WE, Bernad-Ripoll S, Jewell A. Down syndrome and comorbid autism-spectrum disorder: characterization using the aberrant behavior checklist. Am J Med Genet A. 2005;134:373–380. doi:10.1002/ajmg.a.30622
71. Cárdenas A, Paul MA, Correa C, Valderrama S, Cerda J, Lizama M. Morbidity profile in chilean adolescents with down syndrome. Rev Med Chil. 2016;144(8):998–1005. doi:10.4067/S0034-98872016000800006
72. DiGuiseppi C, Hepburn S, Davis JM, et al. Screening for autism spectrum disorders in children with down syndrome: population prevalence and screening test characteristics. J Dev Behav Pediatr. 2010;31(3):181–191. doi:10.1097/DBP.0b013e3181d5aa6d
73. Warner G, Moss J, Smith P, Howlin P. Autism characteristics and behavioral disturbances in ~500 children with down’s syndrome in England and wales. Autism Res. 2014;7(4):433–441. doi:10.1002/aur.1371
74. Ji NY, Capone GT, Kaufmann WE. Autism spectrum disorder in down syndrome: cluster analysis of aberrant behaviour checklist data supports diagnosis. J Intellect Disabil Res. 2011;55:1064–1077. doi:10.1111/j.1365-2788.2011.01465.x
75. Moss J, Richards C, Nelson L, Oliver C. Prevalence of autism spectrum disorder symptomatology and related behavioural characteristics in individuals with down syndrome. Autism. 2013;17(4):390–404. doi:10.1177/1362361312442790
76. Molloy CA, Murray DS, Kinsman A, et al. Differences in the clinical presentation of Trisomy 21 with and without autism. Intellect Disabil Res. 2009;53(2):143–151. doi:10.1111/jir.2009.53.issue-2
77. Carter JC, Capone GT, Gray RM, Cox CS, Kaufmann WE. Autistic-spectrum disorders in down syndrome: further delineation and distinction from other behavioral abnormalities. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(1):87–94. doi:10.1002/ajmg.b.30407
78. Chapman RS, Seung HK, Schwartz S, Kay-Raining Bird E. Language skills of children and adolescents with down syndrome: II. production deficits. J Speech Lang Hear Res. 1998;41:861–873. doi:10.1044/jslhr.4104.861
79. Roberts JE, Price P, Malkin C. Language and communication development in down syndrome. Ment Retard Dev Disabil Res Rev. 2007;13(1):26–35. doi:10.1002/mrdd.20136
80. Capone GT, Chicoine B, Bulova P, et al. Co-occurring medical conditions in adults with down syndrome: a systematic review toward the development of health care guidelines. Am J Med Genet Part A. 2018;176A:116–133. doi:10.1002/ajmg.a.38512
81. Bull MJ. For American academy of pediatrics committee on genetics. Health supervision for children with down syndrome. Pediatrics. 2011;128(2):393–406. doi:10.1542/peds.2010-3664
82. Kusters MA, Verstegen RH, Gemen EF, de Vries E. Intrinsic defect of the immune system in children with down syndrome: a review. Clin Exp Immunol. 2009;156(2):189–193. doi:10.1111/j.1365-2249.2009.03890.x
83. Fidler DJ, Most DE, Guiberson MM. Neuropsychological correlates of word identification in down syndrome. Res Devel Disabil. 2005;26:487–501. doi:10.1016/j.ridd.2004.11.007
84. Devenny DA, Krinsky-McHale SJ, Sersen G, Silverman WP. Sequence of cognitive decline in dementia in adults with down’s syndrome. J Intellect Disabil Res. 2000;44:654–655.
85. Pueschel SM, Romano C, Failla P, et al. A prevalence study of celiac disease in persons with down syndrome residing in the United States of America. Acta Paediatr. 1999;88(9):953–956.
86. Book L, Hart A, Black J, Feolo M, Zone JJ, Neuhausen SL. Prevalence and clinical characteristics of celiac disease in downs syndrome in a U.S. study. Am J Med Gen. 2001;98:70–74. doi:10.1002/1096-8628(20010101)98:1<70::AID-AJMG1002>3.0.CO;2-G
87. Daneshpazhoo M, Nazemi TM, Bigdeloo L, Yoosefi M. Mucocutaneous findings in 100 children with down syndrome. Pediatr Dermatol. 2007;24:317–320. doi:10.1111/j.1525-1470.2007.00412.x
88. Pierce MJ, LaFranchi SH, Pinter JD. Characterization of thyroid abnormalities in a large cohort of children with down syndrome. Horm Res Paediatr. 2017;87(3):170–178. doi:10.1159/000457952
89. Du Y, Shan LF, Cao ZZ, Feng JC, Cheng Z. Prevalence of celiac disease in patients with down syndrome: a meta-analysis. Oncotarget. 2018;9(4):5387–5396. doi:10.18632/oncotarget.23624
90. Whooten R, Schmitt J, Schwartz A. Endocrine manifestations of down syndrome. Curr Opin Endocrinol Diabetes Obes. 2018;25:61–66.
91. Ferrafiat V, Raffin M, Freri B, et al. A causality algorithm to guide diagnosis and treatment of catatonia due to autoimmune conditions in children and adolescents. Schizophr Res. 2018;200:68–76. doi:10.1016/j.schres.2017.06.036
92. Lahutte B, Cornic F, Bonnot O, et al. Multidisciplinary approach of organic catatonia in children and adolescents may improve treatment decision making. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(6):1393–1398. doi:10.1016/j.pnpbp.2008.02.015
93. Denysenko L, Sica N, Penders TM, et al. Catatonia in the medically ill: etiology, diagnosis, and treatment. The academy of consultation-liaison psychiatry evidence-based medicine subcommittee monograph. Ann Clin Psychiatry. 2018;30(2):140–155.
94. Kovaleva NV. Sex ratio in down syndrome. Tsitol Genet. 2002;36(6):54–69.
95. Morris JK, Alberman E, Mutton D, Jacobs P. Cytogenetic and epidemiological findings in down syndrome: England and wales 1989–2009. Am J Med Genet A. 2012;158A:1151–1157.
96. Zhao W, Chen F, Wu M, et al. Postnatal identification of Trisomy 21: an overview of 7,133 postnatal Trisomy 21 cases identified in a diagnostic reference laboratory in China. PLoS One. 2015;10(7):1–13.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.