")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 16
Case Report: Congenital Myasthenic Syndrome Presenting with Bilateral Vocal Cord Paralysis Caused by De-Novel Compound Heterozygous MUSK Mutation
Authors Jiang L, Wang SC, Zhang J, Han FG, Zhao J, Xu Y
Received 24 November 2022
Accepted for publication 21 March 2023
Published 17 April 2023 Volume 2023:16 Pages 373—379
DOI https://doi.org/10.2147/PGPM.S398071
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Lan Jiang,1 Sheng-Cai Wang,2 Jie Zhang,2 Fu-Gen Han,1 Jing Zhao,2 Ying Xu1
1Department of Otorhinolaryngology Head and Neck Surgery, Children’s Hospital Affiliated to Zhengzhou University; Henan Children’s Hospital; Zhengzhou Children’s Hospital, Zhengzhou, 450003, People’s Republic of China; 2National Center for Children’s Health, Department of Otolaryngology Head and Neck Surgery, Beijing Children’s Hospital, Capital Medical University, Beijing, 100045, People’s Republic of China
Correspondence: Ying Xu, Department of Otorhinolaryngology Head and Neck Surgery, Children’s Hospital Affiliated to Zhengzhou University; Henan Children’s Hospital; Zhengzhou Children’s Hospital, Zhengzhou, 450003, People’s Republic of China, Tel/Fax +86 3718939569373, Email [email protected]
Background: We report the genetic etiology of a case of bilateral vocal cord paralysis in a female infant.
Case Description: The female infant developed dyspnea after birth, which improved with treatment, allowing her to be discharged from the local hospital. At 2 months of age, the child experienced a recurrence of dyspnea and was treated in a local hospital with interventions such as tracheal intubation and mechanical ventilation. However, as the child continued to suffer from dyspnea, she was transferred to the neonatal intensive care unit of the Children’s Hospital affiliated to Zhengzhou University for further treatment. A second electronic nasopharyngoscopy examination revealed bilateral vocal cord paralysis. The child underwent a tracheostomy due to a failure to wean from mechanical ventilation; after surgery, the respirator was effectively removed, and oxygen delivery ceased. The child and her parents underwent genetic testing with next-generation sequencing technology, which revealed that the child had two heterozygous variants in the MUSK gene, namely the c.2287G>A heterozygous mutation (p.Ala763Thr) and the c.790C>T heterozygous mutation. In addition, Sanger sequencing was performed, which confirmed that these two mutations were, respectively, inherited from the mother and father.
Conclusion: Congenital myasthenic syndrome caused by MUSK gene mutations can present clinically as bilateral vocal cord paralysis in neonates.
Keywords: congenital, gene, MUSK, myasthenic syndrome, vocal cord paralysis
Introduction
Congenital myasthenic syndrome (CMS) is a group of rare genetic disorders characterized by neuromuscular transmission disorders.1–3 There are already over 30 identified CMS mutations with differing disease severity, muscle groups affected, onset ages, and therapies. Chevessier et al were the first to identify mutations in the MUSK gene as the cause of a congenital myasthenic syndrome;4 they also discovered that the muscle-specific receptor tyrosine kinase (MuSK) encoded by the muscle skeletal receptor tyrosine (MUSK) gene plays a crucial role in the formation and stabilization of neuromuscular junctions. Several studies have since established that several site mutations in the MUSK gene are linked to CMS. The gene is located on chromosome 9 and consists of 8294 base pairs with 15 exons. It is an autosomal-recessive genetic disorder.5–9
In this paper, we report a case of MUSK gene mutation-induced CMS in a female neonate who carries two mutations of c.2287G>A and c.790C>T and manifested clinically only with bilateral vocal cord paralysis. Although the c.2287G>A mutation has been described previously, the c.790C>T pathogenic mutation has been detected for the first time. Therefore, this study can provide a new theoretical foundation for genetic illness identification and treatment.
Case Description
The female infant, G2P1, was delivered at 41 weeks by cesarean section due to “fetal malposition.” The amniotic fluid was contaminated to a grade III level, and the Apgar score was unknown. The parents denied any history of asphyxia at childbirth. The child experienced laryngeal stridor and dyspnea immediately after birth and was crying feebly. The child was admitted to a local hospital, treated with nasal cannula oxygenation, and discharged when dyspnea improved. Later, feeding was difficult, and the dyspnea gradually worsened. The child was again admitted to a local hospital at the age of 2 months due to dyspnea, laryngeal stridor, and diarrhea. Despite therapies including tracheal intubation and mechanical ventilation, dyspnea was not adequately alleviated. The child was then transferred to the neonatal intensive care unit of the Children’s Hospital affiliated to Zhengzhou University for mechanical ventilation treatment. The parents of the child were in good health and denied that their marriage was consanguineous.
The child’s crying remained weak; however, the sucking reflexes, eye movements, and facial expressions were normal. There were no indications of ptosis, ophthalmoplegia, fatigue, or muscle weakness. Multiple nasopharyngoscopies revealed bilateral vocal cord paralysis (BVCP). Due to the failure of weaning off ventilator-assisted breathing, the child underwent a tracheostomy, and the evacuation of the respirator and termination of oxygen administration were effectively accomplished after surgery. Post-discharge monitoring revealed a consistent rhythm of spontaneous breathing, no oxygen inhalation, and no requirement for mechanical ventilation. Initially, the child required repeated invasive and noninvasive ventilator-assisted ventilation and nasal feeding. After the tracheostomy, the child was fed orally.
Method
Electronic Laryngoscopy
During the examination, the child was kept in a lying position and mucosal surface anesthetic was administered. The child was maintained in a lying position during the examination, and mucosal surface anesthesia was performed. Electronic laryngoscopy was performed to observe whether there was any inflammation or mass in the laryngeal mucosa or on the surface of the vocal cords, and whether the vocal cord movement was normal.
Whole Genome DNA Extraction from Peripheral Blood
Gene DNA was extracted using a whole-genome DNA extraction kit (German Qiagen company), and the DNA concentration and purity were determined using NanoDrop 2000 UV-Vis (Thermo).
High-Throughput Sequencing Detection Technology
DNA extracted from peripheral blood was fragmented and libraries were produced. Gene exons and adjacent regions were captured with probes and then sequenced using high-throughput family whole-exome sequencing platforms. These detected mutation sites were annotated using molecular biology. The pathogenic mutation database, normal human genome database, clinical characteristics, and gene data analysis algorithms were combined, and finally, the variants with clinical reference significance were screened.
Ethical Considerations
The study was conducted in accordance with the Declaration of Helsinki (as was revised in 2013). The study was approved by Ethics Committee of the Children’s Hospital Affiliated to Zhengzhou University (2022K-041), and institution has approved to publish the case details. Written informed consent has been provided by the legal guardian to have the case details and any accompanying images published.
Results
Electronic Laryngoscopy Results
The bilateral vocal cords were adducted and hence could not be retracted abducted (Figure 1).
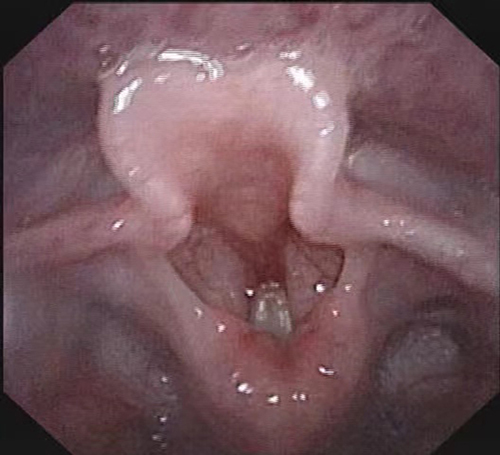 |
Figure 1 Bilateral vocal cord paralysis. |
Genetic Test Results
The results of family whole-exome sequencing revealed that the child possessed two heterozygous mutations in the MUSK gene, namely the c.2287G>A heterozygous mutation on exon 15, resulting in missense mutation of amino acid 763 that transforms alanine into threonine, and the c.790C>T heterozygous mutation on exon 7, resulting in a nonsense mutation that leads to early termination of the polypeptide chain at amino acid 264. The results suggested that the child possessed a complex heterozygous mutation (Figure 2). Except for wild type, Sanger sequencing demonstrated that these two mutations were complex heterozygous mutations inherited from the mother and father, respectively (Figure 3). According to ACMG guidelines, both c.2287G>A and c.790C>T mutations are regarded as potentially pathogenic.
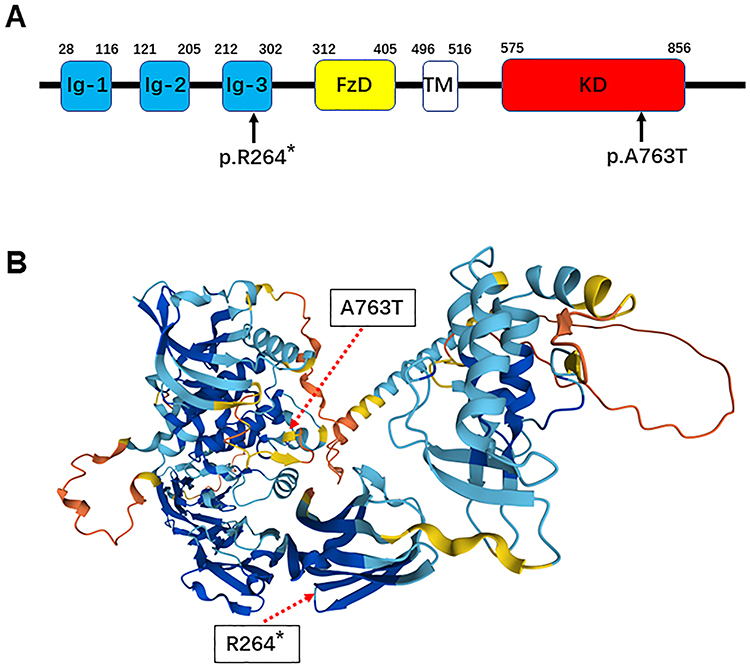 |
Figure 2 Pattern map of MUSK gene mutation sites of the child. (A) Pattern map of MUSK gene mutation sites of the child. (B) Pattern map of mutated amino acid sites in the MUSK protein of the child. (Arrows represent the location of mutation sites, The “*” in “p.R264*” represents the site of the mutated amino acid). |
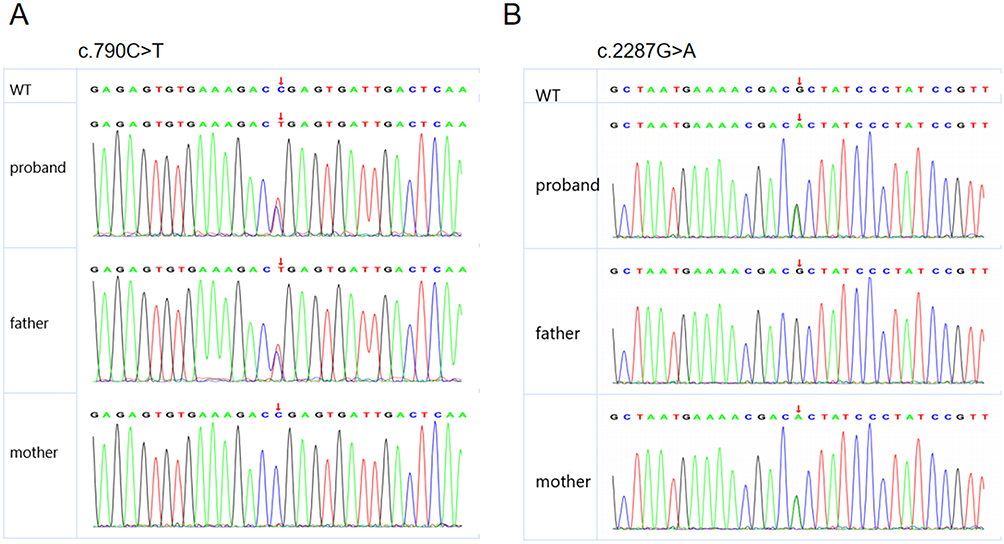 |
Figure 3 Results of Sanger sequencing. (A) c.790C>T sequencing plot; (B) c.2287G>A sequencing plot. The site indicated by the arrow is the mutation site; Top panel: the child; Middle panel: father; Bottom panel: mother. |
Discussion
MUSK mutation-induced CMS is very rare and is typically characterized by laryngeal stridor/vocal cord paralysis and respiratory distress, necessitating mechanical ventilation or tracheostomy to support breathing, especially in neonates and during infection. Posteriorly, it manifests as ptosis, facial weakness, and fluctuating fatigue and proximal limb muscular weakness.10 In addition, fetal akinesia deformation sequence syndrome (FADS),11 congenital vocal cord paralysis (CVCP),12 or late-onset limb-girdle weakness (LOLGW)13 may be present.
In this study, we present a case report of a female neonate with MUSK heteroallelic mutation-associated CMS with bilateral vocal cord paralysis caused by a missense mutation (c.2287G>A) and a nonsense pathogenic mutation (c.790C>T). Nonsense mutations result in protein truncation, whereas missense mutations induce the expression of a defective form of MUSK.
Liu et al14 found that patients with nonsense, splicing, or frameshift mutations in the MUSK gene have an earlier age of onset and skeletal muscles including bulbar and respiratory muscles are more likely to be impacted. This suggests that these mutations result in deletions of the encoded protein or large fragments of the protein, disrupting the interaction between MUSK and Dok7, thereby resulting in signaling interruption. Some MUSK missense mutations can influence MUSK kinase activity. In addition, the expression and stability of MUSK is reduced, and the protein kinase domain is affected, which may lead to a delayed onset of the disease and a reduction in the aggregation of Agrin-dependent AChR, which is a key step in the formation of neuromuscular connections. MUSK is known to be involved in the regulation of AChR aggregation in embryos and AChR expression during development and adulthood at neuromuscular junctions.15,16
In this study, we reviewed the clinical manifestations and gene mutations of six children with MUSK gene mutation-associated CMS accompanied by bilateral vocal cord paralysis from different countries (Table 1). All these neonates presented with bilateral vocal cord paralysis and underwent tracheostomy due to repeated failure of ventilator weaning.6,12,17 There are 11 MUSK gene mutations, of which 10 mutations (c.2561G>A, c.79+2T>G, c.2158A>G, c.308A>g, c.496C>T, c.2165T>C, c.220insC, c.2368G>A, c.2287G>A, and c.2446C>T) have been previously reported,6 while one novel pathogenic mutation (c.790C>T) has not been reported. Murali et al6 reported a case of an Asian male child who presented with vocal cord paralysis after birth, and gene mutations c.2287G>A and c.2446C>T were found on exon 15, which were not classified as nonsense, splicing, frameshifting, or missense mutations. They concluded that biallelic MUSK mutation is the genetic etiology of vocal cord paralysis in patients with no other symptoms. In our study, we note that exon 15 c.2287G>A is a missense mutation and exon 7 c.790C>T is a nonsense mutation. Heteroallelic MUSK mutation may be the genetic etiology of vocal cord paralysis in patients who presented with solely this symptom.
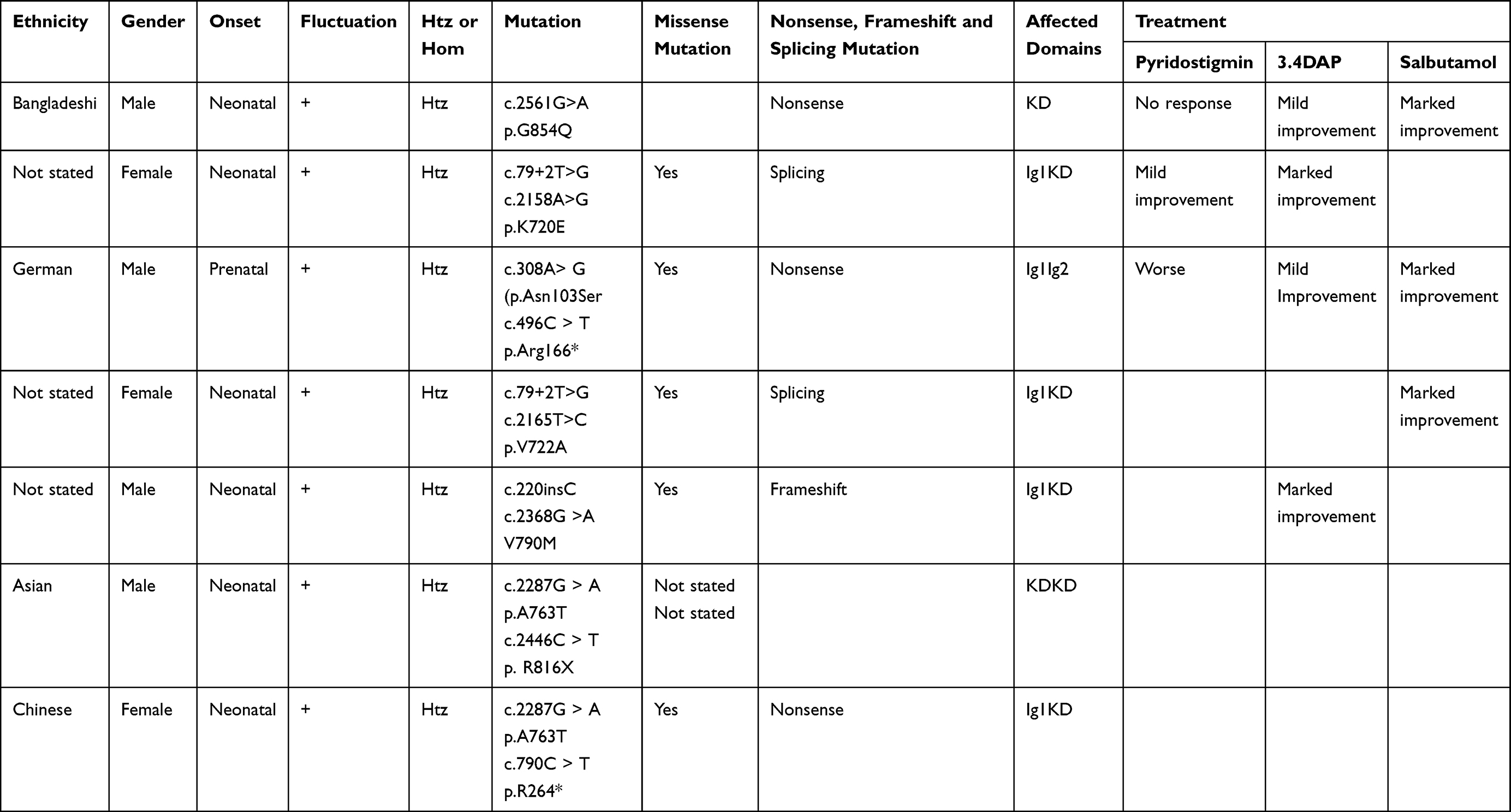 |
Table 1 Clinical Features of Patients with Vocal Cord Paralysis in MUSK-Congenital Myasthenic Syndrome |
Concerning the treatment of MUSK-associated CMS,6,12,18 patients frequently do not respond to anticholinesterase inhibitor therapy or even deteriorate as a result of treatment. In our review of these six MUSK-associated CMS with bilateral vocal cord paralysis (Table 1), except for two children who received no medication, the condition of one child worsened after treatment with anticholinesterase inhibitors, four children had mild to significant improvement with 3,4-diaminopyridine treatment, and three children had mild to significant improvement with salbutamol treatment. In this study, the condition manifested in the child at an early age. With the exception of bilateral vocal cord paralysis and repeated failures of ventilator weaning that led to a tracheostomy, there were no other visible symptoms of muscle weakness. The parents denied permission for drug treatment.
Data Sharing Statement
All data generated or analysed during this study are included in this article. Further enquiries can be directed to the corresponding author.
Ethics Approval
The study was conducted in accordance with the Declaration of Helsinki (as was revised in 2013). The study was approved by Ethics Committee of the Children’s Hospital Affiliated to Zhengzhou University (2022K-041), and institution has approved to publish the case details.
Consent for Publish
Written informed consent has been provided by the legal guardian to have the case details and any accompanying images published.
Acknowledgments
We are particularly grateful to everyone who has given us help on our article.
Funding
There is no funding to report.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Engel AG, Shen XM, Selcen D, Sine SM. Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment [published correction appears in Lancet Neurol. 2015 May;14(5):461. Lancet Neurol. 2015;14(4):420–434. doi:10.1016/S1474-4422(14)70201-7
2. Finsterer J. Congenital myasthenic syndromes. Orphanet J Rare Dis. 2019;14(1):57. PMID: 30808424. doi:10.1186/s13023-019-1025-5
3. Kaplan JC, Hamroun D. The 2016 version of the gene table of monogenic neuromuscular disorders (nuclear genome). Neuromuscul Disord. 2015;25(12):991–1020. PMID: 27563712. doi:10.1016/j.nmd.2015.10.010
4. Chevessier F, Faraut B, Ravel-Chapuis A, et al. MUSK, a new target for mutations causing congenital myasthenic syndrome. Hum Mol Genet. 2004;13(24):3229–3240. PMID: 15496425. doi:10.1093/hmg/ddh333
5. Ben Ammar A, Soltanzadeh P, Bauché S, et al. A mutation causes MuSK reduced sensitivity to agrin and congenital myasthenia. PLoS One. 2013;8(1):e53826. PMID: 23326516. doi:10.1371/journal.pone.0053826
6. Murali C, Li D, Grand K, Hakonarson H, Bhoj E. Isolated vocal cord paralysis in two siblings with compound heterozygous variants in MUSK: expanding the phenotypic spectrum. Am J Med Genet A. 2019;179(4):655–658. PMID: 30719842. doi:10.1002/ajmg.a.61060
7. Gallenmüller C, Müller-Felber W, Dusl M, et al. Salbutamol-responsive limb-girdle congenital myasthenic syndrome due to a novel missense mutation and heteroallelic deletion in MUSK. Neuromuscul Disord. 2014;24(1):31–35. PMID: 24183479. doi:10.1016/j.nmd.2013.08.002
8. Burden SJ, Yumoto N, Zhang W. The role of MuSK in synapse formation and neuromuscular disease. Cold Spring Harb Perspect Biol. 2013;5(5):a009167. PMID: 23637281. doi:10.1101/cshperspect.a009167
9. Herbst R. MuSk function during health and disease. Neurosci Lett. 2020;716:134676. doi:10.1016/j.neulet.2019.134676
10. Shen Y, Wang B, Zheng X, Zhang W, Wu H, Hei M. A neonate with MuSK congenital myasthenic syndrome presenting with refractory respiratory failure. Front Pediatr. 2020;8:166. PMID: 32373561. doi:10.3389/fped.2020.00166
11. Wilbe M, Ekvall S, Eurenius K, et al. MuSK: a new target for lethal fetal akinesia deformation sequence (FADS). J Med Genet. 2015;52(3):195–202. PMID: 25612909. doi:10.1136/jmedgenet-2014-102730
12. Al-Shahoumi R, Brady LI, Schwartzentruber J, Tarnopolsky MA. Two cases of congenital myasthenic syndrome with vocal cord paralysis. Neurology. 2015;84(12):1281–1282. PMID: 25695962. doi:10.1212/WNL.0000000000001396
13. Owen D, Töpf A, Preethish-Kumar V, et al. Recessive variants of MuSK are associated with late onset CMS and predominant limb girdle weakness. Am J Med Genet A. 2018;176(7):1594–1601. PMID: 29704306. doi:10.1002/ajmg.a.38707
14. Liu Y, Qiao K, Yan C, et al. Congenital myasthenia syndrome in a Chinese family with mutations in MUSK: a hotspot mutation and literature review. J Clin Neurosci. 2020;76:161–165. PMID: 32253145. doi:10.1016/j.jocn.2020.03.036
15. Rodríguez Cruz PM, Cossins J, Cheung J, et al. Congenital myasthenic syndrome due to mutations in MUSK suggests that the level of MuSK phosphorylation is crucial for governing synaptic structure. Hum Mutat. 2020;41(3):619–631. doi:10.1002/humu.23949
16. Petrov KA, Girard E, Nikitashina AD, et al. Schwann cells sense and control acetylcholine spillover at the neuromuscular junction by α7 nicotinic receptors and butyrylcholinesterase. J Neurosci. 2014;34(36):11870–11883. doi:10.1523/JNEUROSCI.0329-14.2014
17. Giarrana ML, Joset P, Sticht H, et al. A severe congenital myasthenic syndrome with “dropped head” caused by novel MUSK mutations. Muscle Nerve. 2015;52(4):668–673. PMID: 25900532. doi:10.1002/mus.24687
18. Pinto MV, Saw JL, Milone M. Congenital vocal cord paralysis and late-onset limb-girdle weakness in MuSK-congenital myasthenic syndrome. Front Neurol. 2019;10:1300. PMID: 31920924. doi:10.3389/fneur.2019.01300
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.