")
Back to Journals » Cancer Management and Research » Volume 13
Case Analysis of 14 Children with Malignant Rhabdoid Tumor of the Kidney
Authors Li J, Zhang W, Hu H, Zhang Y, Wang Y, Gu H, Huang D
Received 2 March 2021
Accepted for publication 17 May 2021
Published 21 June 2021 Volume 2021:13 Pages 4865—4872
DOI https://doi.org/10.2147/CMAR.S309274
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Eileen O'Reilly
Jing Li,* Weiling Zhang,* Huimin Hu, Yi Zhang, Yizhuo Wang, Huali Gu, Dongsheng Huang
Department of Pediatrics, Beijing Tongren Hospital of China Capital Medical University, Beijing, 100176, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Dongsheng Huang
Department of Pediatrics, Beijing Tongren Hospital of China Capital Medical University, No. 1 Dongjiao Lane, Dongcheng District, Beijing, 100730, People’s Republic of China
Tel +86 10 5826 6032
Email [email protected]
Objective: This study aims to summarize the clinical features and prognoses of the malignant rhabdoid tumor of the kidney (MRTK) in children. It further aims to analyze the high-risk factors affecting MRTK prognosis.
Methods: Clinical data from 14 children with MRTK treated in Paediatrics of Beijing Tongren Hospital from January 2010 to December 2019, along with the high-risk factors affecting prognosis, were retrospectively analyzed.
Results: There were 14 children with MRTK included in the study, with a median onset age of 13 (3– 46) months. Thirteen patients had distant metastases, the most common site for metastases being inside the lung. A comprehensive treatment protocol combined with chemotherapy was mainly applied during the surgery. A surgical resection of primary tumors was performed on 13 (13/14) patients, and all 14 children received chemotherapy with ifosfamide + carboplatin + etoposide, ifosfamide + etoposide, and vincristine + pirarubicin + cyclophosphamide regimens, alternately. Three patients received radiotherapy and two received oral targeted drugs after partial response. The median follow-up was after 16.5 months (3– 53 months) and the four-year overall survival (OS) was 41.8%. In children aged ≤ 24 months and children aged > 24 months, the two-year OS was 67.2% and 100% (χ2 = 108.998, P< 0.05), respectively. In children with Ki 67 > 70% and children with Ki 67 < 70%, the two-year OS was 52.6% and 86.9% (χ2 = 8.544, P = 0.003), respectively. In children with distant metastases and children without distant metastasis, the two-year OS was 70% and 100% (χ2 = 14.239, P< 0.05), respectively.
Conclusion: The most common MRTK distant metastasis site is the lung. Risk factors for poor MRTK prognoses include an age of < 24 months, Ki 67 > 70%, and distant metastases.
Keywords: children, malignant rhabdoid tumor of the kidney, MRTK, treatment, prognosis
Introduction
The malignant rhabdoid tumor (MRT) is a class of highly invasive tumors1 mainly occurring in children. It is relatively rare in clinical practice. Most commonly, the MRT occurs in kidneys and accounts for 1.5%–4% of renal malignancies.2,3 It was formerly considereda4 special subtype of the renal Wilms tumor; however, in 1981, Haas et al5 clarified that the tumor is a type of malignant renal tumor occurring in children and is completely independent of nephroblastoma. It was given the name MRTK. Due to the new understanding of the disease, other MRTs6–9 in children with different primary sites, such as the central nervous system, liver, abdomen, retroperitoneum, or other soft tissues, have been reported. According to its different anatomical sites, the tumor can be divided into MRTK, atypical teratoid/rhabdoid tumor (AT/RT), extra-renal noncerebral rhabdoid tumor.10 The MRT is a highly invasive tumor commonly occurring in children. It was first described14 in children with renal tumors in 1978 and named “MRTK” by Haas et al in 1981.5 With the development of research, it has been found that the MRT can occur in many tissues and organs of the whole body, of which the MRTK is the most common, accounting for 0.9%–2%15 of renal tumors in children. The MRTK’s main pathological features are rhabdoid cells, and the deletion or mutation of SMARCBl (also known as INll) expression in immunohistochemical staining is a sensitive and specific diagnostic marker.20 In most cases, skeletal muscle markers, such as desmin and myogenin, have detectable expression, while CK, EMA, and Vimentin have positive expression. Generally, the Ki67 proliferation index score is >50%. The expression of S-100 protein, NSE, and myoglobin varies. The immunohistochemical characteristics of this study are consistent with the literature reports in terms of INI1, desmin, and myogenin having detectable expression and the Vimentin and EMA having positive expression. A recent study shows that the Ki67 score is a good prognostic factor21 in adult soft tissue sarcoma.
This study aims to improve the clinical understanding of MRTK by summarizing its clinical and biological characteristics as well as the prognoses in children with MRTK who have been diagnosed and treated in our hospital in the past 10 years.
Methods
Subjects
A total of 14 children were enrolled in this study. All of them were admitted to Beijing Tongren Hospital from January 1, 2010–December 31, 2019 and clearly diagnosed with MRTK by surgical resection or biopsy pathology, ie, the postoperative pathology or biopsy pathology was MRTK. Imaging findings in the children, including B ultrasound, computed tomography (CT), magnetic resonance imaging (MRI), and positron emission tomography–computed tomography, were retrospectively collected and analyzed in order to evaluate the progression of primary tumors, distant metastases and their recurrence, etc. This study was approved by the Ethical Review Committee of Beijing Tongren Hospital, Capital Medical University. This study was conducted in accordance with the Declaration of Helsinki. All of the children’s guardians provided a signed informed consent form.
Study Methods
Diagnostic Criteria and Staging
All patients underwent either surgical resection or biopsy and were diagnosed with MRTK after two or more pathological consultations. Complete genome sequencing, which required the parents to confirm whether the patients had any mutated genes or not, was performed on the children.
Tumor staging (I, II, III, IV, and V)11 was carried out with all patients according to the staging criteria of the National Wilms Tumor Study (NWTS).
Treatment Protocols
The chemotherapy regimens consisted of the ifosfamide + carboplatin + etoposide (ICE), ifosfamide + etoposide (IE), or vincristine + pirarubicin + cyclophosphamide (VDC) regimen. Treatment was replaced by the adriamycin + vincristine + cisplatin + cyclophosphamide (AVCP) chemotherapy regimen or the European12 Ewing’s sarcoma first-line vincristine + pirarubicin + ifosfamide + etoposide (VIDE) regimen in children who were insensitive to the above-stated chemotherapy. These treatments were also applied in the RS-99 ultra-high-risk group at Shanghai Children’s Medical Center.13 Details of chemotherapy:VDC (vincristine 1.5mg/m2 + doxorubicin (pirubicin 25 mg/m2× 3) + cyclophosphamide 600 mg/m2× 3) and ICE (carboplatin 450 mg/m2 + etoposide 100 mg/m2×4+ cyclophosphamide 1.5mg/m2) ×5) or IE (1.8mg/m2 ×5+ etoposide 100mg/m2×5) alternatively. 8 cases (8/14) had poor response to the above chemotherapy regimen. Shanghai Children’s Medical Center RS-99 super high risk group was replaced with AVCP regimen (pirubicin 25 mg/m2 ×2+ vincristine 1.5 mg/m2 ×2+ cisplatin 90 mg/m2 + cyclophosphamide 300 mg/m2 ×3) and Vide regimen (vincristine 1.5 mg/m2) + isocyphosphamide 3mg/m2 ×3+ pirubicin 25 mg/m2 ×2+ etoposide 150mg/m2 ×3). Due to the positioning and radiotherapy in the specialized radiotherapy department, and the small number of radiotherapy patients, the effect of radiotherapy for this disease was not exact, so we did not further analyze the correlation of radiotherapy. We are following up the children who have received radiotherapy and those who are receiving radiotherapy, to further compare the influence of radiotherapy or not on the survival rate.
Evaluation Criteria for Tumor Lesions
Complete response (CR): All lesions have disappeared and have been completely gone for over four weeks.
Partial response (PR): The primary tumor shrank by ≥64% and the metastatic tumor shrank by ≥30%. There were no new lesions.
Progressive disease (PD): The primary tumor increased by ≥40% after the first visit and the metastatic tumor either increased by ≥20% or there was a new lesion.
Evaluation Criteria for Overall Efficacy
CR: Both primary and metastatic tumors had CR, with no new lesions.
PR: The primary tumor had CR and the metastatic tumor either had CR, PR, or stable disease (SD), or there was a new lesion.
SD: The primary tumor had SD, the metastatic tumor either had CR, PR, or SD, or there was no new lesion.
PD: No primary and metastatic tumors had PD.
Follow-Up Analysis
Outpatient or telephone follow-ups: Regular follow-ups of primary sites were conducted by ultrasound, CT or MRI, as well as by lung CT, abdominal ultrasound, cranial MRI, etc. Metastatic foci were monitored simultaneously in children with distant metastases. The follow-ups ended on June 30, 2020 or at the patient’s time of death.
Statistical Method
The SPSS 21.0 software was used to calculate the enumeration data, which were expressed with a percentage or rate. The rate was obtained through the χ2 test and the survival analysis was conducted using the Kaplan–Meier Method in order to calculate the survival time and draw the survival curve. P < 0.05 was considered a statistically significant difference.
Results
Clinical Characteristics
Among the 14 children with MRTK observed in this study, there were nine boys and five girls, with a median onset age of 13 (3–46) months. In six patients, the lesion was located in the left kidney and in eight patients it was located in the right kidney. The tumors were rather large. Of the patients, 13 had a tumor with the largest known diameter (>5 cm). The first symptoms included abdominal pain in two patients, abdominal mass in six patients and gross hematuria in six patients (one patient had gross post-traumatic hematuria). Lung metastases at the beginning of the disease occurred in 12 patients, of whom two had liver metastases, and four had lymph node metastases. During the treatment, liver metastases only occurred in one patient. Tumor embolus was found in 9/13 patients (Table 1).
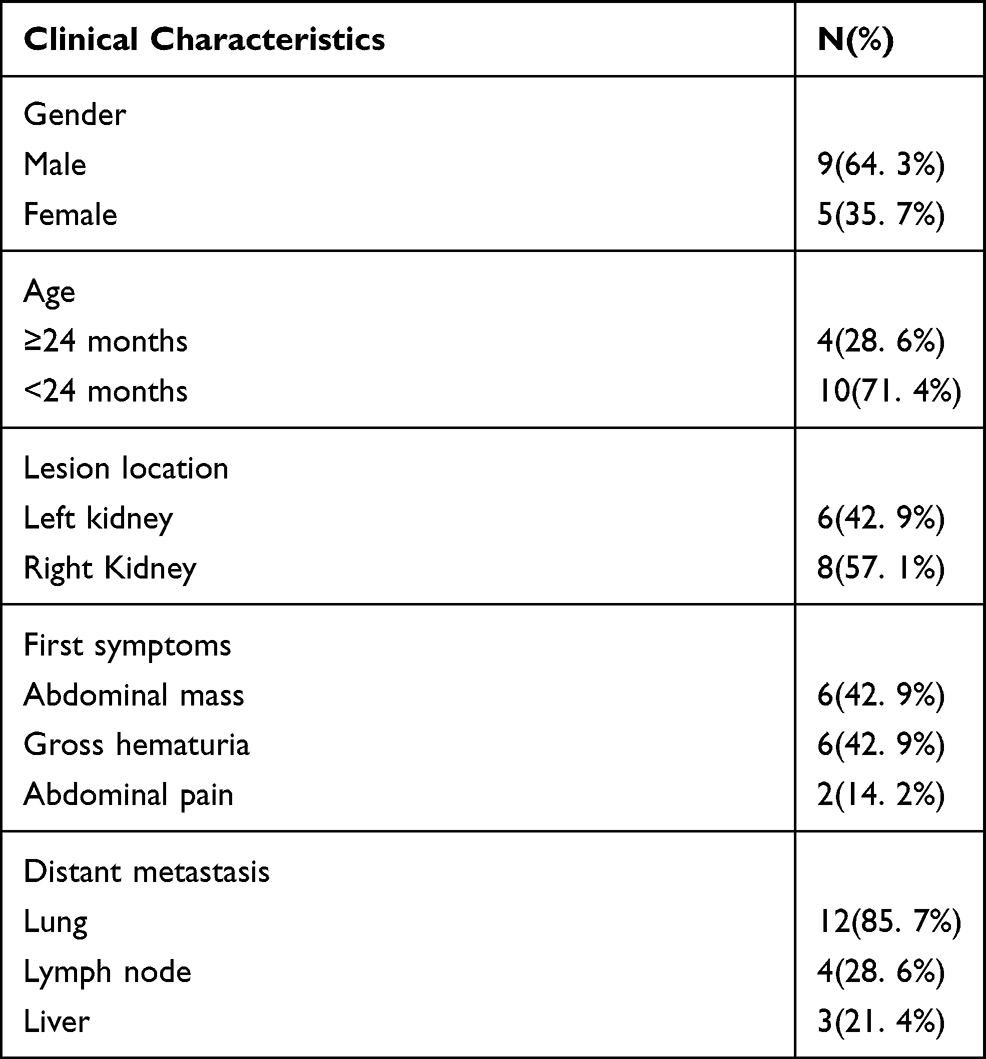 |
Table 1 The Clinical Characteristics of Malignant Rhabdoid Tumor of the Kidney |
Immunohistochemical and Genetic Characteristics
Immunohistochemical results concerning 14 primary tumors showed a INI1 detectable expression present in 11 patients (11/14), desmin detectable expression in eight patients (8/10), myogenin detectable expression in seven patients (7/7), S-100 detectable expression in eight patients (8/10), EMA positive expression in seven patients (7/10), CD99 positive expression in six patients (6/7), and vimentin positive expression in eight patients (8/8). The Ki 67 proliferation index was high (30%–90%). The complete genome sequencing of tumor tissue was performed in four patients, two of whom showed SMARCBl gene abnormalities. Namely, one patient had nonsense mutation and one had gene deletion. Furthermore, one patient showed a presence of SOX2 mutations and one patient showed an absence of tumor-related genotype changes (Table 2).
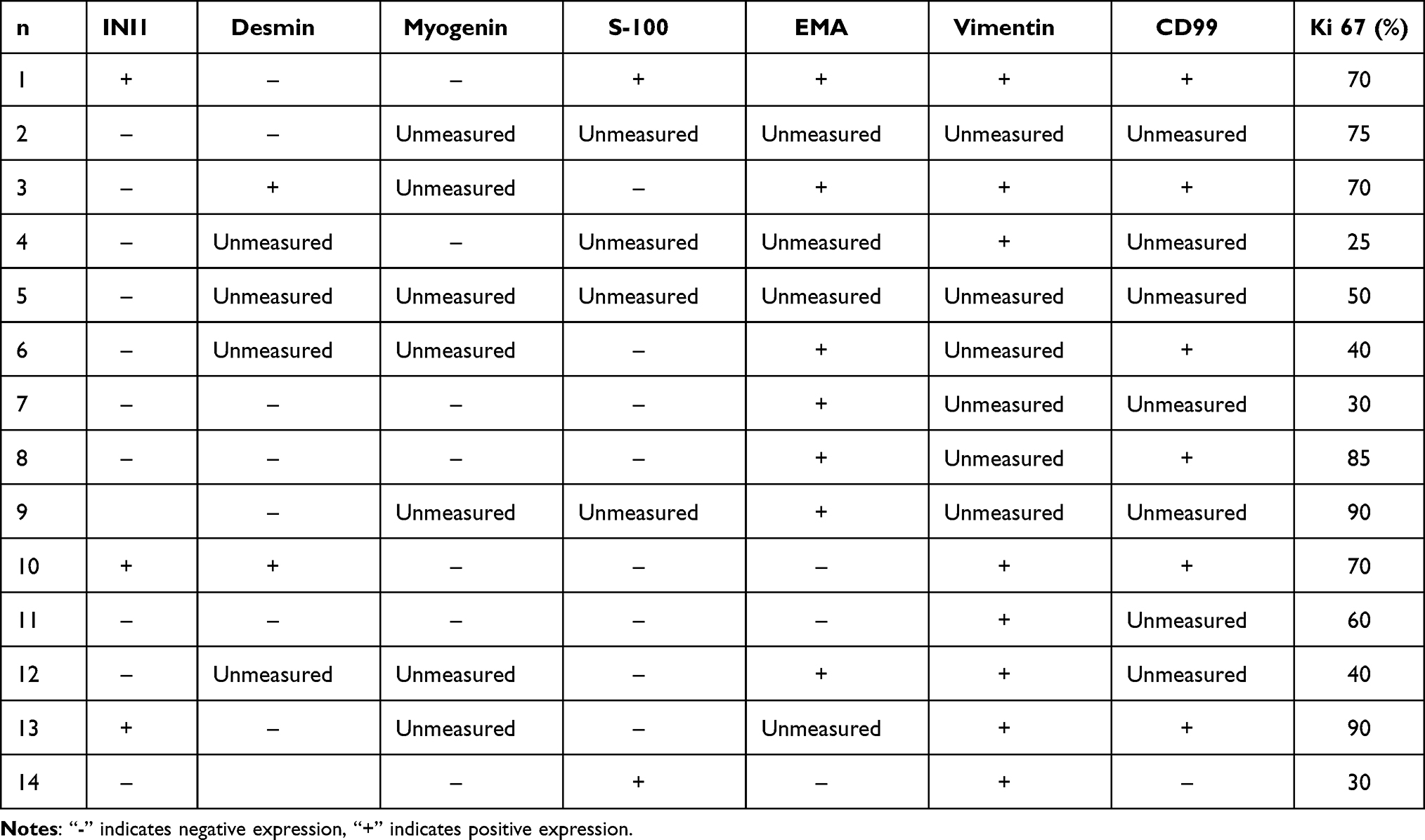 |
Table 2 Immunohistochemical Characteristics of 14 Children with Renal Rhabdomyoidoma |
Treatment and Prognosis
After the follow-up ending on June 30, 2020, the median follow-up time was 19 months (3–53 months). Complete primary resection was performed on 13 patients (13/14), of whom eight underwent direct excision and five received preoperative neoadjuvant chemotherapy after being confirmed to have MRTK by biopsy and undergoing surgical resection after partial regression. One patient (1/14) underwent neoadjuvant chemotherapy after being confirmed to have MRTK by biopsy. The tumor continued to progress until death. All 14 children received postoperative chemotherapy and six of them died. One patient died of tumor progression during the preoperative neoadjuvant chemotherapy, three patients died of tumor progression during postoperative chemotherapy, and two patients recurred after the CR after initial treatment. However, the re-treatment had a poor effect and the patients died after ceasing treatment. Two patients had CR. Both received surgery and achieved CR after chemotherapy. One case of PD was a patient with CR after surgery, chemotherapy, and radiotherapy, who recurred in situ after three months and was still in the progressive phase of the disease after re-chemotherapy and surgery. Among the patients, there were five cases of PR, two of which occurred after the patients received surgery and chemotherapy. At present, one patient is taking sorafenib orally, and one patient is taking anlotinib orally. Two of the remaining three children have received local and primary radiotherapy and continued to maintain chemotherapy. Currently, all patients are in the maintenance stage of chemotherapy. The four-year OS was 41.8% (Figure 1A). In children aged <24 months and children aged >24 months, the two-year OS was 67.2% and 100%, respectively. The difference was statistically significant (χ2 = 108.998, P<0.05) (Figure 1B). In children with Ki67 ≥ 70% and children with Ki67 < 70%, the two-year OS was 52.6% and 86.9%, respectively. The difference was statistically significant (χ2 = 8.544, P = 0.003) (Figure 1C). In children with distant metastasis or children without distant metastasis, the two-year OS was 70% and 100%, respectively. The difference was statistically significant (χ2 = 14.239, P<0.05) (Figure 1D).
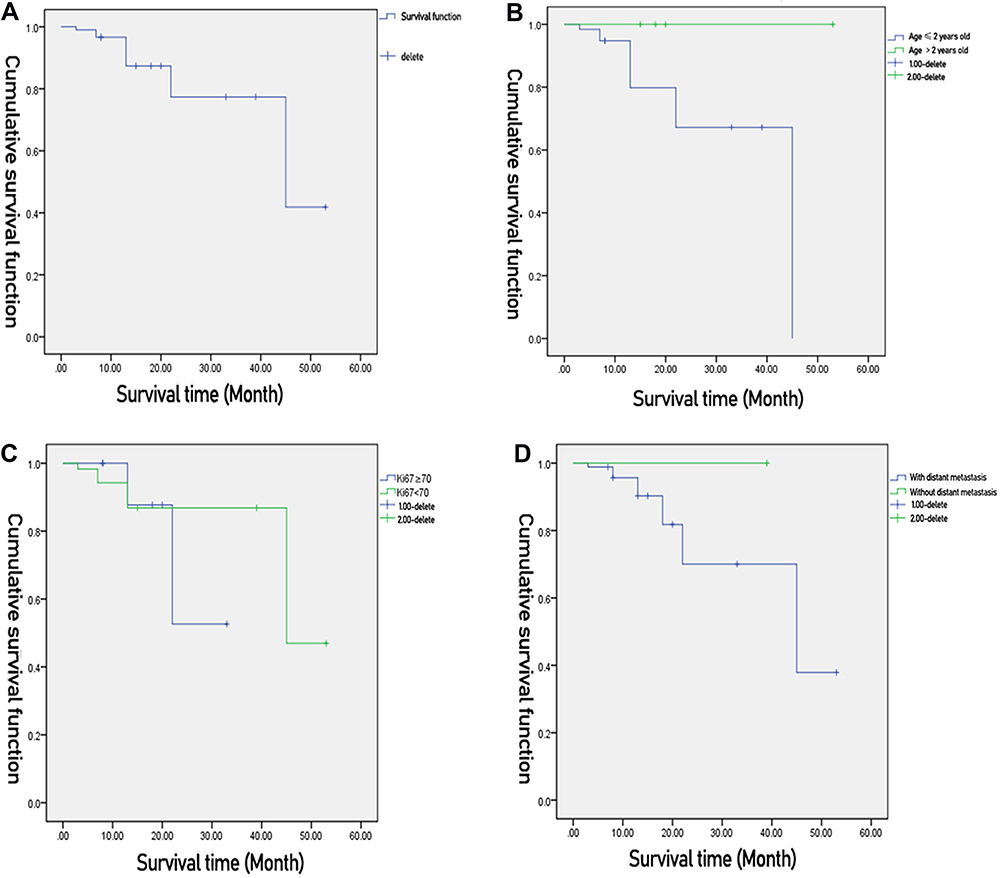 |
Figure 1 Clinical characteristics of malignant renal rhabdomyoid tumor in children. |
Discussion
The median onset age of susceptible MRTK infants and younger children is 18 months, and the male–female ratio is 1.5:1.0.11 The median onset age is 13 (3–46) months, and the male–female ratio is 9:5. Tomlinson et al16 find that the onset age is an independent MRTK prognostic factor. In this study, the two-year OS is 67.2% and 100% in children with MRTK older than 24 months and children with MRTK younger than 24 months, respectively. The difference is statistically significant (χ2 = 108.998, P<0.05). It is suggested that age may be a bad factor affecting prognosis. At the onset of the disease, the common clinical manifestations are gross hematuria, abdominal pain, abdominal distension, fever, etc.15 The first symptoms of this group mainly include the gross hematuria (n = 6) and abdominal mass (n = 6).
The MRTKT staging is adopted from the staging criteria of the American Nephroblastoma Research Collaboration Group. Previous studies have shown that more than 2/3 of children with MRTK are at the progressive stage of the disease (with distant metastasis in the lungs and/or brain)15 when diagnosed. Among 12 patients (12/14) in this study, the lung metastasis occurs when the disease attacks. During this treatment, two patients (2/12) had liver metastasis, four patients (4/12) had lymph node metastasis, and one patient (1/14) had liver metastasis. This confirms that the MRTK has an extremely high level of malignancy. In patients with a presence of distant metastases and patients with an absence of distant metastases, the two-year OS is 70% and 100%, respectively. The difference is statistically significant (χ2 = 14.239, P<0.05). The prognosis of distant metastases is suggested to be poor. It has also been reported that the MRTK may invade the renal veins or inferior vena cava and transfer to the central nervous system.17 In 1989, Weeks et al18 summarized 111 patients with MRTK and found that approximately 10%–15% of children with MRTK also had intracranial tumors, most of them being AT/RT. Follow-up studies also report that MRTK is associated with secondary intracranial tumors, such as medulloblastoma and primitive neuroectodermal tumors.19 However, no intracranial metastases were found in the children observed in this study during the initial diagnosis and treatment. The follow-up time should be prolonged and the head imaging closely monitored.
The MRTK Ki67 score is generally high. In this study’s group, the Ki67 score is 30%–90%. The difference in the two-year survival rates of the >70% group and the <70% group is statistically significant (χ2 = 8.544, P = 0.003), indicating that the higher the Ki67 score, the worse the prognosis.
It is currently believed that the inactivation of the INIl gene is located in the chromosome 22q11.2, leading to the deletion of INll protein expression, which is related to the occurrence of many malignant tumors.22 Studies show that two genes are inactivated in malignant rhabdoid tumors,23 namely, SMARCBl or SMARCA4. When the SMARCBl or SMARCA4 is abnormal, it can lead to the failure of the SWI/SNF complex to play an important role in the process of gene transcription and affect the normal expression of subsequent genes, thereby, resulting in the occurrence of an MRT.24 In this study, the entire tumor tissue genome sequencing was conducted in four patients, including two patients (2/4) with abnormal SMARCBl (one patient with nonsense mutations and one with SMARCBl gene deletion), one patient with SOX2 mutations, and one patient with a reported unknown tumor-related genotype alteration. SOX2 amplification or activation of mutations may promote cancer stem-like behavior, namely, cell renewal. SOX2 overexpression has been reported as a driving event in several cancer types, especially squamous cell carcinoma. Many preclinical studies have also involved SOX2-mediated self-renewal, invasion, and tumorigenesis in non-small cell lung cancer. However, there are no relevant reports on SOX2 and MRT.25–27
Tomlinson et al16 have found that the prognoses of children with MRTK who have SMARCBl and SMARCA4 gene mutations are very poor. However, it is not possible to infer whether or not the children with tumor-related mutant genes have poor prognoses due to the short follow-up time and non-detected deaths.
Current studies show that radical surgery, supplemented by chemotherapy and radiotherapy, is an important basis for MRTK treatment. The previous chemotherapy protocols of nephroblastoma used in MRTK treatment have a poor therapeutic effect as the prognosis.16 NWTS-5 recommends a chemotherapy regimen that includes carboplatin, etoposide, and cyclophosphamide. Subsequent case reports show that the ICE and IE, or, alternately, the VDC, regimens may be effective in MRTK treatment.28 Post-retreatment with carboplatin and thiotepa is also recommended for hematopoietic stem cell transplantation,29 however, there has been no observed significant improvement in the MRTK prognosis, which is still 20%–25%.16 Moreover, the post-retreatment may increase the mortality of chemotherapy-related adverse reactions.30,31 In the group observed in this study, 13 patients underwent complete resection of primary lesions and one patient was confirmed by biopsy. However, the chemotherapy had a poor effect and the tumor died rapidly. All 14 children received chemotherapy. The chemotherapy regimens mainly consist of VDC, ICE, or IE regimens. Eight (8/14) children did not respond well to the above-mentioned chemotherapy regimens. Their treatment was replaced with AVCP and VIDE chemotherapy regimens, alternately, in the RS-99 ultra-high-risk group of Shanghai Children’s Medical Center. Four (4/8) patients were still insensitive to the chemotherapy regimen, had tumor progression and died. Four (4/8) patients experienced tumor reduction after a change in the regimen. Two patients had a poor outcome in re-chemotherapy after recurrence with tumor progression and died. Only three patients received radiotherapy for the primary foci and metastases in this study. However, one patient recurred again three months after CR and two patients are still in the maintenance stage of chemotherapy. This study suggests that radiotherapy may be beneficial to patients aged >3 years with have AT/RT limitations. Nevertheless, for all patients with MRT, the role of adjuvant radiotherapy still requires further clinical studies and validation.32 For these conditions that have an uncertain effect of radiotherapy, the ability to find an effective chemotherapy regimen directly affects the survival rate.
Because of the poor prognosis of traditional treatment, the MRT also has obvious characteristics of epigenetics and gene expression heterogeneity. The targeted therapy has become a new study protocol. However, there are currently no FDA-approved anti-tumor drugs targeting SMARCBL. Tazemetostat, a histone methylation inhibitor [enhancer of zeste homolog (EZH) 2 inhibitor], is used to treat the SMARCBL-expressed negative epithelioid sarcoma (NCT02601950) in the Phase II clinical trial, which has a total of 31 patients. The results show that four patients have PR and six patients have been stable for more than 32 weeks.33 In a different Phase I clinical trial (NCT01897571) of Tazemetostat for treating SMARCBL or SMARCA4-expressed epithelioid sarcoma, eight patients have a negative SMARCBL expression. Among them, one patient suffering from MRT with negative SMARCBL expression has CR and has lasted 65 weeks and two patients with SMARCBL-expressed negative epithelioid sarcoma are showing a stable condition, lasting 25 and 24 weeks, respectively. Although no gene detection was carried out in two of the study’s patients, the disease is partially alleviated after oral administration of targeted drugs. Due to the small sample size, it should not be concluded that such targeted drugs are effective for MRTK treatment. This research data are very limited by the retrospective single-center research character, especially by the erroneous presentation of the data and results and unfortunately also by the tiny patient cohort.
To sum up, with the gradual understanding and research regarding the MRTK, the disease has always been considered rare, highly invasive and fatal. Age <24 months, Ki67 ≥ 70%, and the presence of distant metastases may be poor prognosis risk factors. There are still many unknown areas to explore, from pathogenesis to treatment options. Large sample size data may be obtained by clinical centers working together closely in order to comprehensively analyze the treatment and prognosis of this disease. Targeted therapy based on pathogenesis is becoming a hot spot in MRTK therapy and is expected to be a breakthrough in improving disease prognosis.
Conclusion
The most common MRTK distant metastasis site is the lung. Risk factors for poor MRTK prognoses include an age of <24 months, Ki 67 > 70%, and distant metastases.
Disclosure
Jing Li and Weiling Zhang are co-first authors for this study. The authors report no conflicts of interest in this work.
References
1. Yasui N, Yoshida A, Kobayashi E, et al. Successful treatment of extra-renal noncerebral rhabdoid tumors with VIDE. Pediatr Blood Cancer. 2016;63(2):352–354. doi:10.1002/pbc.25777
2. Tomlinson GE, Breslow NE, Dome J, et al. Rhabdoid tumor of the kidney in the National Wilms’ Tumor Study: age at diagnosis as a prognostic factor. J Clin Oncol. 2005;23:7641–7765. doi:10.1200/JCO.2004.00.8110
3. Mitchell C, Jones PM, Kelsey A, et al. The treatment of Wilms’ tumour: results of the United Kingdom Children’s cancer study group (UKCCSG) second Wilms’ tumour study. Br J Cancer. 2000;83:602–608. doi:10.1054/bjoc.2000.1338
4. Venkatramani R, Shoureshi P, Malvar J, et al. High dose alkylator therapy for extracranial malignant rhabdoid tumors in children. Pediatr Blood Cancer. 2014;61(8):1357–1361. doi:10.1002/pbc.25093
5. Joel E. Ultrastructure of malignant rhabdoid tumor of the kidney: a distinctive renal tumor of children. Hum Pathol. 1981.
6. Parham DM, Weeks DA, Beckwith JB. The clinicopathologic spectrum of putative extrarenal rhabdoid tumors. An analysis of 42 cases studied with immunohistochemistry or electron microscopy. Am J Surg Pathol. 1994;18(10):1010–1029. doi:10.1097/00000478-199410000-00005
7. Gururangan S, Bowman LC, Parham DM, et al. Primary extracranial rhabdoid tumors. Clinicopathologic features and response to ifosfamide. Cancer. 1993;71:2653.
8. Helmke L, Engler S, Mattke A, Henne-Bruns D. Extrarenal malignant rhabdoid tumors in childhood. Med Pediatr Oncol. 2001;36:317–319. doi:10.1002/1096-911X(20010201)36:2<317::AID-MPO1073>3.0.CO;2-W
9. Kodet R, Newton WA, Sachs N, et al. Rhabdoid tumors of soft tissues: a clinicopathologic study of 26 cases enrolled on the Intergroup Rhabdomyosarcoma Study. Hum Pathol. 1991;22:674–684. doi:10.1016/0046-8177(91)90289-2
10. Uwineza A, Gill H, Buckley P, et al. Rhabdoid tumor: the Irish experience 1986–2013. Cancer Genet. 2014;207(9):398–402. doi:10.1016/j.cancergen.2014.05.015
11. van den Heuvel—Eibrink MM, van Tinteren H, Rehorst H. Malignant rhabdoid tmours of the kidney(MRTKs), registered on recent SlOP protocols from 1993 to 2005:a report of the SIOP renal tumour study group. Pediatr Blood Cancer. 2011;56::733–737. doi:10.1002/pbc.22922
12. Wang Y, Zhang J, Wang Y, et al. Rhabdomyosarcoma in children: a clinical report. Chine J Practical Pediatrics. 2003;18(4):208–211.
13. Ladenstein R, Pötschger U, Le Deley MC, et al. Primary disseminated multifocal Ewing sarcoma: results of the Euro‐EWING 99 trial. J Clin Oncol. 2010;10:3284–3291. doi:10.1200/JCO.2009.22.9864
14. Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms tumors: results from the First National Wilms’ Tumor Study. Cancer. 1978;41(5):1937–1948. doi:10.1002/1097-0142(197805)41:5<1937::AID-CNCR2820410538>3.0.CO;2-U
15. Farmakis SG, Siegel MJ. Rhabdoid tumor: an aggressive renalmedullary tumor of childhood. J Comput Assist Tomogr. 2015;39(1):44–46. doi:10.1097/RCT.0000000000000195
16. Tomlinson GE, Breslow NE, Dome J, et al. Rhabdoid tumor of the kidney in the National Wilms’ Tumor Study: age at diagnosis as a prognostic factor. J Clin Oncol Official J Am Soc Clin Oncol. 2005;23(30):7641–7645.
17. Cai M, Tang J. Research progress of malignant rhabdomyosarcoma in children. Int J Blood Transfusion Hematol. 2016;39(2):169–173.
18. Weeks DA, Beckwith JB, Mierau GW, et al. Rhabdoid tumor of kidney. A report of 1 1 1 cases from the National Wilms’Tumor Study Pathology Center. Am J Surg Patrol. 1989;13:439458.
19. Savla J, Chen TT, Schneider NR, et al. Mutations of the hSNF5/INll gene in renal rhabdoid tumors with second primary brain tumors. J Natl Cancer Inst. 2000;92:648–650. doi:10.1093/jnci/92.8.648
20. Zhang N, He LJ. Clinicopathological features of malignant rhabdomyosarcoma in children. Chine J Pediatric Hematol Oncol. 2018;1(3):113–117.
21. Hasegawa T, Yamamoto S, Yokoyama R, et al. Prognostic significance of grading and staging systems using MIB-1 score in adult patients with soft tissue sarcoma of the extremities and trunk. Cancer. 2002;95(4):843–851. doi:10.1002/cncr.10728
22. Kohashi K, Oda Y. Oncogenic roles of sMARcBlINll and its deficienttumors. cancer Sci. 2017;108(4):547–552. doi:10.1111/cas.13173
23. Ramalingam P. Loss of expression of SMARCA4 (BRG1), SMARCA2 (BRM) and SMARCB1 (INI1) in undifferentiated carcinoma of the endometrium is not uncommon and is not always associated with rhabdoid morphology. Histopathol Official J Br Division Int Acad Pathol. 2017;70:359–366.
24. Bahrami A, Lee S, Caradine KD, et al. SMARCB1 deletion by a complex three-way chromosomal translocation in an extrarenal malignant rhabdoid tumor. Cancer Genet. 2014;207(9):437–440. doi:10.1016/j.cancergen.2014.08.002
25. Siegle JM, et al. SOX2 is a cancer-specific regulator of tumour initiating potential in cutaneous squamous cell carcinoma. Nat Commun. 2014;5:4511. doi:10.1038/ncomms5511
26. Justilien V, et al. The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer Cell. 2014;25:139–151. doi:10.1016/j.ccr.2014.01.008
27. Schröck A, Bode M, Göke FJM, et al. Expression and role of the embryonic protein SOX2 in head and neck squamous cell carcinoma. Carcinogenesis. 2014;35(7):1636–1642. doi:10.1093/carcin/bgu094
28. Geller JI, Roth JJ, Biegel JA. Biology and treatment of rthabdoid tumor. Crit Rev Oncog. 2015;20(3–4):199–216. doi:10.1615/CritRevOncog.2015013566
29. Kerl K, Holsten T, Fruhwald MC. Rhabdoid tumors: clinical approaches and molecular targets for innovative therapy. Pediatr Hematol Oncol. 2013;30(7):587–604. doi:10.3109/08880018.2013.791737
30. Hong CR, Kang HJ, Ju HY, et al. Extra-Cranial Malignant Rhabdoid Tumor in Children:A Single Institute Experience. Cancer ResTreat. 2015;47(4):889–896.
31. Furtwangler R, Kager L, Melchior P, et al. High-dosetreatment for malignant rhabdoid tumor of the kidney:No evidence for improved survival-The Gesellschaftfür P diatrische Onkologie und H matologie (GPOH) experience. Pediatr Blood Cancer. 2018;65(1):e26746. doi:10.1002/pbc.26746
32. Panandiker ASP, Merchant TE, Beltran C, et al. Sequencing of Local Therapy Affects the Pattern of Treatment Failure and Survival in Children With Atypical Teratoid Rhabdoid Tumors of the Central Nervous System. Int J Radiat Oncol Biol Phys. 2012;82(5):1756–1763. doi:10.1016/j.ijrobp.2011.02.059
33. Gounder MM, Stacchiotti S, Patrick S, et al. Phase 2 multicenter study of the EZH2 inhibitor tazemetostat in adults with INI1 negative epithelioid sarcoma (NCT02601950). J Clin Oncol. 2017;35(15_suppl):11058. doi:10.1200/JCO.2017.35.15_suppl.11058
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.