")
Back to Archived Journals » Research and Reports in Forensic Medical Science » Volume 5
Cardiac genetic investigation of sudden cardiac death: advances and remaining limitations
Received 9 March 2015
Accepted for publication 29 April 2015
Published 30 July 2015 Volume 2015:5 Pages 7—15
DOI https://doi.org/10.2147/RRFMS.S72063
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Henrik Druid
Jonathan Robert Skinner,1,3 Paul Lowell Morrow2
1Green Lane Paediatric and Congenital Cardiac Service/Cardiac Inherited Disease Group, Starship Children's Hospital, 2Department of Forensic Pathology, LabPLUS, Auckland City Hospital, 3Department of Paediatrics: Child and Youth Health, University of Auckland, Auckland, New Zealand
Abstract: Directing a thorough postmortem investigation of a young sudden unexpected death is one opportunity that a forensic pathologist has to save lives. Achieving a diagnosis of an inherited heart condition in the decedent means that family members can be screened for the condition and effective protective therapies can be put into place. Since these conditions are almost always autosomal dominant, 50% of family members are at risk. Diagnosis is achieved through a thorough high-quality autopsy examination, storage and analysis of DNA, and involvement of the family in the investigation, including cardiac tests upon them. Forensic pathologists should form a partnership with a cardiac genetic service and liaise with them in cases of sudden unexplained death, particularly in 1- to 40-year olds, and when cardiomyopathy is diagnosed at autopsy. The molecular autopsy in sudden unexplained death should include the most common long QT genes and RyR2, the cardiac ryanodine gene linked to catecholaminergic polymorphic ventricular tachycardia. Alternatively or in addition, genetic testing can be guided by cardiac findings in first-degree relatives. With such an approach, a diagnosis can be achieved in up to 50%. Most sudden death in young people occurs at sleep, rest or light activity, and not during sport. Pathologists should be aware that long QT and catecholaminergic polymorphic ventricular tachycardia can be misdiagnosed as epilepsy in life. These ion channelopathies and the cardiomyopathies such as hypertrophic cardiomyopathy and arrhythmogenic right ventricular cardiomyopathy can present with drowning in a competent swimmer, straight road car accidents, and death that can be triggered by certain medications or toxins.
Keywords: sudden death, genetic testing, long QT syndrome, CPVT, hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy
Introduction
Directing a thorough postmortem investigation of a young sudden unexpected death is one opportunity that a forensic pathologist has to save lives. Achieving a diagnosis of an inherited heart condition in the decedent means that family members can be screened for the condition and effective protective therapies can be put into place. Since these conditions are almost always autosomal dominant, 50% of family members are at risk.
It has been known for decades that inherited cardiac ion channelopathies and cardiomyopathies cause sudden death through malignant ventricular arrhythmia. The ion channelopathies include long QT syndrome (LQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), Brugada syndrome and the cardiomyopathies hypertrophic cardiomyopathy (HCM), and arrhythmogenic right ventricular cardiomyopathy (ARVC). Many of the genes underlying these conditions have been identified, and genetic testing is now commonly used to identify family members at risk of the condition. This process of following the diagnosis through the families is known as family cascade testing. However, it is only in the last 10 years that the full potential of cardiac genetic investigation of sudden death has been realized.
Cardiac genetic investigation of autopsy-negative sudden death in the young
In 2005, a multidisciplinary group in the Netherlands described a technique of investigating the family members of young sudden death victims in whom no cause had been found.1 They used mostly simple cardiac tests such as the electrocardiography (ECG), exercise testing, and echocardiography. In all, 40% were found to have inherited heart disease, LQTS being the most common. They noted that the more family members they saw, the more likely they would find the diagnosis.
All of these conditions have variable clinical expression. Gene carriers may die suddenly at a young age, while others have no symptoms or even clinical signs at all during their life. Since this time, a similar series from the UK has described a diagnostic rate of >50%.2
At the Mayo Clinic, during the same time, they used genetic testing for LQTS and CPVT from DNA of the decedents, introducing the so-called molecular autopsy. They reported a series of 173 cases in which they also found that 40% had these conditions, mostly in 1- to 40-year olds.3 They and others also identified that between 2% and 12% of Sudden Infant Death Syndrome (SIDS) cases were caused by mutations in the LQTS genes.4–7
In New Zealand, an LQTS molecular autopsy was first tested in a retrospective cohort of individuals ≤35 years old, with DNA from the long-deceased victims being extracted from the archived neonatal screening (Guthrie) card.8 Twenty-two percent of them were positive. LQTS autopsy genetic testing was introduced in 2006 as a clinical service, and over the first 26 months in a nonselected population of sudden unexplained death in the young (SUDY) victims aged 1–40 years, this molecular autopsy was positive in 15%. A further 15% were diagnosed from family investigations, including cases of ARVC.9 Thus, an overall diagnostic rate of 30% was achieved.
Postmortem examination
Experience from New Zealand revealed that the quality of autopsies was quite variable among pathologists. Autopsy reports were often very brief and lacking sufficient detailed descriptions to allow the investigating cardiac genetic team to have confidence that anatomical diagnoses such as cardiomyopathies or coronary artery anomalies were adequately searched for.10 In one family with three young sudden deaths, none of the three autopsies described the right ventricle. Subsequent tests have revealed the cause as ARVC.
Thus, it was clear that such investigations need to have proper protocols. With this in mind, a multidisciplinary New Zealand/Australian group known as Trans-Tasman Response Against Sudden Death in the Young (TRAGADY) wrote best practice guidelines for the appropriate investigations of sudden death in young people.11,12
The four key TRAGADY guidelines were as follows:
- A high-quality thorough autopsy should be done by an experienced pathologist, including a minimum number of appropriate sections (for example, the right ventricle).
- The family should be involved early in the process (a thorough family history can reveal diagnostic clues).
- Tissue suitable for DNA extraction should be saved (whole blood in EDTA, snap frozen tissue, or RNA later). Formalin-fixed paraffin-embedded tissue is not suitable.
- Referral should be made to a cardiac genetic service.
Autopsy
There is good evidence that special expertise is often required to diagnose subtle features of cardiomyopathies, particularly ARVC, as well as to assess subtle potentially misleading findings, such as ventricular fibrosis or minor coronary artery disease.13 Pathologists should have a low threshold for seeking experts’ second opinion. Pathologists may well have a sense of pressure to achieve a diagnosis. We have seen cases where death has been ascribed to myocarditis, with unconvincing histological evidence, as well as coronary artery disease when there was only moderate disease and no definitive evidence of infarction. When there is uncertainty, it is better to give an honest description and invite cardiac genetic investigation or at least case review. A thorough family history may indicate a red flag for inherited heart disease. When the heart is significantly enlarged, familial dilated cardiomyopathy (DCM) is a possibility. Genetic testing is still finding its place here but improving all the time and involves a large number of genes. Full engagement of the family is particularly important prior to genetic testing to permit phenotype/genotype testing of unclassified variants.
Documenting positive findings clearly is important, but equally important is documenting significant negative findings and how thorough the examination was. For example, simply stating normal coronary arteries is inadequate. Description should include which arteries were examined, did they arise normally, and was there or not stenosis (and to what degree) or atherosclerosis. Details of how the heart was examined (valves, chamber dilation, myocardium, weight, and measurements), how many histological sections were taken and from where, and which tissue (if any) was kept are essential. The cardiac genetic team must have confidence in the autopsy, particularly if thousands of dollars and many clinical hours are to be spent in investigating families or doing genetic tests.
A full toxicology report is essential. Consideration should be given to the conduction system, although histological screening is not always practical or essential.
Cardiac genetic services
At the time the TRAGADY guidelines were written in 2008, there were remarkably few cardiac genetic services, but in New Zealand, the Cardiac Inherited Disease Group had already been established for 8 years, with a nationwide multidisciplinary clinical and scientific network. The molecular autopsy was and still is funded by government, through the Ministry of Justice (Coronial Service). The Cardiac Inherited Disease Group has now been involved in almost 800 cases. The positive diagnostic rate in SIDS was low, less than 5%. In New Zealand, the well-recognized risk factors of co-sleeping (bed sharing), accidental asphyxiation, and other environmental risk factors are particularly common among Maori and Pacific peoples, in whom SIDS predominates.5 However, such features were also common in a large US series with a likely diagnostic rate of 13%.7
A good cardiac genetic service is multidisciplinary, usually led by cardiology or pediatric cardiology, with access to electrophysiological expertise, clinical and molecular genetics, genetic counseling, and psychological support.14 In New Zealand, a senior forensic pathologist forms part of the team.
As well as autopsy negative cases, referrals should include those in which a likely inherited cardiac condition has been detected by the pathologist, such as HCM, suspected ARVC, and idiopathic DCM. In these cases, we will often perform genetic testing aimed only at the particular condition concerned, for example HCM.
Genetic diagnosis
There are a few key messages for pathologists working in this area.
- Genetic testing is a family issue. Families should be involved and counseled at the outset, either prior to or at least parallel with genetic testing.15 The aim of the test is not only to bring a diagnosis of cause of death and some closure for the bewildered and distressed family, but also to potentially find others with the same condition. It is much better to explain the possible consequences and strengths and weaknesses of the test at the beginning so that they know what to expect.
- Genes are complex noisy things. Genes vary normally between individuals. Such changes are called common polymorphisms. Very often, we detect a genetic variant that has never been seen before in a gene of interest. The current practice is to call these rare unclassified variants and only use the term mutations, when we are confident of its pathogenicity. We can prove that it is rare, but proving that it is disease causing can be very difficult. Prediction software is used, and sometimes in vitro testing of the mutation, but the best method is to see that the gene-positive individuals in the family have the condition, whereas others do not. This is termed phenotype–genotype cosegregation, and engagement of the family with proper genetic counseling is essential.
- Mutations may be unique to the family concerned. Many hundreds of mutations have been described in each of the LQTS genes for example.
- The diagnostic rate varies by condition.16 In people with definite phenotype, genetic mutations can be found in more than half with CPVT,17 75% of those with LQTS,16 50% of those with HCM, 20%–40% of those with Brugada syndrome, and about 25% of those with ARVC, although this condition may be polygenic in many instances.
- Genetic ancestry matters. Genetic variants in one population may be malignant, while being benign in others. For example, R1193Q in the gene SCN5A is known to cause Brugada syndrome and sudden death in whites;18 yet, it is present in 10% of the Han Chinese.19 In the study of rare variants, it is essential to have normative population controls.
Triggers for sudden death
Although the medical literature has abundant references to HCM being the most common cause of sudden cardiac death in the young, it is not. The ion channelopathies are far more common.20 HCM and ARVC remain common causes of sudden death in the older athlete, but death during sport is very uncommon, though it often reaches the media.
Most sudden unexpected deaths in 1- to 40-year olds occur during sleep (>50%) or light daily activity.7,9 Death during strenuous or athletic activity is rare. The group most likely to have a genetic diagnosis of LQTS is 25- to 40-year-old females who die in their sleep; long QT type 2 predominates (KCNH2). If death does occur during exercise or swimming, it is most likely to be due to CPVT (the gene is RyR2; the cardiac ryanodine gene) or long QT type 1 (KCNQ1).21,22 In Asian countries, death during sleep in young males is typically due to Brugada syndrome, sometimes linked to the cardiac sodium channel gene SCN5A.16
Which cases should be referred?
By establishing a regular dialog and a referral for opinion approach with your cardiac genetic service, you will develop a sense of what is most likely to be a good referral. It is better to refer too many. When you send a referral, include appropriate police or scene reports and the full autopsy report.
Cases we consider that should always be referred to the cardiac genetic service are:
- sudden unexplained (autopsy-negative) death in 1- to 40-year olds;
- SIDS in an infant without known risk factors;
- sudden unexplained death in epilepsy (SUDEP) unless there is a documented structural brain or developmental abnormality (LQTS and CPVT are often misdiagnosed as epilepsy in life);23
- drowning in a strong swimmer (LQTS and CPVT classically present this way); and
- where an idiopathic cardiomyopathy (HCM, ARVC, idiopathic DCM) has been identified.
Consider also cases where some other medical history or pathology is present but there may be doubt as to whether it caused the death, for example, sudden death in asthma, sudden death in bed with diabetes or alcohol intoxication, minor or borderline coronary atherosclerosis, or minor illnesses. We have a number of infants having either Brugada syndrome or LQTS who died suddenly during febrile illness.
Generally, we have not investigated those with morbid obesity unless there was a suggestive family history or a suggestive cardiac abnormality on histology.
Choosing who to test and restricting costs
Genetic testing and family cardiac investigation are expensive, and the multidisciplinary team needs to take a gatekeeping role to gain highest yield and avoid unnecessary tests. Overall, we performed a molecular autopsy in about 20% of cases referred to us and have seen and performed cardiac investigations on about 50% of the families. The cases where we will almost automatically do a molecular autopsy are sudden young deaths (1–40 years) where there are no confounding factors (medications/medical conditions/morbid obesity/coronary artery disease) with a high-quality autopsy being completely negative. Otherwise, we will start the investigation with the clinical records and the family or decide that cardiac genetic testing is not warranted.
Resuscitated sudden cardiac death
With increasing awareness of cardiopulmonary resuscitation and availability of advisory defibrillators, many young people now reach the intensive care unit after their cardiac arrest. Since it is possible to do cardiac tests on such people, the diagnostic rate for inherited heart conditions is >60%, when coronary artery disease and myocarditis have been excluded.24
The pathologist may become involved if such patients have severe brain injury and life support is withdrawn. The same general principles of investigation should be applied as in those who die suddenly in the community, except that dialog with the attending physicians may direct attention to a particular question. Expert review of the cardiological tests prior to death is essential.
Which genes should be tested?
The Heart Rhythm Society guidelines suggest that LQTS genes 1, 2, 3, 5, 6, and 7 and RyR2 (the CPVT gene) should be tested in autopsy-negative sudden death.25 The gene for long QT type 3, SCN5A, is the most common gene also to be linked to Brugada syndrome and will likely have particular significance in Asian countries. This is the currently accepted standard molecular autopsy and is a pragmatic approach based on the rarity of other types of LQTS (there are at least 15 genotypes recognized to date). The largest series to date using this strategy, from the US state of New York, was published last year. Diagnostic yield in 144 infants was 13.5% and in 133 noninfants was 19.5%.7 Not one individual died during strenuous activity. Cost and availability will vary by region, but in general costs are coming down rapidly, and the number of genes we can test is going up exponentially. It is even possible to test the whole human exome or genome.26 Gene panels are becoming increasingly common, comprising 20–100 genes.
The problem is that the more the genes you test, the more the variants of unknown significance you will get back and the more likely you will create anxiety and uncertainty among the family members. Whole exome studies of normal populations have revealed variants originally thought to be pathogenic in HCM that have now been found in up to one in seven healthy individuals.27 Genes like titin, linked to DCM, are massive and almost always reveal unique or rare variants.
In 2015, we recommend for practical clinical purposes using the standard molecular autopsy for autopsy-negative deaths, only moving to other genes when a cardiomyopathy is established either in the deceased (eg, HCM seen by the pathologist) or in the family (such as ARVC seen on cardiac Magnetic Resonance Imaging [MRI] of one of the parents).
Notes on the inherited heart conditions
A brief summary of key features is included in Table 1 and Figures 1 and 2. Figure 1 displays ECGs from the cardiac ion channel disorders, and Figure 2 demonstrates some anatomical cardiomyopathy specimens. There is an excellent review of the key genetics by Wilde and Behr.16 Note, in particular, that the familial component of DCM is often overlooked. In life and after death, it may be described as idiopathic, postpartum, or secondary to alcohol, whereas in fact, it is a familial disease, recognized after examining relatives. If in doubt, referral for cardiac genetic investigation is recommended.
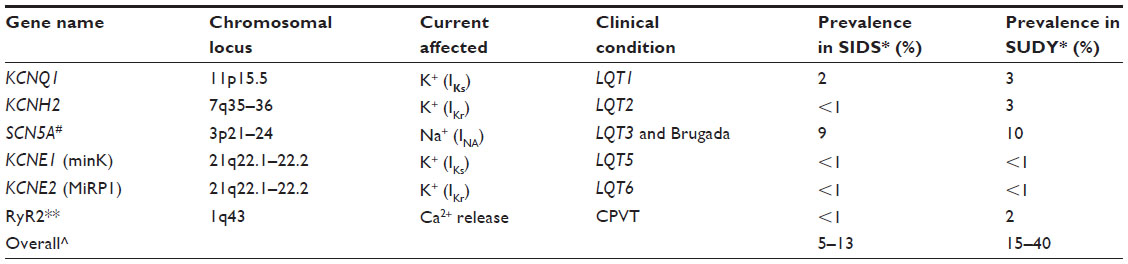 | Table 1 Genes in the recommended molecular autopsy list for SUDY |
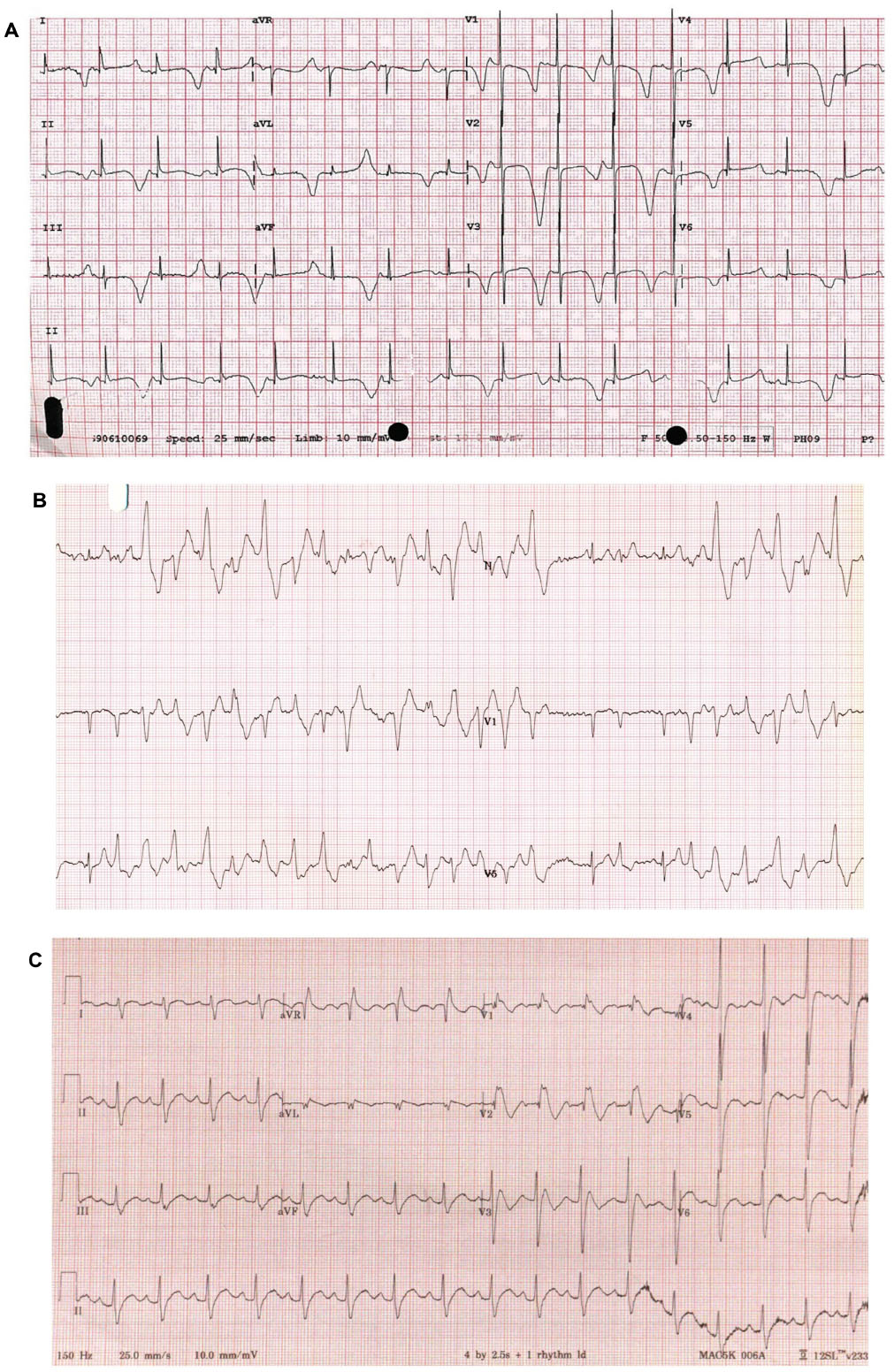 | Figure 1 (A) Long QT syndrome: ECG from a 2-year-old patient with severe QT prolongation. It also shows T-wave alternans; each consecutive T-wave goes up or down. This typically precedes torsades de pointes ventricular tachycardia and syncope or death. There are at least 15 genotypes for long QT syndrome, but types 1, 2 and 3 predominate. Each has certain characteristic T-wave morphologies and modes of typical presentation. Recurrent syncope and misdiagnosis as epilepsy are common. Types 1 and 2 are the most common in those who are alive, but type 3 is present in about 8% of living cohorts and >50% among the gene-positive deceased. Long-term beta blocker therapy reduces risk by up to 75% in types 1 and 2. Intracardiac defibrillators are used in those who have survived a cardiac arrest. All must avoid medications that prolong the QT interval (https://www.crediblemeds.org). (B) CPVT: Three-lead ECG during exercise in a 29-year-old woman with recurrent collapse during exercise. There are runs of polymorphic ventricular tachycardia. In lead II, in the middle of the trace, the classical bidirectional ventricular beats are seen, consecutively upward and downward. This is a highly malignant condition, and the first presentation is often catastrophic. About one-third of cases are de novo, ie, not present in the parents. (This is a feature of conditions that are not survivable to reproductive years.) Beta blockers in high doses are effective, but mortality remains high. Flecainide, which specifically blocks the calcium release at the sarcoplasmic level, is a powerful adjuvant therapy. (C) Brugada syndrome: ECG from a 1-year-old child with Brugada syndrome. Note the right bundle branch appearance in V1–V3 with ST elevation. This is a complex condition, which, in about one-third of cases, is truly familial with autosomal dominant inheritance and most commonly related to a deficient cardiac sodium channel. Triggers are fever and a large meal, but most events occur at night. Treatment is aggressive management of fever and intracardiac defibrillators in the survivors of cardiac arrest. |
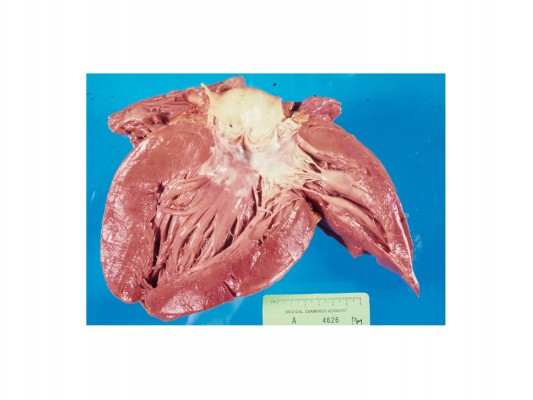
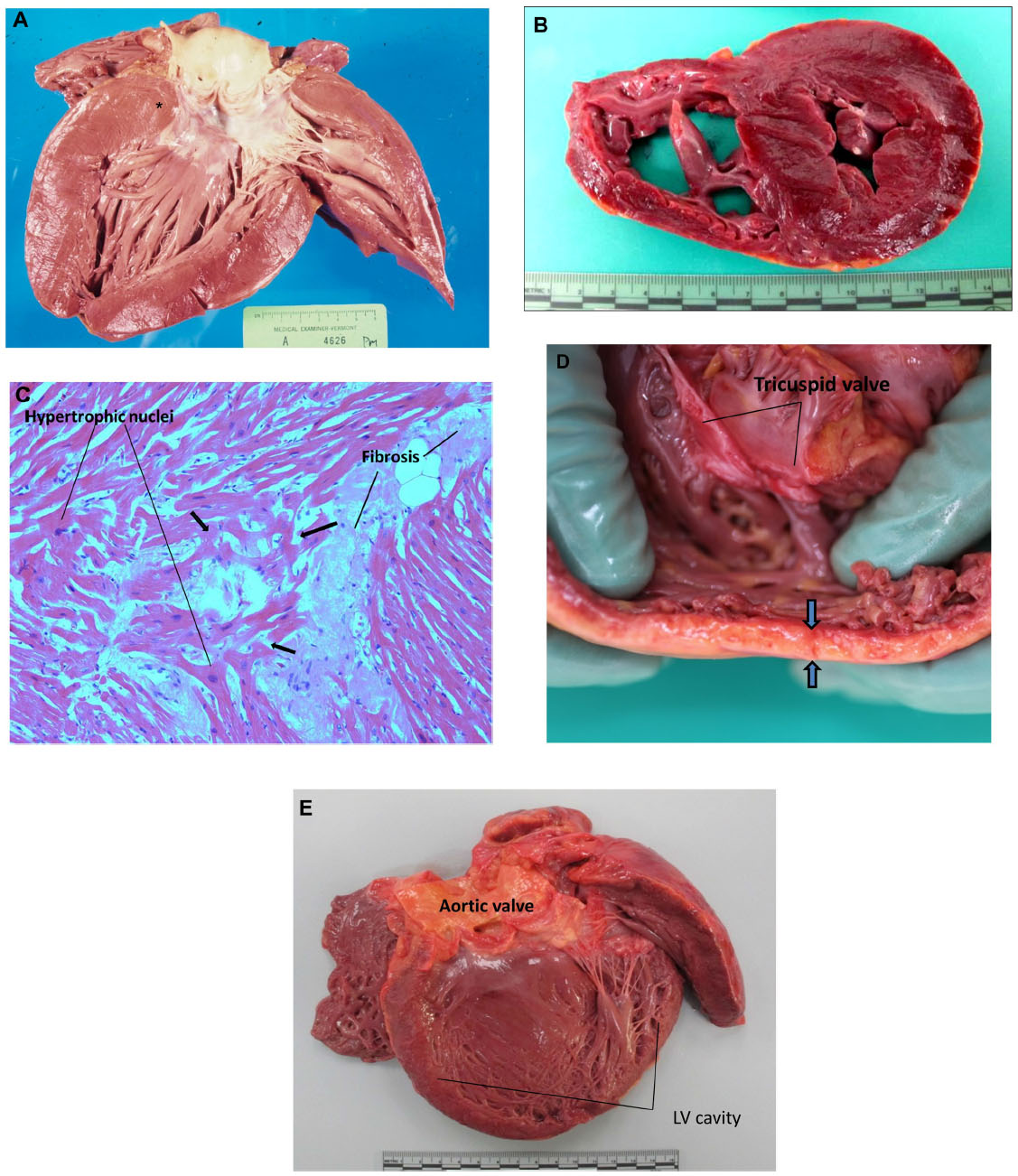 | Figure 2 (A) HCM, subaortic stenosis: longitudinal section through the LV from a 36-year-old man with sudden death. Prominent thickening of interventricular septum below aortic valve (*). Note endocardial thickening of aortic outflow tract. (B) HCM, left ventricular hypertrophy: short axis cross-section through the LV from a 42-year-old sudden death victim with concentric HCM. Note the virtual cavity (LV lumen) obliteration and thickened muscular appearing myocardium. Note endocardial thickening of aortic outflow tract. (C) HCM, histology: microscopic section of left ventricular myocardium of patient who died suddenly of HCM. Hypertrophic nuclei of myocytes reflect widespread myofiber hypertrophy. Note interstitial fibrosis. Myocyte disarray is indicated by short arrows. H&E stain, ×20. (D) ARVC: dilated right ventricle being held open of a 37-year-old woman who died suddenly with ARVC. There is extensive fatty infiltration and fatty replacement of myocardium of right ventricular wall (between arrows). (E) Dilated cardiomyopathy: marked left ventricular dilation with u-shaped configuration in a 48-year-old man found dead in bed. This is a nonspecific pathological picture, but if other reasonable causes (eg, alcohol, myocarditis, toxic, etc) can be ruled out, then consider a familial etiology. |
What does the future hold?
We still can not find a cause of death in more than half of our SUDY cases. A few may be due to inherited heart conditions hitherto undetected or undescribed and the others remain undetermined and need further research. In future, genetic testing will become cheaper, quicker, and more complex. Understanding the genotype–phenotype link will take a long time, however, and will depend on robust registries for these conditions.
The minimally invasive autopsy is emerging. The combination of static MRI with tissue biopsy, toxicology, and DNA analysis is likely to be societally and culturally more acceptable to many.28 What role this will have in sudden death investigation is still to be determined.
The final word
Autopsy in sudden unexpected death in the young represents a real chance to protect others in the community. A multidisciplinary team approach with appropriate protocols is required. When families learn that they may have an inherited heart issue, almost all are exceedingly grateful for the care taken in a thorough investigation. It is not possible to overstate the heart-rending desperate devastation felt by families in the position of sudden loss of a child, brother, or wife, totally out of the blue. A diagnosis can help to bring closure, and sympathetic efforts to find the cause of sudden death, even when they fail, are generally greatly appreciated.
Parents will always blame themselves even when it appears irrational to do so. The mother of a child found to have died with LQTS, some years after her death stated, “Thank you doctor. I don’t have to blame myself anymore”.
Acknowledgment
We would like to thank Charlene Nell, Desktop Support Administrator, Green Lane Cardiovascular Services/Cardiology Department, for excellent secretarial assistance.
Disclosure
Dr Skinner receives salary support from Cure Kids and Dr Morrow has no conflicts of interest to declare.
References
Tan HL, Hofman N, van Langen IM, van der Wal AC, Wilde AA. Sudden unexplained death: heritability and diagnostic yield of cardiological and genetic examination in surviving relatives. Circulation. 2005;112(2):207–213. | |
Behr ER, Dalageorgou C, Christiansen M, et al. Sudden arrhythmic death syndrome: familial evaluation identifies inheritable heart disease in the majority of families. Eur Heart J. 2008;29(13):1670–1680. | |
Tester DJ, Medeiros-Domingo A, Will ML, Haglund CM, Ackerman MJ. Cardiac channel molecular autopsy: insights from 173 consecutive cases of autopsy-negative sudden unexplained death referred for postmortem genetic testing. Mayo Clin Proc. 2012;87(6):524–539. | |
Ackerman MJ, Siu BL, Sturner WQ, et al. Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome. JAMA. 2001;286(18):2264–2269. | |
Glengarry JM, Crawford J, Morrow PL, Stables SR, Love DR, Skinner JR. Long QT molecular autopsy in sudden infant death syndrome. Arch Dis Child. 2014;99(7):635–640. | |
Arnestad M, Crotti L, Rognum TO, et al. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation. 2007; 115(3):361–367. | |
Wang D, Shah KR, Um SY, et al. Cardiac channelopathy testing in 274 ethnically diverse sudden unexplained deaths. Forensic Sci Int. 2014;237:90–99. | |
Gladding PA, Evans CA, Crawford J, et al. Posthumous diagnosis of long QT syndrome from neonatal screening cards. Heart Rhythm. 2010;7(4):481–486. | |
Skinner JR, Crawford J, Smith W, et al; Cardiac Inherited Disease Group New Zealand. Prospective, population-based long QT molecular autopsy study of postmortem negative sudden death in 1 to 40 year olds. Heart Rhythm. 2011;8(3):412–419. | |
Wilms HR, Midgley DJ, Morrow P, Stables S, Crawford J, Skinner JR. Evaluation of autopsy and police reports in the investigation of sudden unexplained death in the young. Forensic Sci Med Pathol. 2012;8(4):380–389. | |
Skinner JR, Duflou JA, Semsarian C. Reducing sudden death in young people in Australia and New Zealand: the TRAGADY initiative. Med J Aust. 2008;189(10):539–540. | |
TRAGADY. Post-Mortem in Sudden Unexpected Death in the Young: Guidelines on Autopsy Practice 2008; 2008. Available from: https://www.rcpa.edu.au/Library/Publications/Joint-and-Third-Party-Guidelines/Guidelines/Guidelines-on-Autopsy-Practice. | |
Papadakis M, Raju H, Behr ER, et al. Sudden cardiac death with autopsy findings of uncertain significance: potential for erroneous interpretation. Circ Arrhythm Electrophysiol. 2013;6(3):588–596. | |
Ingles J, Semsarian C. Sudden cardiac death in the young: a clinical genetic approach. Intern Med J. 2007;37(1):32–37. | |
Ingles J, Yeates L, Semsarian C. The emerging role of the cardiac genetic counselor. Heart Rhythm. 2011;8(12):1958–1962. | |
Wilde AA, Behr ER. Genetic testing for inherited cardiac disease. Nat Rev Cardiol. 2013;10(10):571–583. | |
Medeiros-Domingo A, Bhuiyan ZA, Tester DJ, et al. The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: a comprehensive open reading frame mutational analysis. J Am Coll Cardiol. 2009;54(22):2065–2074. | |
Huang H, Zhao J, Barrane FZ, Champagne J, Chahine M. Nav1.5/R1193Q polymorphism is associated with both long QT and Brugada syndromes. Can J Cardiol. 2006;22(4):309–313. | |
Hwang HW, Chen JJ, Lin YJ, et al. R1193Q of SCN5A, a Brugada and long QT mutation, is a common polymorphism in Han Chinese. J Med Genet. 2005;42(2):e7. [author reply e8]. | |
van der Werf C, Hofman N, Tan HL, et al. Diagnostic yield in sudden unexplained death and aborted cardiac arrest in the young: the experience of a tertiary referral center in The Netherlands. Heart Rhythm. 2010;7(10):1383–1389. | |
Albertella L, Crawford J, Skinner JR. Presentation and outcome of water-related events in children with long QT syndrome. Arch Dis Child. 2011;96(8):704–707. | |
Choi G, Kopplin LJ, Tester DJ, Will ML, Haglund CM, Ackerman MJ. Spectrum and frequency of cardiac channel defects in swimming-triggered arrhythmia syndromes. Circulation. 2004;110(15):2119–2124. | |
MacCormick JM, McAlister H, Crawford J, et al. Misdiagnosis of long QT syndrome as epilepsy at first presentation. Ann Emerg Med. 2009;54(1):26–32. | |
Skinner JR. Investigating sudden unexpected death in the young: a chance to prevent further deaths. Resuscitation. 2012;83(10):1185–1186. | |
Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011;8(8):1308–1309. | |
Bagnall RD, Das KJ, Duflou J, Semsarian C. Exome analysis-based molecular autopsy in cases of sudden unexplained death in the young. Heart Rhythm. 2014;11(4):655–662. | |
Andreasen C, Nielsen JB, Refsgaard L, et al. New population-based exome data are questioning the pathogenicity of previously cardiomyopathy-associated genetic variants. Eur J Hum Genet. 2013; 21(9):918–928. | |
Thayyil S, Sebire NJ, Chitty LS, et al; MARIAS Collaborative Group. Post-mortem MRI versus conventional autopsy in fetuses and children: a prospective validation study. Lancet. 2013;382(9888):223–233. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.