")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
Caspase-3 Promotes Diabetic Kidney Disease Through Gasdermin E-Mediated Progression to Secondary Necrosis During Apoptosis
Authors Wen S , Wang ZH, Zhang CX, Yang Y, Fan QL
Received 12 December 2019
Accepted for publication 16 January 2020
Published 10 February 2020 Volume 2020:13 Pages 313—323
DOI https://doi.org/10.2147/DMSO.S242136
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Konstantinos Tziomalos
Si Wen,1,* Zhao-Hua Wang,2,* Cong-Xiao Zhang,1 Ying Yang,1 Qiu-Ling Fan1
1Department of Nephrology, First Hospital of China Medical University, Shenyang, People’s Republic of China; 2Affiliated Dalian Friendship Hospital of Dalian Medical University, Dalian, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qiu-Ling Fan
Department of Nephrology, First Hospital of China Medical University, No. 155 Nanjing Bei Street, Shenyang 110001, People’s Republic of China
Tel +86 13904012680
Email [email protected]
Background: Apoptosis has been repeatedly linked with diabetic kidney disease (DKD), which is a programmed cell death mediated by effector caspases-3, 6 and 7, targeting > 600 substrates. However, the pathophysiologic correlations of this process remain obscure. As a putative tumor suppressor, gasdermin E (GSDME) was recently reported to be cleaved by caspase-3 to produce a GSDME-N fragment which targets the plasma membrane to switch apoptosis to secondary necrosis. However, it remains elusive whether GSDME is involved in the regulation of DKD.
Methods: To evaluate the therapeutic potential of caspase-3 inhibition in DKD, we administered caspase-3 inhibitor Z-DEVD-FMK to STZ-induced diabetic mice for eight weeks. Albuminuria, renal function, pathological changes, and indicators of secondary necrosis and fibrosis were evaluated. In vitro, human tubule epithelial cells (HK-2 cells) were subjected to high-glucose treatment. Secondary necrosis was determined by LDH release, GSDME cleavage, and morphological feature under confocal microscopy. Z-DEVD-FMK and GSDME inhibition by shRNA were administered to suppress the cleavage and expression of GSDME. Flow cytometry, cytotoxicity assay and immunoblot were used to assess cell death and fibrogenesis.
Results: Caspase-3 inhibition by Z-DEVD-FMK ameliorated albuminuria, renal function, and tubulointerstitial fibrosis in diabetic mice. The nephroprotection mediated by Z-DEVD-FMK was potentially associated with inhibition of GSDME. In vitro, molecular and morphological features of secondary necrosis were observed in glucose-stressed HK-2 cells, evidenced by active GSDME cleavage, ballooning of the cell membrane, and release of cellular contents. Here we showed that caspase-3 inhibition prevented GSDME activation and cell death in glucose-treated tubular cells. Specifically, knocking down GSDME directly inhibited secondary necrosis and fibrogenesis.
Conclusion: These data suggest GSDME-dependent secondary necrosis plays a crucial role in renal injury, and provides a new insight into the pathogenesis of DKD and a promising target for its treatment.
Keywords: GSDME, DFNA5, secondary necrosis, diabetic kidney disease, Z-DEVD-FMK
Introduction
Gasdermin E (GSDME), originally called DFNA5, is a deafness gene, and has been implicated as a putative tumour suppressor.1,2 Recently, Wang et al showed that GSDME belongs to the same gasdermin family as GSDMD, and could switch caspase-3-dependent non-inflammatory and immunologically silent apoptosis to a terminal phase, namely secondary necrosis.3 Both mouse and human GSDME possess caspase-3 recognition site at residues 267–270.4 Cleavage of GSDME by caspase-3 at the site of Asp270 releases the inhibitory GSDME-C domain allowing generation of the necrotic GSDME-N domain. The intrinsic ability of GSDME-N is to translocate and form oligomers in the plasma membrane to permeabilize it and trigger secondary necrosis. Consequently, apoptotic cells progressively lose their integrity, resulting in membrane damage, cytoplasmic swelling, and release of cytoplasmic contents, similar to the features of pyroptotic cells.4–6 These observations suggest that secondary necrosis is not accidental during apoptosis, but a controllable process with remarkable therapeutic and pathophysiological significance.7 Researchers have found that pyroptosis had an indispensable role in renal ischemia-reperfusion injury and contrast-induced acute kidney injury.8,9 Whether pyroptotic-like secondary necrosis takes a role in kidney diseases remains unknown.
Markers of apoptosis have repeatedly been linked with diabetic kidney disease (DKD).10 However, specific targeting of caspase-3 as a therapeutic strategy remains controversial, since unequivocal evidence for a causative role of this process is sparse and the pathophysiologic relevance remains obscure.11 Shahzad et al showed that neither CIX (inhibiting caspase-3, −6, −7, −8, and −10) nor caspase-3 deficiency protected mice from diabetic nephropathy.11 To address this question, we employed a selective caspase-3 inhibitor, Z-DEVD-FMK, and found that treatment of diabetic mice with Z-DEVD-FMK ameliorated albuminuria, renal function and tubulointerstitial fibrosis. Here, we found that the renoprotective effect of caspase-3 inhibition is possibly mediated via regulating the activation of GSDME. However, to the best of our knowledge, the involvement of GSDME has not been reported in DKD. The present study reveals the underlying mechanism mediating renoprotection conferred by GSDME regulation in the kidney, specifically in human tubular cells, and provides novel insights into the pathogenesis of DKD.
Materials and Methods
Animal Models
Male CD1 (ICR) mice were obtained from Charles River Laboratories. Eight-week-old male CD1 mice were employed and divided into three groups (n=10): control (Ctrl), diabetic mice (DM), diabetic mice with Z-DEVD-FMK treatment (DM+DEVD). Streptozocin (STZ) (S0130, Sigma Aldrich, US) was administered via intraperitoneal injection to induce diabetes at a dose of 55mg/kg for five consecutive days.12–14 Mice with blood glucose level above 300mg/dl 14-16d after STZ administration were considered diabetic.13,15 Diabetic mice were then treated with Z-DEVD-FMK (A1920, ApexBio, US) or DMSO intraperitoneally for eight weeks. Our work was performed following the NIH Guide for the Care and Use of Laboratory Animals and approved by the Laboratory Animal Welfare and Ethics Committee of China Medical University.
Blood and Urine Examination
Blood samples were collected for subsequent examination after 8 weeks of treatment. Samples were used to detect serum creatinine, blood urea nitrogen and serum albumin. Urine samples were obtained every 2–3 weeks and were used to detect urinary creatinine and albumin using the Mouse Albumin ELISA Kit (ab108791, Abcam, UK) following the manufacturer’s protocol.
Histology and Immunohistochemical Staining
Renal tissues from mice underwent fixation, dehydration, embedding and section, followed by Masson trichrome and Hematoxylin-eosin staining. For immunohistochemical staining, paraffin-embedded sections were prepared and subjected to microwave retrieval in citrate buffer, followed by quenching in 3% hydrogen peroxide, and blocking in 3% bovine serum albumin. The sections were incubated with anti-GSDME and anti-collagen type I antibodies overnight, then stained with a peroxidase-conjugated secondary antibody. Slides were developed using DAB substrate and imaged using DMI4000B Leica Microsystems.
Caspase-3 Activation Assay
Caspase-3 activity in renal cortex lysates was measured using the Caspase-Glo® 3/7 Assay Kit (G8090, Promega, US) and a modified protocol.16,17 Briefly, renal tissues were homogenized in RIPA buffer, and the protein concentration was adjusted to 2 mg/mL. Thereafter, protein extracts (100μg/50μL) were added to a white 96-well plate and mixed with 50μL caspase 3/7 Glo Reagent. After incubation for 1 hr in the dark, the luminescence was measured using a Cytation™ 5 Cell Imaging Multi-Mode Reader (BioTek).
Cell Culture and Treatments
We cultured a human renal proximal tubule epithelial cell line (HK-2 cells; ATCC®CRL-2190™) in DMEM/F12 supplemented with 100U/mL penicillin and streptomycin and 10% FBS (#1009-1148, Gibco) at 37°C in a 5% CO2 environment. We prepared DMEM/F12 medium with DMEM (#11885-084, Gibco), containing 1g/l glucose, and F12 (#11765-054, Gibco) at a 1:1 ratio. The final DMEM/F12 medium contains 5.5mM glucose. HK-2 cells were subjected to glucose (30mM), and treated with 25μM or 50μM Z-DEVD-FMK. Cells and supernatants were collected at indicated times for subsequent experimentation.
Knock-down of GSDME was performed by transfecting HK-2 cells with plasmid vectors encoding shRNA to target GSDME (Sequence1: GATGATGGAGTATCTGATCTTTTCAAGAGAAAGATCAGATACTCCATCATCTTTTTT; Sequence2: GCATGATGAATGACCTGACTTTTCAAGAGAAAGTCAGGTCATTCATCATGCTTTTTT), or a control vector encoding a non-target shRNA sequence utilizing jetPRIME® transfection reagent according to the manufacturer’s instructions (#114-15, PolyPlus). Transfected cells were then treated with glucose (30mM) for 48 hrs. In some experiments HK-2 cells were stimulated with 25μM etoposide at the same time after transfection.
Western Blotting
Cell and renal cortex lysate were obtained using RIPA buffer containing a protease inhibitor cocktail (Roche). Equal amounts (30μg) of protein samples were separated using SDS–polyacrylamide gel electrophoresis (SDS–PAGE), followed by transferred onto Immobilon®-P transfer membranes (Millipore). Membranes were then probed with anti-GSDMD (NBP2-33422, Novus, 1:1000), anti-fibronectin (NBP1-91258, Novus, 1:1000), anti-GSDME (13075-1-AP, Proteintech, 1:1000), anti-PARP (13371-1-AP, Proteintech, 1:1000), anti-collagen type I (ab34710, Abcam, 1:1000) and anti-β-actin (#4970, Cell Signaling Technology, 1:5000). After incubation with secondary antibodies, the blots were visualized with Immobilon Western Chemiluminescent HRP Substrate (Millipore). Images were captured with the ChemiDoc™ Touch Imaging System (Bio-Rad).
Real-Time qPCR Analysis
Total RNA was extracted with Trizol (Invitrogen). The GoScript™ Reverse Transcription System (A5001, Promega) was used for reverse transcribing 1–2μg RNA to generate template cDNA. Fluorogenic quantitative PCR was performed in triplicate utilizing the GoTaq® qPCR and RT-qPCR Systems (A6001, Promega) on the CFX Connect™ Optics Module (Bio-Rad) according to the manufacturer’s protocol. Specific primers were used for GSDME mRNA detection: forward 5′- TTTGCATTCATAGACATGCCAG-3′ and reverse 5′- GTTCATCATCAAATAGGACCGC-3′. Relative mRNA levels were obtained after normalization with internal reference gene, β-actin.
Imaging of Cell Death by Microscopy
HK-2 cells treated with 25μM etoposide for 24 or 48 hrs were imaged using the DMI3000B Leica Microsystems to observe cell death morphology. The images displayed is representative of four randomly selected fields.
Confocal Microscopy
HK-2 Cells were seeded on 35mm glass-bottom confocal culture dishes and allowed to attach overnight, followed by high-glucose treatment for 48 hrs. For confocal microscopy assaying, cells were probed with FAM-DEVD-FMK, propidium iodide (PI) and Hoechst following the manufacturer’s instructions (#94, ImmunoChemistry Technologies, US). Briefly, cells were incubated with FAM-DEVD-FMK, which targets active caspase-3, for 60 mins, followed by washing with wash buffer. Cells were subsequently incubated with PI and Hoechst for 10 mins. The samples were imaged live using Nikon C2 Confocal Microscope System.
Flow Cytometry Analysis of Secondary Necrosis
Following the indicated treatments, HK-2 cells were incubated with FAM-DEVD-FMK and PI for analysis of secondary necrosis using flow cytometry. The percentage of secondary necrotic cells was analyzed using the BD Accuri™ C6 Plus (BD Biosciences). Approximately 2000–5000 cells were analyzed per experimental condition.
LDH Release Assay
Cell culture supernatants were collected after various treatments. The activity of released LDH was detected to evaluate cell death with the CytoTox96 LDH release kit (G1780, Promega) following the manufacturer’s instructions. The LDH activity in the supernatants was expressed as a percentage of total LDH in the cell lysate.
Data Analysis
The Student’s t-test was used for two-group comparisons. Multiple-group comparisons were conducted using ANOVA and Bonferroni t-tests. Statistical analyses for nonparametric test were performed using the Kruskal–Wallis test and Mann–Whitney test. P-values <0.05 were considered statistically significant
Results
Caspase-3 Inhibition Ameliorates Diabetic Nephropathy in Mice
To evaluate the therapeutic potential of caspase-3 inhibition in DKD, we employed Z-DEVD-FMK, which specifically targets caspase-3. At the outset of this study, diabetic mice were administered with 1mg, 5mg, 10mg or 20mg/kg Z-DEVD-FMK for three consecutive days, respectively. Sufficient inhibition of casapase-3 activity by Z-DEVD-FMK was observed at the dose of 20mg/kg (Figure 1A).
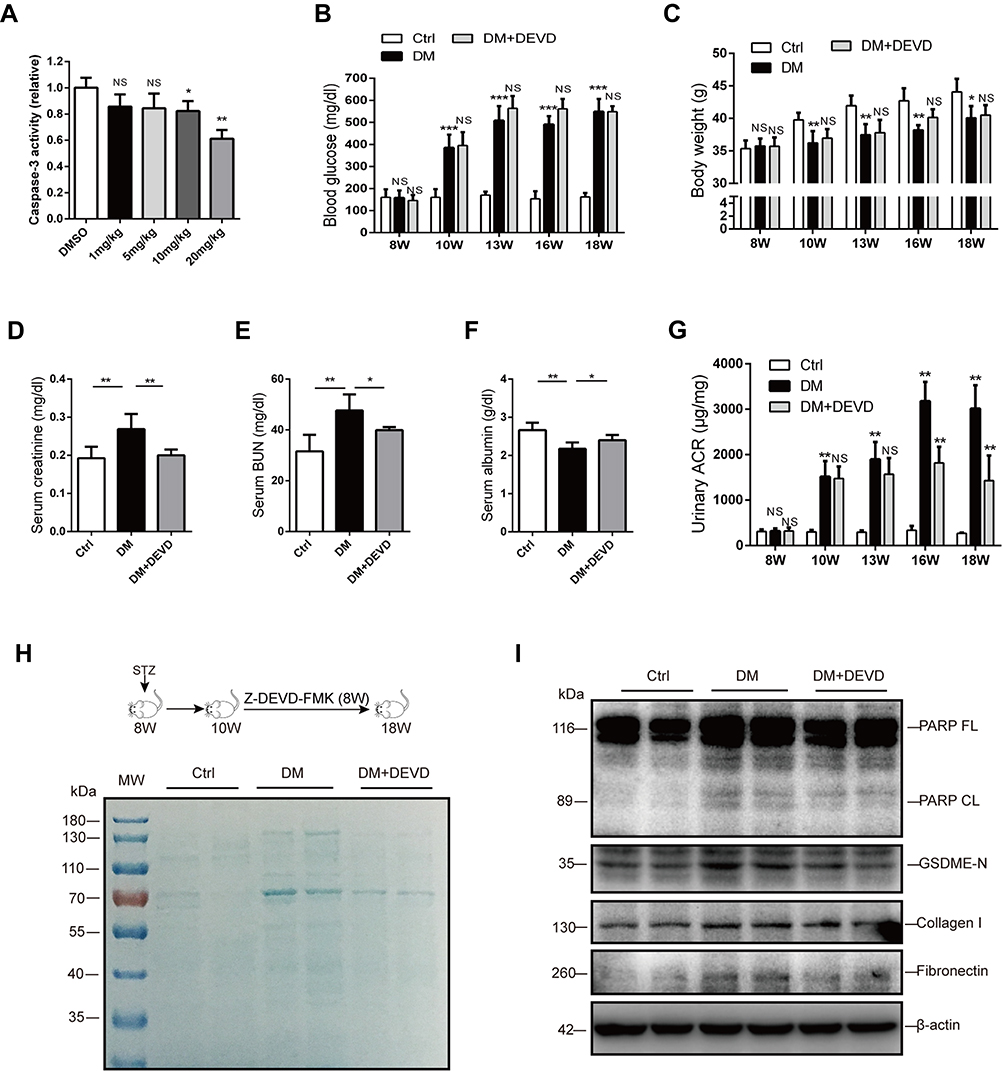 |
Figure 1 Caspase-3 inhibition ameliorates albuminuria and renal function in diabetic mice. (A) 20 mg/kg Z-DEVD-FMK effectively inhibits caspase-3 activity in the renal cortex of diabetic mice. Body weight (C) and blood glucose level (B) are not affected by Z-DEVD-FMK treatment in diabetic mice. Serum creatinine (D), blood urea nitrogen (E), and serum albumin (F) levels of mice. (G) Albuminuria is significantly alleviated after 6–8 weeks of Z-DEVD-FMK treatment in diabetic mice. (H) Urine samples from mice were separated using SDS-PAGE and stained with Coomassie blue. In general, albumin is around 70 kDa. (I) Immunoblotting reveals Z-DEVD-FMK treatment prevents elevation of cleaved PARP, GSDME-N, collagen I and fibronectin expression in diabetic mice. Data are represented as means ± SEM. n=5–7. *p < 0.05, **p < 0.01, ***p < 0.001. Abbreviations: Ctrl, control; DM, diabetic mice; DEVD, Z-DEVD-FMK; ACR, albumin-to-creatinine ratio; BUN, blood urea nitrogen; NS, not significant; W, weeks of age. |
Eight-week-old male CD1 mice were administered with 55mg/kg STZ for 5 days to induce diabetes. Thus, treatment with 20mg/kg Z-DEVD-FMK was initiated two weeks after STZ administration and mice were analyzed after eight weeks of treatment. Mice administrated with STZ developed prominent hyperglycemia within 14 days. Body weight and blood glucose level did not differ between diabetic mice treated with Z-DEVD-FMK and DMSO (Figure 1B and C), indicating that the treatment effects of Z-DEVD-FMK are independent of these metabolic factors. Z-DEVD-FMK improved renal function, and blunted the decreased serum albumin level in diabetic mice (Figure 1D–F). Notably, albuminuria was ameliorated after 6 weeks of treatment (Figure 1G), implying that caspase-3 activation may exert an effect at a later stage of DKD. We also analyzed urinary protein excretion using Coomassie brilliant blue staining of the urine samples, and found that there is more urinary protein in diabetic mice. After Z-DEVD-FMK-treatment, the excretion was decreased in diabetic mice (Figure 1H). Pathological analysis revealed that prominent glomerular mesangial expansion, tubular atrophy and tubulointerstitial fibrosis were observed in diabetic mice compared with nondiabetic mice. Tubular injury and interstitial fibrosis were amendable to Z-DEVD-FMK treatment, as evaluated by tubulointerstitial injury score and semi-quantifying Masson trichrome staining (Figure 2A, C and D).
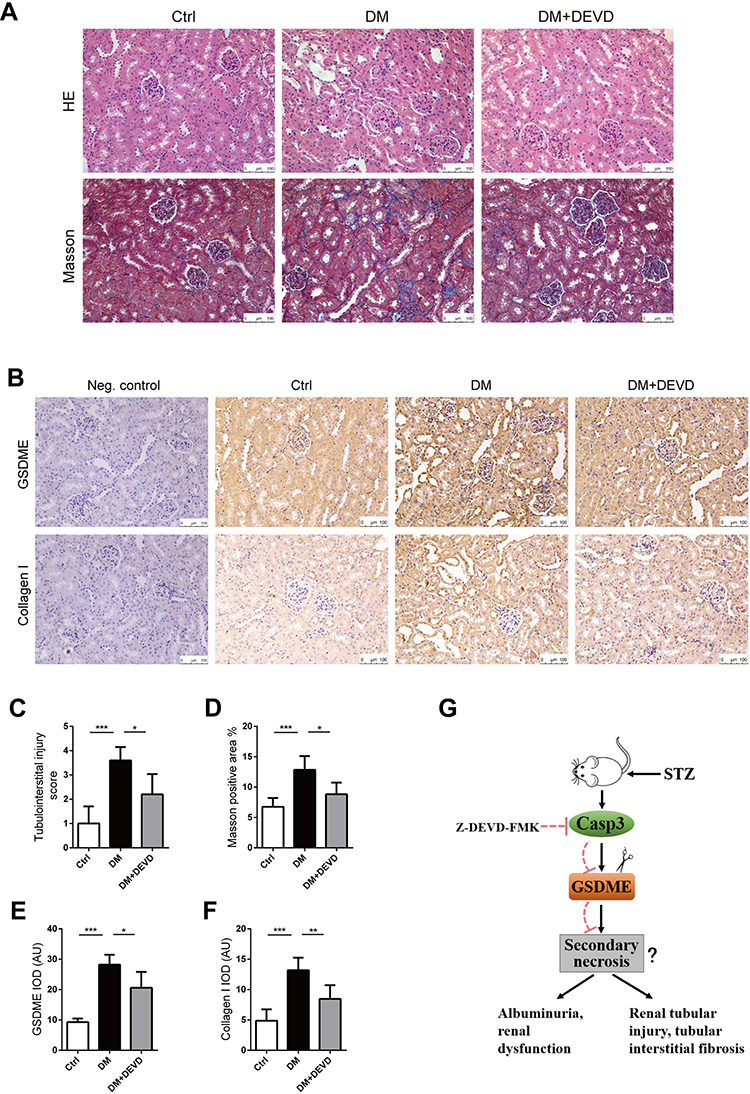 |
Figure 2 Caspase-3 inhibition protects against renal injury in diabetic mice. Kidney slides stained with hematoxylin-eosin solution to assess glomerular mesangial expansion and tubular injury (A), and semi-quantify tubulointerstitial injury (C). Slides underwent Masson trichrome staining for the evaluation of tubulointerstitial fibrosis (A). Graph summarizing results of determining the area of blue staining (D). (B) Representative images of immunohistochemistry stained with anti-GSDME and anti-collagen I antibodies. (Negative control included, using a nonspecific primary antibody; Neg. control). Graphs summarizing data of integrated optical density (IOD) of positive GSDME (E) and collagen I (F) staining. (G) Schematic representation showing that caspase-3 inhibition by Z-DEVD-FMK plays an important role in protecting mice from DN possibly via regulating GSDME cleavage. Scale bar=100 μm. Data are represented as means ± SEM. n=5–7. *p < 0.05, **p < 0.01, ***p < 0.001. Abbreviations: Ctrl, control; DM, diabetic mice; DEVD, Z-DEVD-FMK; AU, arbitrary units. |
As the substrate of active caspase-3, PARP was upregulated in diabetic mice, and the upregulated PARP cleavage was inhibited after Z-DEVD-FMK treatment, which verified the effective block of caspase-3 activity (Figure 1I). Markers for fibrosis, collagen I and fibronectin, both decreased after treatment in diabetic mice (Figure 1I), which was further confirmed by immunohistochemistry staining of anti-collagen I. The expression of collagen I in diabetic mice was increased as compared to nondiabetic controls, which decreased after Z-DEVD-FMK treatment, especially at the tubular level (Figure 2B and F). We observed that aberrant activation of GSDME in diabetic mice was inhibited after Z-DEVD-FMK treatment (Figure 1I). Immunohistochemistry revealed that GSDME is highly expressed at the tubular level in diabetic mice (Figure 2B and E). Thus, the nephroprotective effects of caspase-3 inhibition may be related to the inhibition of GSDME.
Secondary Necrosis of Apoptotic HK-2 Cells in High Glucose Conditions
We next conducted a closer analysis of cell death in vitro. Cell death was evaluated by the release of LDH, a marker of lytic cell death. In addition to the initial LDH release at 3 hrs, we noted that there was also marked LDH release during later stages (24 to 48 hrs) in glucose-stressed HK-2 cells (Figure 3A). Previous literature has reported that glucose-induced caspase-1 activation precedes caspase-3 in podocytes.11 However, comparison of kinetic processes of GSDMD and GSDME activation are lacking. Here, our results demonstrated that GSDME was initially activated at 24 hrs, which occurred far later than the activation of GSDMD (3 hrs), the executor of pyroptosis. (Figure 3B and C). Previous studies by us and others have indicated that pyroptosis occurs at an early stage of DKD. Our present data suggest that the LDH release at a later stage correlates with GSDME activation. Taken together, these observations indicate that the induction of GSDME may play an important role in promoting cell death during later stages in diabetic tubular cells.
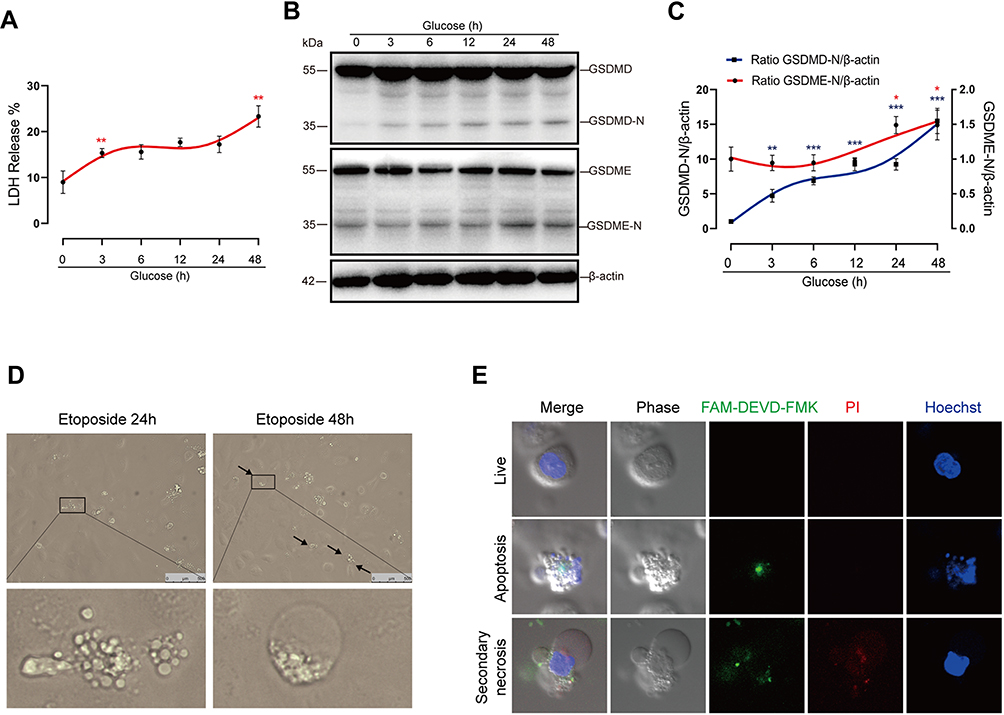 |
Figure 3 High-glucose treatment induces secondary necrosis in HK-2 cells. (A) LDH release of HK-2 cells treated with normal glucose (5.5 mM) for 48 hrs or high glucose (30 mM) for 3, 6, 12, 24 or 48 hrs, respectively. (B) Immunoblots of GSDME and GSDMD in cells treated with glucose for the indicated times at a concentration of 30 mM. Densitometry of the data in C. (D) Microscopic images of HK-2 cells treated with 25 μM etoposide for 24 or 48 hrs. Arrows indicate ballooned cell membrane, characteristic of secondary necrosis. The insets show higher magnification of areas indicated by black lines. (E) Confocal images of high-glucose treated cells stained with FAM-DEVD-FMK (green), PI (red), and Hoechst (blue). The upper panel shows normal cells (double negative cells), the middle panel shows apoptotic cells (PI− and FAM-DEVD-FMK + cells), and the bottom shows secondary necrotic cells (double positive cells). Results are representative of at least three independent experiments. Data from at least three independent experiments are shown as the means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. |
We imaged cells treated with apoptotic inducer etoposide, and found that cells mainly exhibited membrane blebbing (apoptotic body) in 24 hrs. After treatment with etoposide for 48 hrs, apoptotic cells progressively exhibited pyroptotic-like morphological changes, namely ballooning of the cell membrane (Figure 3D). We next performed a closer observation of cell morphologic features after glucose treatment for 48 hrs using confocal microscopy. HK-2 cells were stained with PI and FAM-DEVD-FMK, which specifically targets active caspase-3. Because secondary necrosis is a lytic form of cell death mediated by active caspase-3, secondary necrotic cells were defined as double positive for PI and FAM-DEVD-FMK. We observed that secondary necrotic cells (FAM-DEVD-FMK+ and PI+ cells) exhibited characteristic ballooning of the cell membrane (Figure 3E), confirming the occurrence of secondary necrosis in high-glucose treated tubular cells.
Caspase-3 Inhibition Prevented GSDME Activation and Secondary Necrosis
We next wanted to determine whether caspase-3 inhibition prevented secondary necrosis in vitro. To address this question, HK-2 cells were pretreated with Z-DEVD-FMK for 30 mins before high-glucose exposure. We utilized active caspase-3 probe, FAM-DEVD-FMK, to detect caspase-3 activity and found that upregulated caspase-3 activity in glucose-stressed HK-2 cells was inhibited by Z-DEVD-FMK treatment, with a more effective inhibition at the concentration of 50μM (Figure 4A and B).
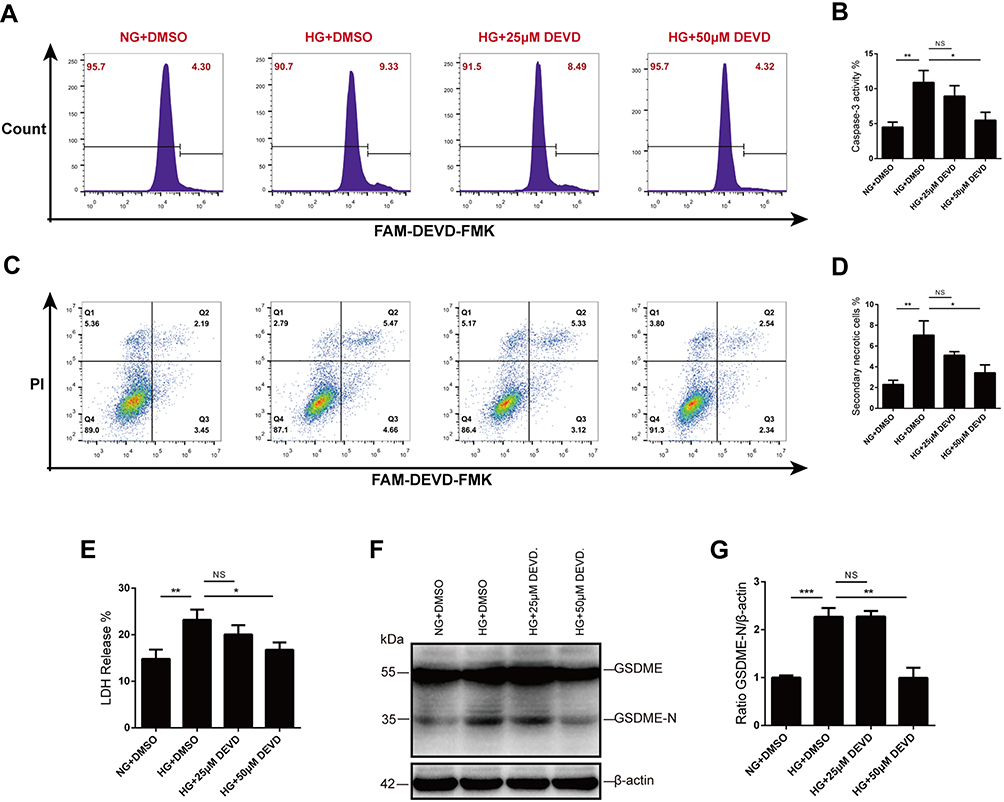 |
Figure 4 Caspase-3 blocker inhibits GSDME activation and secondary necrosis. (A and B) Caspase-3 activity was assessed by flow cytometry after FAM-DEVD-FMK staining. Compared with 25μM Z-DEVD-FMK, 50μM Z-DEVD-FMK is more effective in inhibiting caspase-3 activity. (C and D) Treatment with Z-DEVD-FMK before and during high-glucose stimulation reduced secondary necrosis in HK-2 cells, especially at the concentration of 50μM. (E) Lytic cell death was evaluated by assaying LDH release. Treatment with 50μM Z-DEVD-FMK significantly prevented glucose-induced LDH release of HK-2 cells. (F and G) Glucose stimulation promotes GSDME cleavage in tubular cells, while caspase-3 blocker inhibits the activation of GSDME. Densitometry of the data in G. Results are representative of at least three independent experiments. Data from at least three independent experiments are shown as the means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. Abbreviations: NG, normal glucose; HG, high glucose; DEVD, Z-DEVD-FMK; NS, not significant. |
As secondary necrotic cells were defined as double positive for PI and FAM-DEVD-FMK, we employed flow cytometry to quantitative analysis the rate of secondary necrotic cells. Glucose induced an increased population of secondary necrosis (FAM-DEVD-FMK+ and PI+ cells), and treatment with caspase-3 inhibitor reduced secondary necrosis in HK-2 cells, especially at the concentration of 50μM (Figure 4C and D). We also evaluated cell death by assaying the activity of LDH released into cell culture supernatants. Consistent with the flow cytometry analysis, LDH release was prevented by Z-DEVD-FMK treatment (Figure 4E). Moreover, the caspase-3 inhibitor prevented cleavage and activation of GSDME of tubular cells in high glucose (Figure 4F and G).
Knocking Down GSDME Inhibited Secondary Necrosis and Fibrosis
To further confirm the role of GSDME in the progression to secondary necrosis, we knocked down GSDME using plasmid vectors encoding shRNA targeting GSDME. Immunoblot and qPCR were performed to verify the effective silence, and the shRNA sequence 1 is more efficient than sequence 2 (Figure 5A-C). As expected, secondary necrosis in response to glucose stimulation was alleviated by GSDME knock-down, as analyzed by flow cytometry (Figure 5D and E). Cell death was also evaluated by the release of LDH. Increased LDH release in glucose-stressed HK-2 cells was markedly blunted by GSDME knock-down (Figure 5F). In addition, high-glucose treatment increased the expression of fibrosis indicators, collagen I and fibronectin, in HK-2 cells, while GSDME knock-down strikingly reduced collagen I and fibronectin formation (Figure 5G–I).
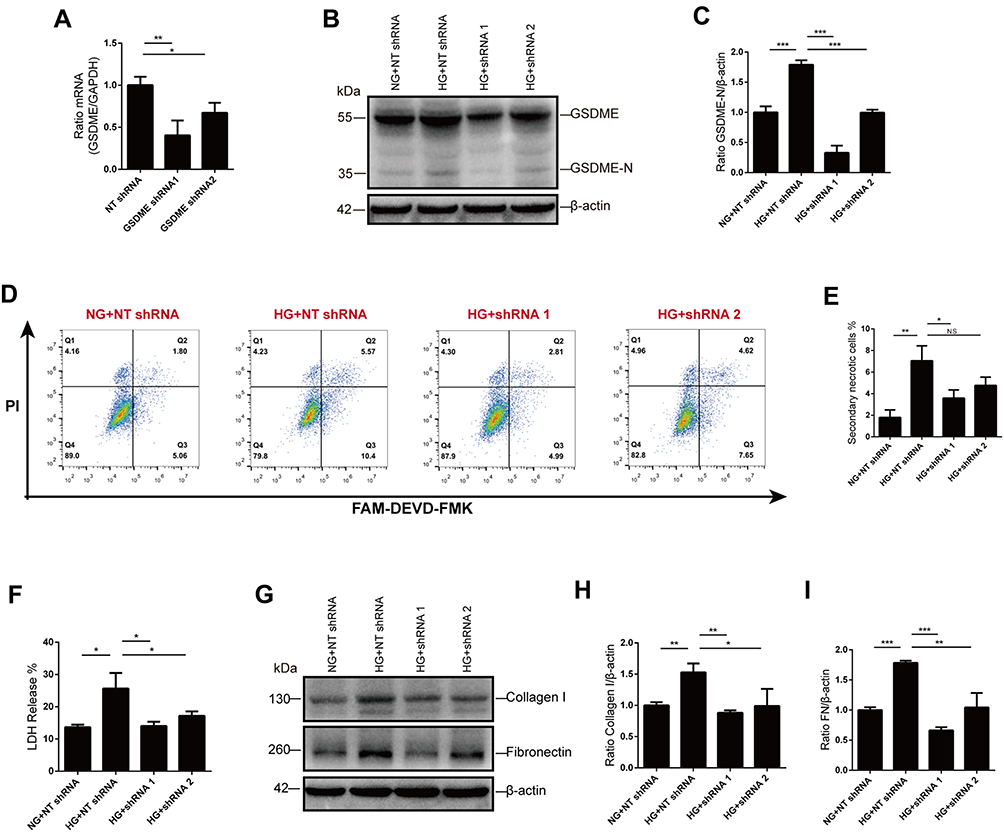 |
Figure 5 GSDME mediates secondary necrosis and fibrosis. (A–C) GSDME knock-down was verified using RT-qPCR and Western blot. (D and E) The percentage of cells dead by secondary necrosis decreases in GSDME shRNA transfected cells, as analyzed by flow cytometry. (F) Cell death is alleviated by GSDME knock-down as assessed by LDH release. (G–I) Immunoblotting reveals elevation of collagen I and fibronectin expression are inhibited in cells transfected with GSDME shRNA. Densitometry of the data in H and I. Results are representative of at least three independent experiments. Data from at least three independent experiments are shown as the means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. Abbreviations: NG, normal glucose; HG, high glucose; NT shRNA, non-target shRNA; NS, not significant. |
Taken together, these data contribute to our understanding of renoprotection conferred by caspase-3 inhibition in response to diabetes, and provide evidence that GSDME-mediated secondary necrosis of HK-2 cells contributes to the manifestation of renal injury in diabetic mice.
Discussion
Currently, strict glucose and blood pressure control, including reducing albuminuria via blocking the renin-angiotensin system, are still the primary treatments for diabetic kidney disease. Thus, much effort has been devoted to revealing the underlying pathogenesis of renal injury in DKD aiming at developing novel treatment targets and therapeutic approaches.18 Chronic sterile inflammation has been proved to be the crucial basis for the progression of DKD.19–21 Our previous study also demonstrated the participation of pyroptosis in diabetic tubular cells.22 However, whether apoptosis could be an effective treatment target remained controversial hitherto. In this study, we employed Z-DEVD-FMK for the treatment of diabetic mice, and our data provide experimental evidence that caspase-3-mediated cell death promotes renal injury and fibrosis in DKD.
However, previous study by Shahzad et al drew different conclusions.11 One of the possible reasons is that Shahzad and collaborators focused their attention on changes in glomeruli rather than tubulointerstitial histopathology. Previously, researches about diabetic kidney disease was once mainly focused on the glomerulus, which is doubtlessly of great importance; however, glomerular lesions are not the primary determinant of the progression of DKD.23 In DKD, tubule injury and interstitial fibrosis correlate more closely with renal dysfunction than glomerular changes.24 The dependent on aerobic metabolism and high energy demand of tubular cells render it particularly susceptible to disorders under diabetic conditions. Here, we showed that Z-DEVD-FMK treatment ameliorated renal function and tubulointerstitial fibrosis. We also found that targeting caspase-3 therapeutically might exert renoprotective effects by inhibiting the cleavage of GSDME. In addition, immunohistochemistry indicated that GSDME is mainly expressed in tubules. Hence, we utilized HK-2 cells for experiments in vitro.
As a non-inflammatory form of programmed cell death, apoptosis is mediated by activation of the intrinsic or extrinsic pathway.25 Both pathways induce apoptosis by activating initiator caspases and converging on the executioner caspases. Effector caspase-3, 6, and 7 cleave hundreds of different cellular substrates, and account for multiple biochemical and morphological changes of apoptosis, including blebbing, cellular shrinkage and chromosomal DNA fragmentation.26,27
For a long time, secondary necrosis has been considered as a final phase of apoptotic cells, especially in vitro or in vivo when the dying cells are not quickly cleared by phagocytes.28 Cellular swelling (ballooning) and membrane rupture are the main characteristics of secondary necrosis, which are also morphological characteristics of necrosis and pyroptosis.29 Like necrosis and pyroptosis, the concomitant release of cytosolic contents of secondary necrosis may drive persistent immune responses and lead to tissue damage. Therefore, identifying its regulatory mechanism may be of important significance. Secondary necrosis was, however, conceived as a non-programmed and nonspecific form of cell death, and the molecular mechanism regulating its onset remained unknown. Until the revealing of GSDME as an executor of secondary necrosis recently,3,4 secondary necrosis is no longer accidental, but a controllable process that stands out as a potential therapeutic target. GSDME has been proposed as a tumor suppressor, and is now believed to be a strategy to boost the immunogenicity of cancer cell death.30,31 Our results demonstrated that GSDME knock-down prevented secondary necrosis and fibrogenesis in diabetic tubular cells. Our work brings about an advance in identifying GSDME as a promising target for the treatment of DKD. Future studies should elucidate whether knock-down or knock-out of GSDME exerts benefits in diabetic animals, and to design novel drugs targeting this pathway.
As Z-DEVD-FMK did not affect glucose levels and body weight in the STZ-induced diabetic mice. The protective effects of Z-DEVD-FMK on DKD were independent of improvements in metabolic anomalies. Previously, it has been reported that caspase-1 or NLRP3 (NOD-like receptor pyrin domain containing protein 3) deficiency attenuated albuminuria and glomerular extracellular matrix accumulation without affecting the glucose levels and body weight in diabetic mice.15 Birnbaum et al reported Saxagliptin ameliorated diabetes-induced inflammasome activation and DKD (at least partial) independent of glucose lowering.32 Indeed, metabolic disorder is the initial cause of DKD. In addition, hemodynamic abnormalities, oxidative stress, and inflammation are also involved in the progression of DKD. Our results indicate that caspase-3 inhibitor protects mice from DKD via the regulation of lytic form of regulated cell death, GSDME-mediated secondary necrosis.
Our previous study showed that pyroptosis occurs at an early stage of DKD and caspase-1 inhibitor treatment could reduce albuminuria within three weeks. In this study, our results demonstrate that GSDME was initially activated at 24 hrs, which is later than the activation of GSDMD in vitro. Besides, albuminuria was ameliorated after six weeks of treatment with a caspase-3 inhibitor in diabetic mice. Based on these results, we propose that GSDME-mediated cell death may play an important role in promoting DKD during later stages.
Since GSDME can form pores on plasma membrane, it can also act on organelle membrane. A recent study pointed out that GSDME augments the activation of the mitochondrial apoptotic pathway via forming pores in the mitochondria to facilitate cytochrome c release.33 Other superfamily members, like GSDMA and GSDMD, seem to share the ability to augment the apoptotic pathway through mitochondria.33,34 Therefore, more sophisticated experiments are required to further clarify the role of GSDME in DKD.
Conclusion
In summary, we showed that GSDME-mediated secondary necrosis, downstream of apoptotic caspase-3, accounts for the progression of renal tubulointerstitial fibrosis. These findings provide a new insight into the pathogenesis of DKD and a promising target for its treatment. Furthermore, small molecules targeting GSDME may provide a promising therapeutic strategy in the treatment of DKD.
Acknowledgments
This work was supported by grants from the Chinese National Key Technology R and D Program, Ministry of Science and Technology (No. 2017YFC0907601, 2017YFC0907602, and 2017YFC0807603), Natural Science Foundation of China (No. 81770724), and Shenyang Science and Technology Bureau (No. RC170172).
Disclosure
The authors declare that they have no conflicts of interest regarding this paper.
References
1. Van Laer L, Huizing EH, Verstreken M, et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat Genet. 1998;20(2):194–197. doi:10.1038/2503
2. Op de Beeck K, Van Camp G, Thys S, et al. The DFNA5 gene, responsible for hearing loss and involved in cancer, encodes a novel apoptosis-inducing protein. Eur J Hum Genet. 2011;19(9):965–973. doi:10.1038/ejhg.2011.63
3. Wang Y, Gao W, Shi X, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547(7661):99–103. doi:10.1038/nature22393
4. Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128. doi:10.1038/ncomms14128
5. Silva MT, do Vale A, dos Santos NM. Secondary necrosis in multicellular animals: an outcome of apoptosis with pathogenic implications. Apoptosis. 2008;13(4):463–482. doi:10.1007/s10495-008-0187-8
6. Vanden Berghe T, Vanlangenakker N, Parthoens E, et al. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17(6):922–930. doi:10.1038/cdd.2009.184
7. Galluzzi L, Kroemer G. Secondary necrosis: accidental no more. Trends Cancer. 2017;3(1):1–2. doi:10.1016/j.trecan.2016.12.001
8. Miao N, Yin F, Xie H, et al. The cleavage of gasdermin D by caspase-11 promotes tubular epithelial cell pyroptosis and urinary IL-18 excretion in acute kidney injury. Kidney Int. 2019;96:1105–1120. doi:10.1016/j.kint.2019.04.035
9. Zhang Z, Shao X, Jiang N, et al. Caspase-11-mediated tubular epithelial pyroptosis underlies contrast-induced acute kidney injury. Cell Death Dis. 2018;9(10):983. doi:10.1038/s41419-018-1023-x
10. Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55(1):225–233. doi:10.2337/diabetes.55.01.06.db05-0894
11. Shahzad K, Bock F, Al-Dabet MM, et al. Caspase-1, but not caspase-3, promotes diabetic nephropathy. J Am Soc Nephrol. 2016;27(8):2270–2275. doi:10.1681/ASN.2015060676
12. Chen J, Chen JK, Harris RC. EGF receptor deletion in podocytes attenuates diabetic nephropathy. J Am Soc Nephrol. 2015;26(5):1115–1125.
13. Isermann B, Vinnikov IA, Madhusudhan T, et al. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med. 2007;13(11):1349–1358. doi:10.1038/nm1667
14. Zhan M, Usman IM, Sun L, Kanwar YS. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J Am Soc Nephrol. 2015;26(6):1304–1321. doi:10.1681/ASN.2014050457
15. Shahzad K, Bock F, Dong W, et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015;87(1):74–84. doi:10.1038/ki.2014.271
16. Kindt N, Menzebach A, Van de Wouwer M, Betz I, De Vriese A, Conway EM. Protective role of the inhibitor of apoptosis protein, survivin, in toxin-induced acute renal failure. FASEB J. 2008;22(2):510–521. doi:10.1096/fj.07-8882com
17. Watson PA, Birdsey N, Huggins GS, Svensson E, Heppe D, Knaub L. Cardiac-specific overexpression of dominant-negative CREB leads to increased mortality and mitochondrial dysfunction in female mice. Am J Physiol Heart Circ Physiol. 2010;299(6):H2056–H2068. doi:10.1152/ajpheart.00394.2010
18. Wang L, Chang JH, Buckley AF, Spurney RF. Knockout of TRPC6 promotes insulin resistance and exacerbates glomerular injury in akita mice. Kidney Int. 2019;95(2):321–332. doi:10.1016/j.kint.2018.09.026
19. Qiu YY, Tang LQ. Roles of the NLRP3 inflammasome in the pathogenesis of diabetic nephropathy. Pharmacol Res. 2016;114:251–264. doi:10.1016/j.phrs.2016.11.004
20. Wada J, Makino H. Innate immunity in diabetes and diabetic nephropathy. Nat Rev Nephrol. 2016;12(1):13–26. doi:10.1038/nrneph.2015.175
21. Zhang C, Zhu X, Li L, et al. A small molecule inhibitor MCC950 ameliorates kidney injury in diabetic nephropathy by inhibiting NLRP3 inflammasome activation. Diabetes Metab Syndr Obes. 2019;12:1297–1309. doi:10.2147/DMSO.S199802
22. Wang Y, Zhu X, Yuan S, et al. TLR4/NF-kappaB signaling induces GSDMD-related pyroptosis in tubular cells in diabetic kidney disease. Front Endocrinol (Lausanne). 2019;10:603. doi:10.3389/fendo.2019.00603
23. Gilbert RE. Proximal tubulopathy: prime mover and key therapeutic target in diabetic kidney disease. Diabetes. 2017;66(4):791–800. doi:10.2337/db16-0796
24. He X, Cheng R, Park K, et al. Pigment epithelium-derived factor, a noninhibitory serine protease inhibitor, is renoprotective by inhibiting the Wnt pathway. Kidney Int. 2017;91(3):642–657. doi:10.1016/j.kint.2016.09.036
25. Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9(3):231–241. doi:10.1038/nrm2312
26. Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001;3(4):339–345. doi:10.1038/35070009
27. Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391(6662):43–50. doi:10.1038/34112
28. Feng S, Fox D, Man SM. Mechanisms of gasdermin family members in inflammasome signaling and cell death. J Mol Biol. 2018;430(18 Pt B):3068–3080. doi:10.1016/j.jmb.2018.07.002
29. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15(2):135–147. doi:10.1038/nrm3737
30. Qiu S, Liu J, Xing F. ‘Hints’ in the killer protein gasdermin D: unveiling the secrets of gasdermins driving cell death. Cell Death Differ. 2017;24(4):588–596. doi:10.1038/cdd.2017.24
31. Xia S, Hollingsworth L, Wu H. Mechanism and regulation of gasdermin-mediated cell death. Cold Spring Harb Perspect Biol. 2019. doi:10.1101/cshperspect.a036400
32. Birnbaum Y, Bajaj M, Qian J, Ye Y. Dipeptidyl peptidase-4 inhibition by saxagliptin prevents inflammation and renal injury by targeting the Nlrp3/ASC inflammasome. BMJ Open Diabetes Res Care. 2016. doi:10.1136/bmjdrc-2016-000227
33. Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10(1):1689. doi:10.1038/s41467-019-09397-2
34. Lin PH, Lin HY, Kuo CC, Yang LT. N-terminal functional domain of gasdermin A3 regulates mitochondrial homeostasis via mitochondrial targeting. J Biomed Sci. 2015;22:44. doi:10.1186/s12929-015-0152-0
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.