")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Carvedilol attenuates carbon tetrachloride-induced liver fibrosis and hepatic sinusoidal capillarization in mice
Authors Wu Y , Li Z , Xiu AY , Meng DX , Wang SN, Zhang CQ
Received 1 April 2019
Accepted for publication 30 June 2019
Published 1 August 2019 Volume 2019:13 Pages 2667—2676
DOI https://doi.org/10.2147/DDDT.S210797
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianbo Sun
Ying Wu,1 Zhen Li,2 Ai-Yuan Xiu,1 Dong-Xiao Meng,1 Si-Ning Wang,1 Chun-Qing Zhang1
1Department of Gastroenterology, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, Shandong Province, People’s Republic of China; 2Department of Health Digestion, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, Shandong Province, People’s Republic of China
Aim: To investigate the effect of carvedilol on liver fibrosis and hepatic sinusoidal capillarization in mice with carbon tetrachloride (CCl4)-induced fibrosis.
Methods: A liver fibrosis mouse model was induced by intraperitoneal CCl4 injection for 8 weeks. The mice were divided into five experimental groups: the normal group, the oil group, the CCl4 group, the CCl4+carvedilol (5 mg/kg/d) group, and the CCl4+carvedilol (10 mg/kg/d) group. The extent of liver fibrosis was evaluated by histopathological staining, and the changes in fenestrations of hepatic sinus endothelial cells were observed by scanning electron microscope (SEM). The expression of α-smooth muscle actin (α-SMA) and vascular endothelial markers was detected by immunohistochemistry and Western blot assays. The effect of carvedilol on cell apoptosis was studied via Terminal deoxynucleotidyl Transferase Mediated dUTP Nick End Labeling (TUNEL) assay, and the serum levels of matrix metalloproteinase-8 (MMP-8), vascular endothelial growth factor (VEGF), and angiopoietin-2 were detected through a Luminex assay.
Results: Liver fibrosis in CCl4-treated mice was attenuated by reduced accumulation of collagen and the reaction of inflammation with carvedilol treatment. Carvedilol reduced the activation of hepatic stellate cells (HSCs) and increased the number of apoptotic cells. The expression of α-SMA, CD31, CD34 and VWF (von Willebrand factor) was significantly decreased after carvedilol treatment. In addition, the number of fenestrae in the hepatic sinusoid showed notable differences between the groups, and the serum levels of MMP-8, VEGF and angiopoietin-2 were increased in the mice with liver fibrosis and reduced by carvedilol treatment.
Conclusion: The study demonstrated that carvedilol could prevent further development of liver fibrosis and hepatic sinusoidal capillarization in mice with CCl4-induced fibrosis.
Keywords: carvedilol, liver fibrosis, hepatic sinusoid, angiogenesis, hepatic stellate cell, mice
Introduction
Hepatic fibrosis, which is a common pathological process involved in the development of various chronic liver diseases, is far from an ideal effect in their effective prevention and treatment. Therefore, efficient and specific antifibrotic drugs are urgently needed.1 In recent years, targeted therapies for liver fibrosis, especially for activated hepatic stellate cells and other fibrogenic effector cells, have made remarkable progress.1–3 Liver fibrosis is the result of a chronic process of inflammation and tissue repair, that involves the accumulation of connective tissue.4,5 Hepatic stellate cell (HSC) is a specific pericyte located in the Disse space. During the development of hepatic fibrosis and portal hypertension, activated HSCs participate in the formation of fibrosis and the reconstruction of the intrahepatic structure through proliferation and secretion of extracellular matrix (ECM), and these cells could also increase the liver sinusoidal pressure through cell contraction.6–8 Therefore, the degradation of ECM and low-density basement membrane, and the clearance of HSCs through apoptosis are very valuable in the treatment of liver fibrosis.9
The normal hepatic sinusoid is composed of a flat layer of liver sinusoidal endothelial cells (LSECs), which have an open and high density fenestration structure. Endothelial transport in the liver sinusoidal endothelium occurs through fenestrae without a diaphragm.10,11 In liver fibrosis and cirrhosis, hepatic sinus capillarization is characterized by LSEC capillarization, phenotypic changes and the formation of a complete subendothelial basement membrane. These effects result in the formation of a new capillary network and thereby lead to changes in liver microcirculation function.12 The disappearance of the normal filtration barrier in the cirrhotic liver leads to an impaired bidirectional exchange between sinusoidal blood and parenchymal cells. Therefore, capillarization of the sinusoidal endothelium might be an important cause of hepatic failure in cirrhosis.11 Moreover, previous studies have revealed that the relationship between liver fibrosis and endothelial-mediated angiogenesis seriously impedes the reversal and remission of liver fibrosis.13,14 Antiangiogenic therapy might be a strategy for the treatment of hepatic fibrosis and portal hypertension.8,15,16
Carvedilol is known as an adrenergic receptor blocker that blocks sympathetic neural activation through the antagonism of β1-, β2- and α1-adrenoreceptors. At present, carvedilol is clinically used to reduce portal vein pressure and prevent esophageal and gastric variceal bleeding, which is a complication of liver cirrhosis.17,18 Studies have shown that carvedilol can reduce oxidative stress, the inflammatory response and fibrosis in alcohol-induced liver injury.19–21 In addition, our previous study indicated that carvedilol might alleviate liver fibrosis by inhibiting angiogenesis in a human umbilical vascular endothelial cell (HUVEC) model.22 The present study further investigated the effects of carvedilol on liver fibrosis and hepatic sinusoidal capillarization in mice with CCl4-induced fibrosis.
Materials and methods
Mouse model of liver fibrosis
Carvedilol was purchased from Solarbio Life Science Company (Beijing, China) and dissolved to different concentrations. Fifty adult male C57BL/6J mice weighing 18–20 g were obtained from the Experimental Animal Center of Shandong University (Jinan, China). The animals were kept in an air-conditioned room at 23–25 °C with a 12 hours dark/light cycle for one week prior to the initiation of the experiments. All the animals received appropriate care during the study with free access to food and water. The study was approved by the Shandong University Animal Care Committee and followed the guiding principles for the care and use of laboratory animals adopted by the committee.
Liver fibrosis in mice was induced through twice weekly intraperitoneal injections of CCl4 (25%, 0.5 ml/100 g) for 8 weeks. The mice were randomly distributed into five groups: 1) normal control group; 2) olive oil group, the mice were intraperitoneally injected with olive oil for 8 weeks; 3) CCl4 group, the mice were intraperitoneally injected with CCl4 for 8 weeks; 4) CCl4+ carvedilol (5 mg/kg/d, by gavage) group, the mice were administered CCl4 alone for 2 weeks and then both carvedilol and CCl4 for 6 weeks; and 5) CCl4+ carvedilol (10 mg/kg/d, by gavage) group, the mice were administered CCl4 alone for 2 weeks and then both carvedilol and CCl4 for 6 weeks. The mice in group 2 were administered olive oil intraperitoneally at the same dose and under the same condition as those used for the CCl4-treated group.
The mice were euthanized, and liver samples were removed and snap-frozen in liquid nitrogen for storage at −80 °C. An additional section was fixed in 4% paraformaldehyde for 24 hours and embedded in paraffin.
Histological examination
The liver tissue sections were stained with hematoxylin and eosin (H&E) for morphological evaluation. Masson trichrome and Sirius Red staining were used to assess the degree of liver fibrosis. Sirius Red staining was analyzed by Image-Pro Plus software 6.0.
Immunohistochemical staining
The liver tissues embedded in paraffin were sliced into 4–6 mm pieces, deparaffinized and serially dehydrated in ethanol. The sections were boiled for 3 minutes while immersed in EDTA-Tris (pH 9.0) for antigen retrieval and then treated with hydrogen peroxide for 30 minutes to inactivate the endogenous peroxidase. The sections were incubated overnight at 4 °C with primary antibodies, which included antibodies against α-SMA (1:400; Abcam, Cambridge, MA, USA), CD31 (1:2000; Abcam), CD34 (1:2500; Abcam) and von Willebrand factor (VWF) (1:200; Proteintech, Wuhan, China). The sections were subsequently incubated with the appropriate biotinylated secondary (goat anti-rabbit IgG and goat anti-mouse IgG; Origene Technologies, Beijing, China) for 30 minutes at room temperature, stained with Diaminobenzidine (DAB) and counterstained with hematoxylin. The positive areas were colored a brownish yellow. Image-Pro Plus software 6.0 was used for the immunohistochemical analysis.
Scanning electron microscope analysis
Freshly collected specimens were immediately immobilized in electron microscope fixation solution (Wuhan Servicebio Technology Ltd., Wuhan, China) for 2 hours and then transferred to 4 °C. The tissues were immobilized in 1% osmic acid buffer for 2 hours, serially dehydrated in ethanol and dried in a critical point drier (K850; Quorum Technologies Ltd, Lewes, United Kingdom). Then images were obtained under a scanning electron microscope (SU8010; Hitachi, Tokyo, Japan) after conductive treatment using an ion sputtering analyzer (MSP-2S; IXRF Sytems, Inc., Austin, TX, USA). The sections were observed by SEM to analyze the fenestrated structure of the hepatic sinus endothelium.
Apoptosis detection by TUNEL staining and immunofluorescent staining
In situ detection of DNA fragmentation was performed using TUNEL staining of the paraffin-embedded liver sections of mice. During apoptosis, DNA is degraded into 50–300 kb fragments by endogenous nucleic acid hydrolase. The breakage of DNA leads to the formation of a series of 3′DNA hydroxyl groups, and these groups serve as substrates for the nucleotide transferase (TdT), which can attach nucleotides to the 3′DNA hydroxyl groups. Apoptosis was detected by labeling nucleotides with TUNEL stain, using in situ cell death detection kit (Roche, Mannheim, Germany) in accordance with the manufacturer’s recommended protocol. Immunofluorescent staining was performed with primary antibodies against α-SMA (1:500; Abcam) and with fluorescence-conjugated secondary antibodies (Wuhan Servicebio Technology Ltd.). The nucleus was stained with 4ʹ,6-diamidino-2-phenylindole (DAPI) reagent (Wuhan Servicebio Technology Ltd.).
Western blot analysis
Liver samples were homogenized with RIPA lysis buffer (Solarbio Life Science, Beijing, China). After centrifugation, the proteins were extracted for Western blot assay. The protein concentrations were detected using the bicinchoninic acid (BCA) protein determination method. The protein samples were electrophoresed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes (Merk Millipore, Burlington, MA, USA). The membranes were blocked with 5% fat-free milk at room temperature for 1 hour, incubated at 4 °C overnight with the primary antibodies, and were incubated with the secondary antibodies for 1 hour at room temperature. The membranes were then subjected to three 10-minutes washes with TBST, and the antibody-bound proteins were detected using an enhanced chemiluminescence assay kit (Merk Millipore). The results were quantified by using ImageJ software.
Luminex analysis
Blood samples were collected from the mice, allowed to clot at room temperature and centrifuged at 4 °C (3000 rpm) for 10 minutes. The supernatants were stored at −80 °C until use. The relevant biomarkers in mouse serum, including MMP-8, VEGF and angiopoietin-2 were measured using a Luminex System (LXSAMSM-09, R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. The data were collected and the significances of the differences among the groups were analyzed.
Statistical analysis
The results are expressed as mean±standard deviation. The data were statistically analyzed by one-way ANOVA and Student’s t-test as appropriate. A significant difference was considered at P<0.05.
Results
Carvedilol attenuated liver fibrosis in mice
The therapeutic effect of carvedilol on hepatic fibrosis was evaluated by H&E (Figure 1A), Masson trichrome (Figure 1B) and Sirius Red staining (Figure 1C and D). After the mice were administered CCl4 for 8 weeks, their liver tissues exhibited hepatocellular degeneration and inflammatory cell infiltration (Figure 1A). Moreover, Masson trichrome and Sirius Red staining showed increased hyperplasia of the fibrous tissues and the formation of fibrous septa in CCl4 group. In contrast, the carvedilol-treated group showed significant decreases in the amounts of fibrosis tissues and inflammatory cells. Moreover, the high dose (10 mg/kg/d) of carvedilol was more effective than the low dose (5 mg/kg/d). Sirius Red staining under polarized light revealed the deposition of collagen type I (in red) and collagen type III (in green) in the CCl4 group (Figure 1C). Nevertheless, the degree of collagen deposition in the interlobular septum and periportal vein was significantly decreased with carvedilol treatment. (Figure 1C, D and E)
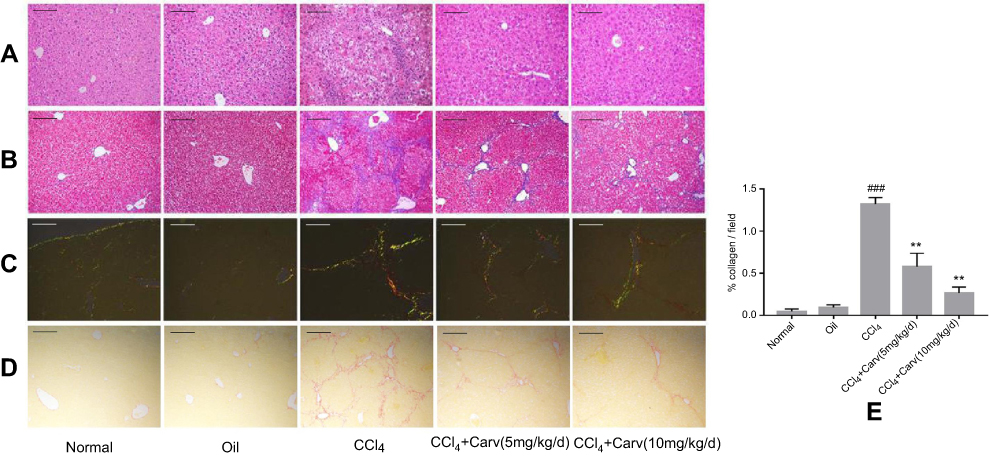 |
Figure 1 Carvedilol attenuated the extent of liver fibrosis in CCl4-treated mice. (A) Representative photomicrographs of liver sections stained with H&E; (B) representative photomicrographs of liver sections stained with Masson trichrome; (C, D) collagen deposition was evaluated by Sirius Red staining under polarized and normal light. Magnification ×100. Scale bars: 200 μm. (E) Collagen deposition in Sirius Red staining was quantified using Image-Pro Plus software. ###P<0.001 vs oil group; **P<0.01 vs CCl4 group; P<0.01 CCl4+Carv (5 mg/kg/d) group vs CCl4+Carv (10 mg/kg/d) group. Group 1, normal; group 2, oil; group 3, CCl4; group 4, CCl4+Carv (5 mg/kg/d); group 5, CCl4+Carv (10 mg/kg/d).Abbreviations: CCl4, carbon tetrachloride; H&E, hematoxylin and eosin; Carv, carvedilol. |
Effect of carvedilol on HSC activation and cell apoptosis
The activation of HSCs can induce α-SMA expression, strengthen their proliferation and increase collagen synthesis. In our study, α-SMA was used for evaluating the effect of carvedilol on HSC activation. Immunohistochemistry staining revealed no obvious positive staining in the normal control and olive oil groups. The number of α-SMA positive cells was significantly increased in the CCl4 group and decreased by carvedilol treatment in a dose-dependent manner (Figure 2). A Western blot analysis further demonstrated that the expression of α-SMA was markedly reduced in the carvedilol-treated group compared with the CCl4 group (Figure 3B). The number of apoptotic cells was evaluated by a TUNEL assay and immunofluorescent analysis, and the results showed that the apoptosis of HSCs in the carvedilol-treated group was higher than that in the CCl4 group (Figure 4). The levels of Bax and Bcl-2, which are used as apoptosis-related indexes, were detected by Western blot assay, and the data showed that carvedilol significantly promoted the expression of the pro-apoptotic factor Bax and inhibited the expression of the anti-apoptotic factor Bcl-2. Moreover, with the increase of the dose, the pro-apoptotic effect of carvedilol was more significant (P<0.05) (Figure 3C and D). The results demonstrated that the mitochondrial apoptosis pathway regulated by Bcl-2 family proteins was involved in the carvedilol-induced process of apoptosis. In summary, the results suggested that carvedilol suppressed the activation of HSCs and promoted cell apoptosis.
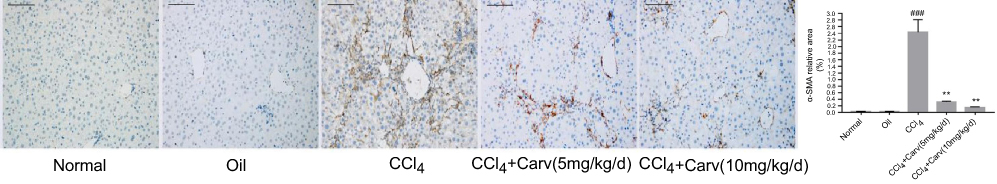 |
Figure 2 The expression of α-SMA was detected by immunohistochemical staining (magnification ×200). Scale bars: 100 μm. Carvedilol effectively inhibited the expression of α-SMA. ###P<0.001 vs oil group; **P<0.01 vs CCl4 group; P<0.01 CCl4+Carv (5 mg/kg/d) group vs CCl4+Carv (10 mg/kg/d) group.Abbreviations: α-SMA, α-smooth muscle actin; CCl4, carbon tetrachloride; Carv, carvedilol. |
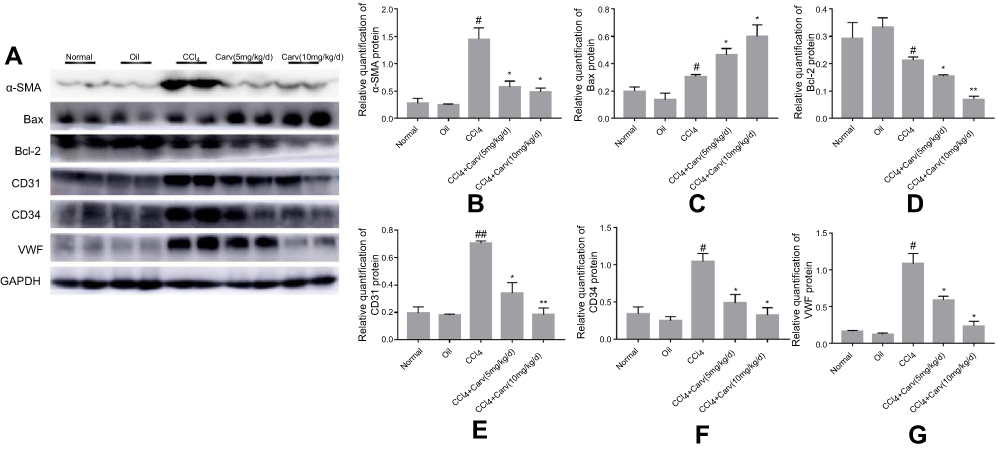 |
Figure 3 Western blot analyses of α-SMA, Bax, Bcl-2, CD34, CD31 and VWF. (A) Protein expression bands of each index detected by Western blot. (B) The expression of α-SMA was significantly decreased after carvedilol treatment. #P<0.05 vs oil group; *P<0.05 vs CCl4 group. (C) The expression of Bax was increased after carvedilol treatment. #P<0.05 vs oil group; *P<0.05 vs CCl4 group. (D) The expression of Bcl-2 was decreased after carvedilol treatment. #P<0.05 vs oil group; *P<0.05 vs CCl4 group; **P<0.01 vs CCl4 group. (E) The expression of CD31 was decreased after carvedilol treatment. ##P<0.01 vs oil group; *P<0.05 vs CCl4 group; **P<0.01 vs CCl4 group. (F, G) The expression of CD34 and VWF was decreased after carvedilol treatment. #P<0.05 vs oil group; *P<0.05 vs CCl4 group.Abbreviations: α-SMA, α-smooth muscle actin; CCl4, carbon tetrachloride; Carv, carvedilol; Bcl-2, B-cell lymphoma-2; VWF, von Willebrand factor. |
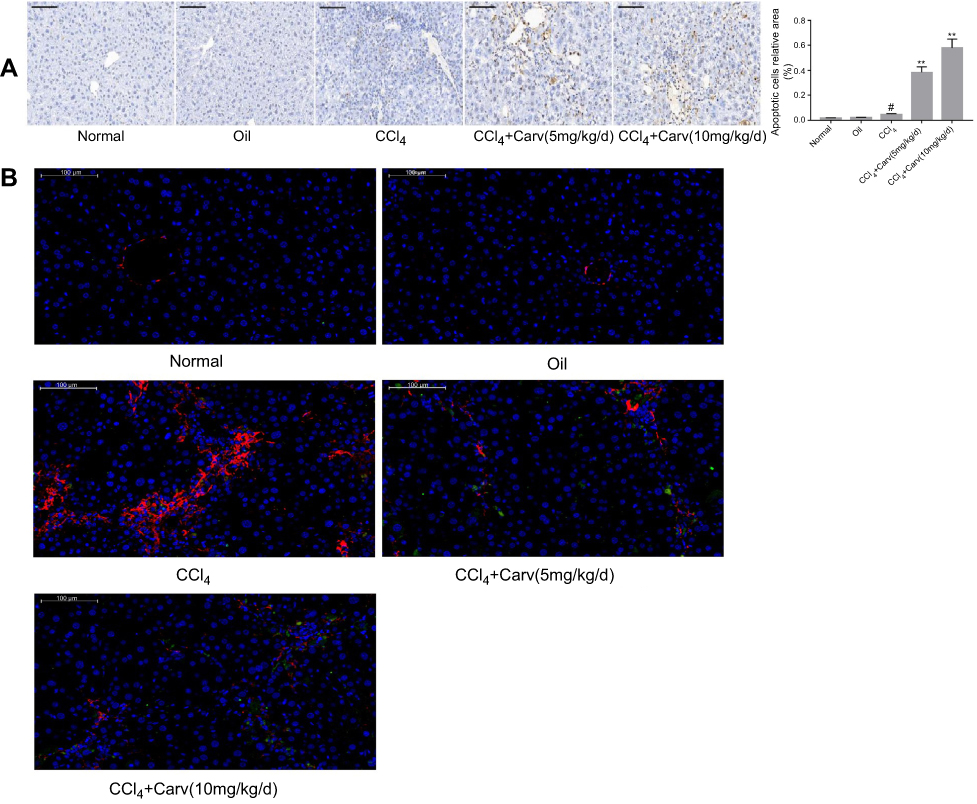 |
Figure 4 (A) Apoptotic cells were detected by TUNEL staining (magnification ×200). The number of apoptotic cells was markedly increased in carvedilol-treated group. #P<0.05 vs oil group; **P<0.01 vs CCl4 group; P<0.05 CCl4+Carv (5 mg/kg/d) group vs CCl4+Carv (10 mg/kg/d) group. (B) Immunofluorescent double staining of TUNEL and α-SMA (magnification ×200). The nucleus stained by DAPI was blue, the tunel kit was labeled with FITC luciferin, the positive apoptotic cells were green, and α-SMA displayed red. Scale bars: 100μm. Abbreviations: TUNEL, terminal deoxynucleotidyl transferase mediated dUTP nick end labeling; α-SMA, α-smooth muscle actin; CCl4, carbon tetrachloride; Carv, carvedilol; DAPI, 4',6-diamidino-2-phenylindole; FITC, fluorescein isothiocyanate. |
The changes of liver sinusoidal endothelial cell fenestrae under SEM
The SEM results showed that the surface of normal LSECs had fenestrae in a sieve arrangement. The number of fenestrae was significantly reduced and even disappeared in the mice with CCl4-induced liver fibrosis. The results also showed that the number of fenestrae in the carvedilol group was increased compared with that in the CCl4 group. (Figure 5)
 |
Figure 5 Low magnification scanning electron micrograph of LSEC from mice liver showing the fenestrae. Magnification ×3500. Scale bars: 10 μm.Abbreviations: LSEC, liver sinusoidal endothelial cell. |
Assessment of the effect of carvedilol on the expression of CD34, CD31 and VWF by immunohistochemistry assay
CD34, CD31 and VWF are commonly used as vascular endothelial markers and these are rarely expressed in normal liver. However, the expression of these indexes increases after hepatic sinusoid capillarization. We detected the expression of CD34, CD31, and VWF by immunohistochemistry staining. The results showed slight positive staining in the normal and olive oil groups. In contrast, the injection of CCl4 for 8 weeks significantly increased the positive staining areas, whereas carvedilol treatment effectively decreased the expression of these markers (Figure 6). Furthermore, the result showed that the inhibitory effect of carvedilol was enhanced with the increase of doses.
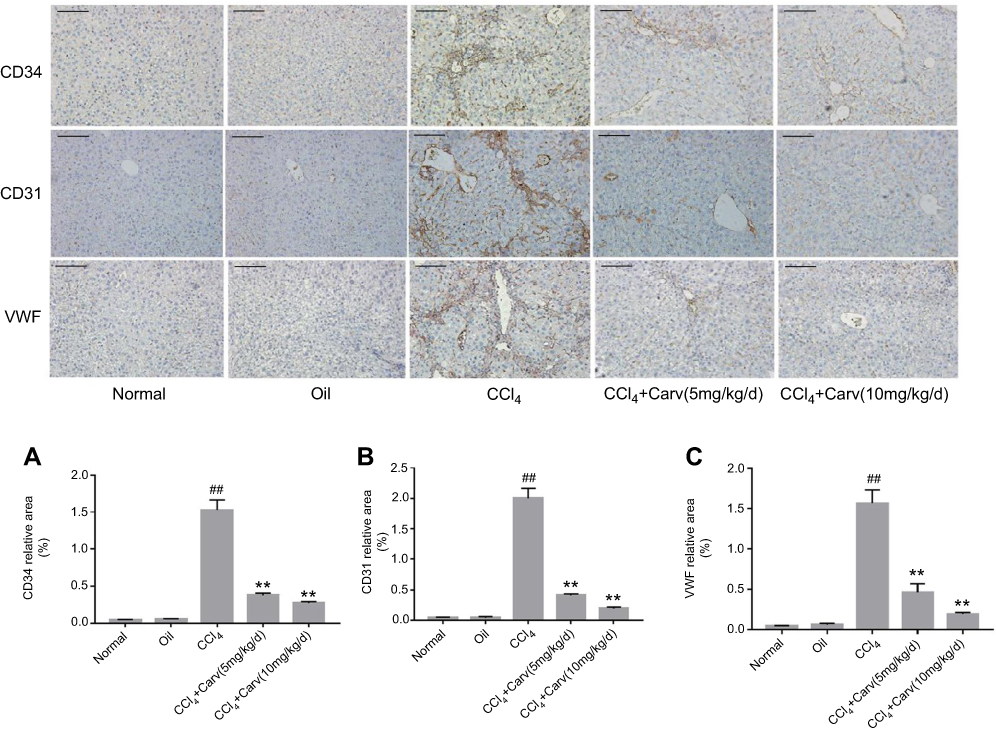 |
Figure 6 (A) Expression of CD34 by immunohistochemical staining (magnification ×200). (B) Expression of CD31 by immunohistochemical staining (magnification ×200). (C) Expression of VWF by immunohistochemical staining (magnification ×200). Scale bars: 100 μm. ##P<0.01 vs oil group; **P<0.01 vs CCl4 group; P<0.01 CCl4+Carv (5 mg/kg/d) group vs CCl4+Carv (10 mg/kg/d) group. Group 1, normal; group 2, oil; group 3, CCl4; group 4, CCl4+Carv (5 mg/kg/d); group 5, CCl4+Carv (10 mg/kg/d). Abbreviations: CCl4, carbon tetrachloride; Carv, carvedilol; VWF, von Willebrand factor. |
Assessment of the effect of carvedilol on the expression of CD34, CD31 and VWF by Western blot analysis
The present study showed that the protein expression levels of CD34, CD31 and VWF were significantly higher in the CCl4 group than in the control group, but they were markedly decreased in the carvedilol-treated group (Figure 3A, E, F and G). The results suggested that these vascular endothelial markers were clearly increased in fibrotic liver tissues and that carvedilol could inhibit the expression of these proteins.
Analysis of serum biomarkers by luminex assay
MMP-8 is a member of the MMP family of neutral proteinases and exerts an effect on the digestion of type I and type III collagens.23 VEGF can promote the proliferation of endothelial cells and has been demonstrated to play an important role in angiogenesis.24 In addition to VEGF, the angiopoietin family is also involved in vascular remodeling.25 The present results showed that the MMP-8, VEGF and angiopoietin-2 levels were significantly increased in the mice with CCl4-induced liver fibrosis compared with the levels in the olive oil and control groups (P<0.001). In contrast, the MMP-8, VEGF and angiopoietin-2 levels in the CCl4+carvedilol (10 mg/kg/d) group were lower than those in the CCl4 group (P<0.05) (Figure 7).
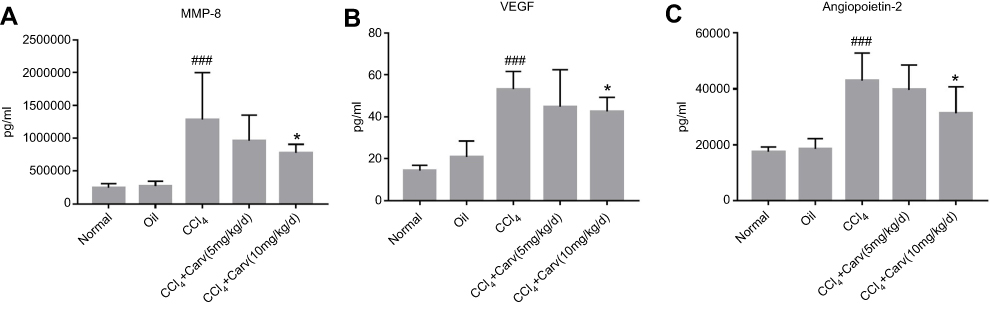 |
Figure 7 Serum levels of relevant biomarkers were detected in a luminex assay. (A) MMP-8; (B) VEGF; (C) angiopoietin-2. ###P<0.001 vs oil group; *P<0.05 vs CCl4 group. Abbreviations: MMP-8, matrix metalloproteinase-8; VEGF, vascular endothelial growth factor; CCl4, carbon tetrachloride. |
Discussion
Liver fibrosis is a complex pathological process that leads to the development of chronic liver disease and cirrhosis.26,27 Liver cirrhosis not only leads to hepatic functional failure but also causes a series of complications.28 Various drugs have been introduced to cure liver fibrosis, but these treatments are not highly effective. Thus, the development of novel therapies for suppressing liver fibrosis is urgently needed. Carvedilol, a nonselective beta blocker (NSBB), is currently recommended for reducing portal venous pressure in chronic liver diseases.17,29,30 Previous studies have shown that carvedilol exerts anti-inflammatory and antioxidant effects in ethanol-induced liver injury.19,20 In the present study, a model of liver fibrosis induced by CCl4 was established in mice, and the effects of carvedilol at different concentrations on hepatic fibrosis and hepatic sinusoidal capillaries were studied.
The establishment of a liver fibrosis model was verified by pathological staining. H&E and Masson trichrome staining revealed that successful establishment of liver fibrosis in the mice that were administered intraperitoneal injections of CCl4 for 8 weeks. After six weeks of carvedilol administration via gavage, the extent of hepatocyte inflammation, collagen deposition and hepatic fibrosis was significantly reduced. Then, we further investigated the anti-fibrosis mechanism of carvedilol. It is known that HSCs play a pivotal role in the progression of liver fibrosis. HSCs are localized in the space of Disse and account for 5–8% of total liver cells. The occurrence of liver injury activates HSCs, and these activated cells then begin to proliferate and release a large number of extracellular matrix components, such as collagen, which results in modification of the hepatic structure.31–33 The activation of HSCs induces changes in their morphology, the loss of their retinoid droplets and increases in the α-SMA expression.34 Therefore, α-SMA is considered an important marker for the evaluation of HSC activation and proliferation.35 In the present study, the expression of α-SMA was detected by immunohistochemical staining and Western blot assay. The results demonstrated that HSC activation was inhibited by carvedilol administration in a dose-dependent manner. On the other hand, induction of apoptosis can clear activated HSCs, thereby alleviating liver fibrosis.36 HSC apoptosis is key to the reversal of liver fibrosis, and TUNEL staining showed that carvedilol increased the apoptosis of cells, mainly including HSCs. In addition, α-SMA was used to localize HSCs in immunofluorescence analysis, and the results showed that the apoptosis rate of HSCs in the carvedilol-treated group was significantly higher than that in the CCl4 group. The study indicated that carvedilol could alleviate liver fibrosis by inducing HSCs apoptosis. The Bcl-2 family proteins located at the outer membrane of mitochondria play an important role in apoptosis. The detection of Bcl-2 family proteins by Western blot analysis further confirmed the pro-apoptotic effect of carvedilol. Based on the results of the present study, we believe that carvedilol might prevent the progression of hepatic fibrosis by reducing the activation and proliferation of HSCs and increasing apoptosis.
In the early stage of chronic liver disease, dysfunction of liver microcirculation occurs due to inflammation and oxidative stress, and the phenotype of LSECs changes.37 Fenestrae are considered a characteristic feature of LSECs. In various liver injuries such as liver fibrosis, LSECs lose their specialized morphology, resulting in the disappearance of fenestrae, and develop capillarization.38,39 It has been reported that LSEC capillarization promotes HSC activation and fibrosis and the reversal of LSEC capillarization could promote the quiescence of HSCs.40 The interaction between LSECs and HSCs increases collagen deposition, promotes fibrogenesis and angiogenesis.33 Furthermore, studies have shown that sinusoidal remodeling and intrahepatic angiogenesis are related to sinusoidal resistance, fibrosis and portal hypertension.16,41 In the present study, LSEC fenestrae within the sinusoids were reduced or lost in the CCl4-treated mice. Moreover, immunohistochemistry and Western blot analyses demonstrated that the expression levels of vascular endothelial markers, such as CD31, CD34 and VWF, were markedly increased in the CCl4-treated group. However, carvedilol decreased the expression of vascular endothelial markers and effectively attenuated sinusoidal capillarization. Therefore, the results suggested that carvedilol could improve sinusoidal remodeling, inhibit intrahepatic angiogenesis, and thus alleviate the pressure in the hepatic sinus.
In addition, MMP-8 was considered to promote collagen breakdown.42 The data confirmed that, along with more HSCs were activated in CCl4-treated mice, the level of MMP-8 in these mice was increased to degrade the collagen. Nevertheless, with carvedilol treatment, the degree of collagen deposition was decreased and thus the level of MMP-8 was reduced. In addition, liver fibrosis can induce a hypoxic environment, which promotes the production of angiogenic factors, including VEGF and angiopoietin, and these factors lead to increased angiogenesis.43–45 Excessive levels of ECM deposition and angiogenic factors lead to LSEC capillarization, which results in increased vascular resistance and portal hypertension.46 The data demonstrated that the levels of angiogenic factors, including VEGF and angiopoietin-2, were significantly increased in the serum of CCl4-treated mice. However, the production of VEGF and angiopoietin-2 was reduced by carvedilol treatment, which suggested that the processes of angiogenesis and LSEC capillarization was inhibited in the carvedilol-treated mice.
Conclusion
In summary, carvedilol can prevent further development of CCl4-induced liver fibrosis in mice by inhibiting HSC activation and increasing cell apoptosis. Moreover, carvedilol can alter the LSEC phenotype and reduce sinusoidal capillarization, which might further alleviate liver fibrosis and portal hypertension. Thus, carvedilol exerts potential anti-fibrosis and anti-angiogenic effects.
Acknowledgment
The research was supported by National Natural Science Foundation of China, No. 81370590.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Schuppan D, Ashfaq-Khan M, Yang AT, Kim YO. Liver fibrosis: direct antifibrotic agents and targeted therapies. Matrix Biol. 2018;68–69:435–51.
2. Zhao Z, Li Y, Jain A, et al. Development of a peptide-modified siRNA nanocomplex for hepatic stellate cells. Nanomedicine. 2018;14(1):51–61. doi:10.1016/j.nano.2017.08.017
3. Jain A, Barve A, Zhao Z, Jin W, Cheng K. Comparison of avidin, neutravidin, and streptavidin as nanocarriers for efficient siRNA delivery. Mol Pharm. 2017;14(5):1517–1527. doi:10.1021/acs.molpharmaceut.6b00933
4. Ueha S, Shand FH, Matsushima K. Cellular and molecular mechanisms of chronic inflammation-associated organ fibrosis. Front Immunol. 2012;3:71. doi:10.3389/fimmu.2012.00198
5. Munoz-Ortega MH, Llamas-Ramirez RW, Romero-Delgadillo NI, et al. Doxazosin treatment attenuates carbon tetrachloride-induced liver fibrosis in hamsters through a decrease in transforming growth factor beta secretion. Gut Liver. 2016;10(1):101–108. doi:10.5009/gnl14459
6. Kisseleva T. The origin of fibrogenic myofibroblasts in fibrotic liver. Hepatology. 2017;65(3):1039–1043. doi:10.1002/hep.v65.3
7. Mimche PN, Lee CM, Mimche SM, et al. EphB2 receptor tyrosine kinase promotes hepatic fibrogenesis in mice via activation of hepatic stellate cells. Sci Rep. 2018;8(1):2532. doi:10.1038/s41598-018-20926-9
8. Thabut D, Shah V. Intrahepatic angiogenesis and sinusoidal remodeling in chronic liver disease: new targets for the treatment of portal hypertension? J Hepatol. 2010;53(5):976–980. doi:10.1016/j.jhep.2010.01.032
9. Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134(6):1655–1669. doi:10.1053/j.gastro.2008.01.074
10. Cogger VC, McNerney GP, Nyunt T, et al. Three-dimensional structured illumination microscopy of liver sinusoidal endothelial cell fenestrations. J Struct Biol. 2010;171(3):382–388. doi:10.1016/j.jsb.2010.06.001
11. Braet F, Wisse E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: a review. Comp Hepatol. 2002;1(1):1. doi:10.1186/1476-5926-1-1
12. Yao Q, Lin Y, Li X, Shen X, Wang J, Tu C. Curcumin ameliorates intrahepatic angiogenesis and capillarization of the sinusoids in carbon tetrachloride-induced rat liver fibrosis. Toxicol Lett. 2013;222(1):72–82. doi:10.1016/j.toxlet.2013.06.240
13. Decaris ML, Li KW, Emson CL, et al. Identifying nonalcoholic fatty liver disease patients with active fibrosis by measuring extracellular matrix remodeling rates in tissue and blood. Hepatology. 2017;65(1):78–88. doi:10.1002/hep.28860
14. Kantari-Mimoun C, Castells M, Klose R, et al. Resolution of liver fibrosis requires myeloid cell-driven sinusoidal angiogenesis. Hepatology. 2015;61(6):2042–2055. doi:10.1002/hep.27635
15. Li Z, Ding Q, Ling LP, et al. Metformin attenuates motility, contraction, and fibrogenic response of hepatic stellate cells in vivo and in vitro by activating AMP-activated protein kinase. World J Gastroenterol. 2018;24(7):819–832. doi:10.3748/wjg.v24.i7.819
16. Kong LJ, Li H, Du YJ, et al. Vatalanib, a tyrosine kinase inhibitor, decreases hepatic fibrosis and sinusoidal capillarization in CCl4-induced fibrotic mice. Mol Med Rep. 2017;15(5):2604–2610. doi:10.3892/mmr.2017.6325
17. Bhardwaj A, Kedarisetty CK, Vashishtha C, et al. Carvedilol delays the progression of small oesophageal varices in patients with cirrhosis: a randomised placebo-controlled trial. Gut. 2017;66(10):1838–1843. doi:10.1136/gutjnl-2016-311735
18. de Franchis R. Revising consensus in portal hypertension: report of the Baveno V consensus workshop on methodology of diagnosis and therapy in portal hypertension. J Hepatol. 2010;53(4):762–768. doi:10.1016/j.jhep.2010.01.032
19. Araujo Junior RF, Garcia VB, Leitao RF, et al. Carvedilol improves inflammatory response, oxidative stress and fibrosis in the alcohol-induced liver injury in rats by regulating kuppfer cells and hepatic stellate cells. PLoS One. 2016;11(2):e0148868. doi:10.1371/journal.pone.0148868
20. Hakucho A, Liu J, Liu X, Fujimiya T. Carvedilol improves ethanol-induced liver injury via modifying the interaction between oxidative stress and sympathetic hyperactivity in rats. Hepatol Res. 2014;44(5):560–570. doi:10.1111/hepr.2014.44.issue-5
21. Liu J, Takase I, Hakucho A, Okamura N, Fujimiya T. Carvedilol attenuates the progression of alcohol fatty liver disease in rats. Alcohol Clin Exp Res. 2012;36(9):1587–1599. doi:10.1111/j.1530-0277.2012.01773.x
22. Ding Q, Tian XG, Li Y, Wang QZ, Zhang CQ. Carvedilol may attenuate liver cirrhosis by inhibiting angiogenesis through the VEGF-Src-ERK signaling pathway. World J Gastroenterol. 2015;21(32):9566–9576. doi:10.3748/wjg.v21.i10.2937
23. Baig MS, Yaqoob U, Cao S, Saqib U, Shah VH. Non-canonical role of matrix metalloprotease (MMP) in activation and migration of hepatic stellate cells (HSCs). Life Sci. 2016;155:155–160. doi:10.1016/j.lfs.2016.04.031
24. Fernandez M, Semela D, Bruix J, Colle I, Pinzani M, Bosch J. Angiogenesis in liver disease. J Hepatol. 2009;50(3):604–620. doi:10.1016/j.jhep.2008.12.011
25. An YA, Sun K, Joffin N, et al. Angiopoietin-2 in white adipose tissue improves metabolic homeostasis through enhanced angiogenesis. eLife. 2017;6. doi:10.7554/eLife.24071
26. Jiang H, Qin XJ, Li WP, Ma R, Wang T, Li ZQ. Effects of Shu Gan Jian Pi formula on rats with carbon tetrachlorideinduced liver fibrosis using serum metabonomics based on gas chromatographytime of flight mass spectrometry. Mol Med Rep. 2017;16(4):3901–3909. doi:10.3892/mmr.2017.7078
27. Popov Y, Schuppan D. Targeting liver fibrosis: strategies for development and validation of antifibrotic therapies. Hepatology. 2009;50(4):1294–1306. doi:10.1002/hep.23123
28. Nakano Y, Nakao S, Sumiyoshi H, et al. Identification of a novel alpha-fetoprotein-expressing cell population induced by the Jagged1/Notch2 signal in murine fibrotic liver. Hepatol Commun. 2017;1(3):215–229. doi:10.1002/hep4.1103
29. Tripathi D, Hayes PC. Beta-blockers in portal hypertension: new developments and controversies. Liver Int. 2014;34(5):655–667. doi:10.1111/liv.2014.34.issue-5
30. Li T, Ke W, Sun P, et al. Carvedilol for portal hypertension in cirrhosis: systematic review with meta-analysis. BMJ Open. 2016;6(5):e010902. doi:10.1136/bmjopen-2015-010902
31. Ding Q, Li Z, Liu B, Ling L, Tian X, Zhang C. Propranolol prevents liver cirrhosis by inhibiting hepatic stellate cell activation mediated by the PDGFR/Akt pathway. Hum Pathol. 2018;76:37–46. doi:10.1016/j.humpath.2018.02.018
32. Deleve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology. 2008;48(3):920–930. doi:10.1002/hep.v48:3
33. Gracia-Sancho J, Maeso-Diaz R, Fernandez-Iglesias A, Navarro-Zornoza M, Bosch J. New cellular and molecular targets for the treatment of portal hypertension. Hepatol Int. 2015;9(2):183–191. doi:10.1007/s12072-015-9613-5
34. Lee TF, Mak KM, Rackovsky O, et al. Downregulation of hepatic stellate cell activation by retinol and palmitate mediated by adipose differentiation-related protein (ADRP). J Cell Physiol. 2010;223(3):648–657.
35. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88(1):125–172.
36. Zhang X, Zhang F, Kong D, et al. Tetramethylpyrazine inhibits angiotensin II-induced activation of hepatic stellate cells associated with interference of platelet-derived growth factor beta receptor pathways. Febs J. 2014;281(12):2754–2768. doi:10.1111/febs.12758
37. DeLeve LD, Wang X, Hu L, McCuskey MK, McCuskey RS. Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am J Physiol Gastrointest Liver Physiol. 2004;287(4):G757–763. doi:10.1152/ajpgi.00017.2004
38. Elvevold K, Smedsrod B, Martinez I. The liver sinusoidal endothelial cell: a cell type of controversial and confusing identity. Am J Physiol Gastrointest Liver Physiol. 2008;294(2):G391–400. doi:10.1152/ajpgi.00167.2007
39. Xie G, Choi SS, Syn WK, et al. Hedgehog signalling regulates liver sinusoidal endothelial cell capillarisation. Gut. 2013;62(2):299–309. doi:10.1136/gutjnl-2011-301494
40. DeLeve LD. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology. 2015;61(5):1740–1746. doi:10.1002/hep.27376
41. Huebert RC, Jagavelu K, Liebl AF, et al. Immortalized liver endothelial cells: a cell culture model for studies of motility and angiogenesis. Lab Invest. 2010;90(12):1770–1781. doi:10.1038/labinvest.2010.132
42. Prystupa A, Boguszewska-Czubara A, Bojarska-Junak A, Torun-Jurkowska A, Rolinski J, Zaluska W. Activity of MMP-2, MMP-8 and MMP-9 in serum as a marker of progression of alcoholic liver disease in people from Lublin Region, eastern Poland. Ann Agric Environ Med. 2015;22(2):325–328. doi:10.5604/12321966.1152088
43. Lin HC, Huang YT, Yang YY, et al. Beneficial effects of dual vascular endothelial growth factor receptor/fibroblast growth factor receptor inhibitor brivanib alaninate in cirrhotic portal hypertensive rats. J Gastroenterol Hepatol. 2014;29(5):1073–1082. doi:10.1111/jgh.12480
44. Bona S, Rodrigues G, Moreira AJ, et al. Antifibrogenic effect of melatonin in rats with experimental liver cirrhosis induced by carbon tetrachloride. JGH Open. 2018;2(4):117–123. doi:10.1002/jgh3.12055
45. Novo E, Cannito S, Zamara E, et al. Proangiogenic cytokines as hypoxia-dependent factors stimulating migration of human hepatic stellate cells. Am J Pathol. 2007;170(6):1942–1953. doi:10.2353/ajpath.2007.060031
46. Mallat A, Lotersztajn S. Cellular mechanisms of tissue fibrosis. 5. Novel insights into liver fibrosis. Am J Physiol Cell Physiol. 2013;305(8):C789–799. doi:10.1152/ajpcell.00026.2013
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.