")
Back to Journals » Cancer Management and Research » Volume 10
Capsaicin-induced TRIB3 upregulation promotes apoptosis in cancer cells
Authors Lin RJ, Wu IJ, Hong JY, Liu BH, Liang RY, Yuan TM, Chuang SM
Received 12 January 2018
Accepted for publication 16 July 2018
Published 4 October 2018 Volume 2018:10 Pages 4237—4248
DOI https://doi.org/10.2147/CMAR.S162383
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Kenan Onel
Rong-Jaan Lin,1,2 I-Jung Wu,1,2 Jo-Yu Hong,3 Bang-Hung Liu,4 Ruei-Yue Liang,4 Tein-Ming Yuan,5 Show-Mei Chuang4,6,7
1College of Life Sciences, National Chung Hsing University, 2Taichung Hospital, Ministry of Health and Welfare, 3Bachelor Program of Biotechnology, College of Agriculture and Natural Resources, 4Institute of Biomedical Sciences, College of Life Sciences, National Chung Hsing University, 5Surgery Department, Feng-Yuan Hospital, Ministry of Health and Welfare, 6Department of Nursing, Asia University, 7Department of Medical Research, China Medical University Hospital, Taichung, Taiwan
Background: Capsaicin (8-methyl-N-vanillyl-6-nonenamide) is one of the main pungent components of chili peppers and has been shown to exert various effects on numerous physiological processes. Recent studies have focused on the chemopreventive effects of capsaicin, which can combat growth in various human cancer cell systems. The tribbles-related protein 3 (TRIB3) is evolutionarily conserved from Drosophila to humans. In the latter, TRIB3 is a key determinant in numerous cellular processes, including apoptosis.
Purpose: The aim of this study was to examine the importance of TRIB3 in the antitumor efficacy of capsaicin in human cancer cells, and further assess potential mechanism(s) underlying the capsaicin-induced upregulation of TRIB3.
Methods: Human cancer cell lines were treated with capsaicin, then evaluated for levels of TRIB3 and molecules related to apoptosis or signaling pathways. The impact of TRIB3 on capsaicin-induced apoptosis was investigated using si-RNA or overexpression of TRIB3.
Results: It is the first time to show that TRIB3 is targeted by capsaicin to promote apoptosis. Capsaicin promotes apoptotic cell death by upregulating TRIB3 expression in cancer cells. Overexpression of TRIB3 enhances capsaicin-induced apoptosis, and TRIB3 knockdown experiments demonstrate that the effect of capsaicin in apoptotic cell death is correlated with the induction of TRIB3 in cancer cells. Finally, enhancements in gene expression and protein stability are involved in the capsaicin-induced upregulation of TRIB3.
Conclusion: Our results show that the capsaicin-induced upregulation of TRIB3 triggers apoptosis and thereby contributes to the suppression of cell growth in cancer cell lines.
Keywords: capsaicin, TRIB3, apoptosis, cancer, Protein stability
Introduction
Capsaicin (8-methyl-N-vanillyl-6-nonenamide) is one of the main pungent components of chili peppers and has been shown to exert various effects on numerous physiological processes. It is commonly used as an anticancer compound owing to its inhibitory effects on cell proliferation. Recent studies focusing on the chemopreventive and chemotherapeutic effects of capsaicin have demonstrated that it can decrease growth in various human cancer cell systems (eg, liver, lung, colon, pancreas, stomach, prostate, leukocyte, breast, esophagus, and tongue) through its effects on cell cycle arrest and apoptosis.1–12 As a principal natural chemopreventive compound, capsaicin has been shown to have a chemosensitizing activity that improves cytotoxicity among 5-fluorouracil-treated gastric cancer cells.13 In the cisplatin-resistant Korean human gastric cancer cell line, SNU-668, co-treatment of cells with capsaicin plus cisplatin induced higher apoptotic cell death than treatment with either agent alone, suggesting that this strategy could potentially overcome cisplatin resistance.14
Cancer cells show differential susceptibility to capsaicin, perhaps reflecting the ability of this agent to modulate diverse signaling pathways that contribute to cell death or survival. However, we do not yet fully understand the molecular mechanism(s) underlying capsaicin-induced growth inhibition and apoptosis, nor have we fully elucidated the complicated cross-talk among the involved signaling pathways. Thus, sustained research efforts are being made to understand the action mechanisms of capsaicin and explore its potential therapeutic use in cancer.
Several mechanisms are thought to be involved in capsaicin-induced apoptosis. For example, capsaicin has been shown to exert cytotoxic effects in an array of mammalian cell lines via various signaling pathways, including the AMPK,15,16 c-Jun N-terminal kinase 1 (JNK1),17 p38 MAPK,17 extracellular signal-regulation kinase (ERK),18 phosphoinositide 3-kinase (PI3K)/Akt,19 and interleukin (IL)-620 pathways. Capsaicin has also been shown to exert its chemopreventive activities via its multitargeting property. For instance, capsaicin enhances p53 gene expression in SNU-1 stomach cancer cells21 and stabilizes p53 protein stability in human colon cancer cells,22 triggering apoptosis in both cases. Under hypoxia, capsaicin enhances the stability and functional activity of the p53 protein, which downregulates hypoxia-inducible factor-1α by facilitating its degradation and inhibiting its transcription, and thereby decreases the expression/function of vascular endothelial growth factor.23 Capsaicin has also been shown to augment the protein stability of the NF-κB inhibitor, IκB, thereby inhibiting NF-κB activation,16,24–26 and it has an antiproliferative effect on human lung cancer cells via the modulation of E2F.11
The multifunctional protein, TRIB3, was recently identified as a scaffold-like regulator of various signaling pathways and has been implicated in several cellular processes.27–30 Of particular relevance, TRIB3 binds AKT and prevents its phosphorylation at Ser473 and Thr308, thereby blocking its activation.31 TRIB3 acts as a molecular switch to regulate the activation of three classes of MAPK signaling cascades,32 and has been shown to negatively regulate NF-κB signaling through a direct interaction that suppresses the transcriptional activity of NF-κB.33 The signals/stresses known to induce TRIB3 expression include nutrient starvation,34 hypoxia,35 endoplasmic reticulum (ER) stress,36 nerve growth factor deprivation,37 and several antitumor drugs such as tetrahydrocannabinoids, salinomycin, or the lipid derivative ABTL0812.38–40 Interestingly, several studies have shown that TRIB3 protein levels are the combined result of a number of regulatory feedback loops and temporally distinct events. Upon nerve growth factor withdrawal, for example, TRIB3 is essential for the nuclear translocation of FoxO1a, which in turn binds the TRIB3 promoter region and is required for the transcriptional induction of TRIB3.37 Other regulatory feedback interactions include the TRIB3–AKT and the ATF4–CHOP–TRIB3 loops.28,41 Although the biological roles of TRIB3 have been widely investigated, conflicting reports suggest that it may both evoke and prevent cell apoptosis.28,31,34,42,43
The role of TRIB3 in apoptosis regulation is not well defined, and more importantly, there is a lack of information on the effects of capsaicin on TRIB3. In this regard, here, we investigated the antitumor efficacy of capsaicin in human cancer cells, examined the role of TRIB3 in this activity, and assessed potential mechanism(s) underlying the capsaicin-induced upregulation of TRIB3. Our data show that capsaicin enhances the protein expression of TRIB3 in various human cancer cells and significantly increases the mRNA and protein stability of TRIB3, and that these effects are accompanied by increased apoptotic cell death. TRIB3 knockdown experiments further demonstrated that capsaicin-induced apoptotic cell death is correlated with the induction of TRIB3 in cancer cells. We also report that the apoptosis associated with capsaicin-mediated induction of TRIB3 suppresses cell growth in cancer cell lines in vitro. It is clear that TRIB3 acts as a critical factor for capsaicin-promoted apoptosis in cancer cells; however, JNK, p38 and PI3K–AKT signaling pathways are not associated with this capsaicin-enhanced upregulation of TRIB3.
Materials and methods
Chemicals
The MAPK inhibitors, U0126, SB203580, and SP600125, and the proteasome inhibitor, MG132, were purchased from TOCRIS Bioscience (Bristol, UK). The protease inhibitor cocktail was a product of Roche Applied Science (Mannheim, Germany). Cycloheximide was obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Capsaicin (8-methyl-N-vanillyl-6-nonenamide) was purchased from Enzo Life Sciences (Farmingdale, NY, USA). All other chemicals were purchased from Sigma-Aldrich Co. (St Louis, MO, USA) or Amresco (Solon, OH, USA).
Antibodies
The antibody against TRIB3 was obtained from Novus Biologicals (Littleton, CO, USA). The antibodies against PARP, caspase 3, caspase 8, caspase 9, caspase 12, phospho-p38, phospho-ERK1/2, phospho-JNK, and phosphor-AKT were products of Cell Signaling Technology (Beverly, MA, USA). The antibodies against ERK2, JNK, p38, and AKT, beta-actin, and the peroxidase-conjugated secondary antibodies against mouse and rabbit IgG were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). The antibodies against phospho-p90RSK (Thr573), Bid and Bax were from Abcam (Cambridge, UK). The antibody against the Myc tag was from EMD Millipore (Billerica, MA, USA).
Cell culture
The human gastric carcinoma cell lines, AGS, SNU-1, and KATO III, were obtained from Bioresource Collection and Research Center (BCRC; Hsinchu, Taiwan). MKN-45 was purchased from Japanese Collection of Research Bioresources (JCRB) Cell Bank (Osaka, Japan). Human gastric cancer cell line TMK-1, human cervical squamous cell carcinoma SiHa, and human colorectal adenocarcinoma cell line SW480 were gifted by Dr Wu (Department of Education and Research and Division of Gastroenterology, Taichung Veterans General Hospital, Taichung, Taiwan; current address is Taipei Veterans General Hospital and Faculty of Medicine, School of Medicine, National Yang-Ming University, Taipei, Taiwan) and their DNA short tandem repeats (STR) profile was confirmed at BCRC. Cells were cultured in RPMI-1640 (Thermo Fisher Scientific, Waltham, MA, USA). Cultures were supplemented with 10% fetal bovine serum, sodium bicarbonate (2%, w/v), l-glutamine (0.29 mg/mL), penicillin (100 units/ml), and streptomycin (100 µg/mL) (Thermo Fisher Scientific), and grown at 37°C in a humidified 5% CO2 incubator.
Plasmid
The full-length human TRIB3 cDNA was cloned into the pcDNA3.1-Myc-His vector using primers designed to introduce Hind III and Xho I restriction sites, as follows: sense, 5′-AAC TCG AGG CCA CCA TGC GAG CCA CCC CTC TG-3′ and antisense, 5′-AAA AGC TTG CCA TAC AGA ACC ACT TC-3′. The obtained TRIB3 cDNA was confirmed by sequencing. Cells were transfected with 2 µg TRIB3-Myc plasmid using jetPEI (Polyplus, Illkirch, France) according to the manufacturer’s recommendations.
Cell viability assay
Cells (5×103/well) were seeded in a 96-well plate. After 24 hours, cells were treated with different concentrations of capsaicin (three wells per concentration) for 48 hours. Cell viability was detected using the WST-1 reagent, according to the manufacturer’s recommendations (Roche Applied Science).
Reverse-transcription polymerase chain reaction
Total RNA was isolated from cells using an RNeasy Mini kit (Qiagen Inc. Germantown, MD, USA) and quantified. One microgram of total RNA was quantified and applied to synthesize single-stranded cDNA using an ImProm-II Reverse Transcriptase kit (Promega Corporation, Madison, WI, USA), followed by amplification reactions using primers: sense, 5′-ATG ATT CCC TGT GGG ACA AG-3′ and antisense, 5′-CTG TCT CCC TCC TCT TCC CT-3′. Reaction conditions consisted of 28 cycles of 95°C for 1 minute 30 seconds, 51°C for 1 minute 10 seconds, and 72°C for 2 minutes 10 seconds, followed by a final extension of 5 minutes at 72°C. Polymerase chain reaction (PCR) products were resolved by electrophoresis on 1% agarose gels and visualized by ethidium bromide staining.
RNA interference
For transient siRNA-mediated knockdown, 30 nM double-stranded RNA duplexes targeting the TRIB3 gene (ON-TARGET plus SMARTpool, Cat. No. L-003754) and non-targeting siRNA duplexes (Cat. No. D-001810-10-05, both from Dharmacon Research, Lafayette, CO, USA) were transfected to the indicated cells using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s recommendations. After 24 hours, the cell extracts were prepared and the levels of TRIB3 were determined by Western blot analysis.
Cell lysis and Western blotting
Cell pellets were lysed in Buffer A (50 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 50 mM NaF, 1 mM Na3VO4, and 10% glycerol plus a protease inhibitor cocktail), and extracts were collected by centrifugation at 10,000×g and 4°C for 10 minutes. Protein concentrations were determined by Bradford protein assay (Bio-Rad Laboratories Inc., Hercules, CA, USA). Equal amounts of protein were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and the resolved proteins were transferred to a polyvinylidene difluoride (EMD Millipore) membrane. The membrane was blocked with 5% nonfat dry milk and probed with primary antibodies, and the signals were detected using an enhanced chemiluminescence system (EMD Millipore). For the detection of phosphoproteins, membrane was blocked with bovine serum albumin (Sigma-Aldrich) or EasyBlocker (GeneTex, Hsinchu, Taiwan).
Quantitative real-time PCR (qPCR)
Total RNA was isolated using an RNeasy Mini kit (Qiagen) and quantified. Single-stranded cDNA was synthesized from 2 µg of RNA using an ImProm-II Reverse Transcriptase kit (Promega). Specific primers were designed using the Probe Finder software from Roche Applied Science, which is available online at the Universal ProbeLibrary Assay Design Center (Roche Applied Science) and is based on the Minimum Information for publication of Quantitative real-time PCR Experiments guidelines. The probes were obtained from the Universal ProbeLibrary collection (Roche Applied Science). The qPCR analysis was performed utilizing a LightCycler Nano instrument (Roche Applied Science), and the results of each experiment were normalized with respect to the expression of the control gene, TBP.
Real-time cell analysis (RTCA)
To measure the electrical impedance of touched cell proliferation, cells (7.5×103/well) were seeded onto E-plates (Roche Applied Science) and incubated for 30 minutes. The plates were placed onto the RTCA station (xCELLigence Real-Time Cell Analysis System, Roche, Germany) and incubated overnight, and then the cells were exposed to capsaicin. Thereafter, impedance was measured every hour over a period of 99 hours. Impedance is presented as the cell index (CI)=(Zi−Z0) (Ohm)/15 (Ohm), where Z0 is the background resistance and Zi is the resistance at an individual time point. A normalized cell index was determined as the cell index at a certain time point (CIti) divided by the cell index at the normalization time point (CInml_time).
Statistical analysis
All data were obtained from at least three independent experiments. Values are presented as the mean±SD from at least three independent experiments, unless otherwise indicated. The SigmaPlot 10.0 software was used to analyze the data and calculate significant differences between the control and experimental groups, as assessed by the Student’s t-test. The results presented in the graphs are representative of multiple independent experiments, and * indicates P<0.05, which was considered statistically significant.
Results
Capsaicin induces the protein expression of TRIB3 in cancer cell lines
We first examined the expression levels of TRIB3 in seven different cancer cell lines: AGS, SW480, MKN-45, SNU-1, TMK-1, SiHa, and KATO III. As shown in Figure 1A, the TRIB3 protein levels were dramatically different in the various cancer cell lines, with relatively low levels in AGS, SNU-1, MKN-45, SiHa, and KATO III cells and high-level expression in SW480 and TMK-1 cells. Similar expression profiles were obtained from our analysis of TRIB3 mRNA expression (Figure 1A, lower panel). Given that AGS cells possess the lowest malignancy and MKN-45 cells are very malignant, our results suggest that the expression level of TRIB3 may not correlate with the malignancy of cancer cell lines in vitro. Next, to test whether the endogenous level of TRIB3 contributed to the susceptibility of cancer cells to capsaicin, we treated AGS, MKN-45, and TMK-1 cells with the indicated concentrations of capsaicin for 48 hours and then determined cell viability. However, we did not identify any significant difference in the viability of the treated cell lines at 100 µM (P>0.05; Figure 1B). To investigate whether capsaicin induces TRIB3 expression in cancer cells, we examined TRIB3 expression in AGS, MKN-45, TMK-1, and SiHa cells exposed to different concentrations of capsaicin for 24 hours, or to a consistent amount of capsaicin for various time periods. Our results revealed that capsaicin dose- (Figure 1C) and time-dependently (Figure 1D) increased TRIB3 expression in different cancer cell lines.
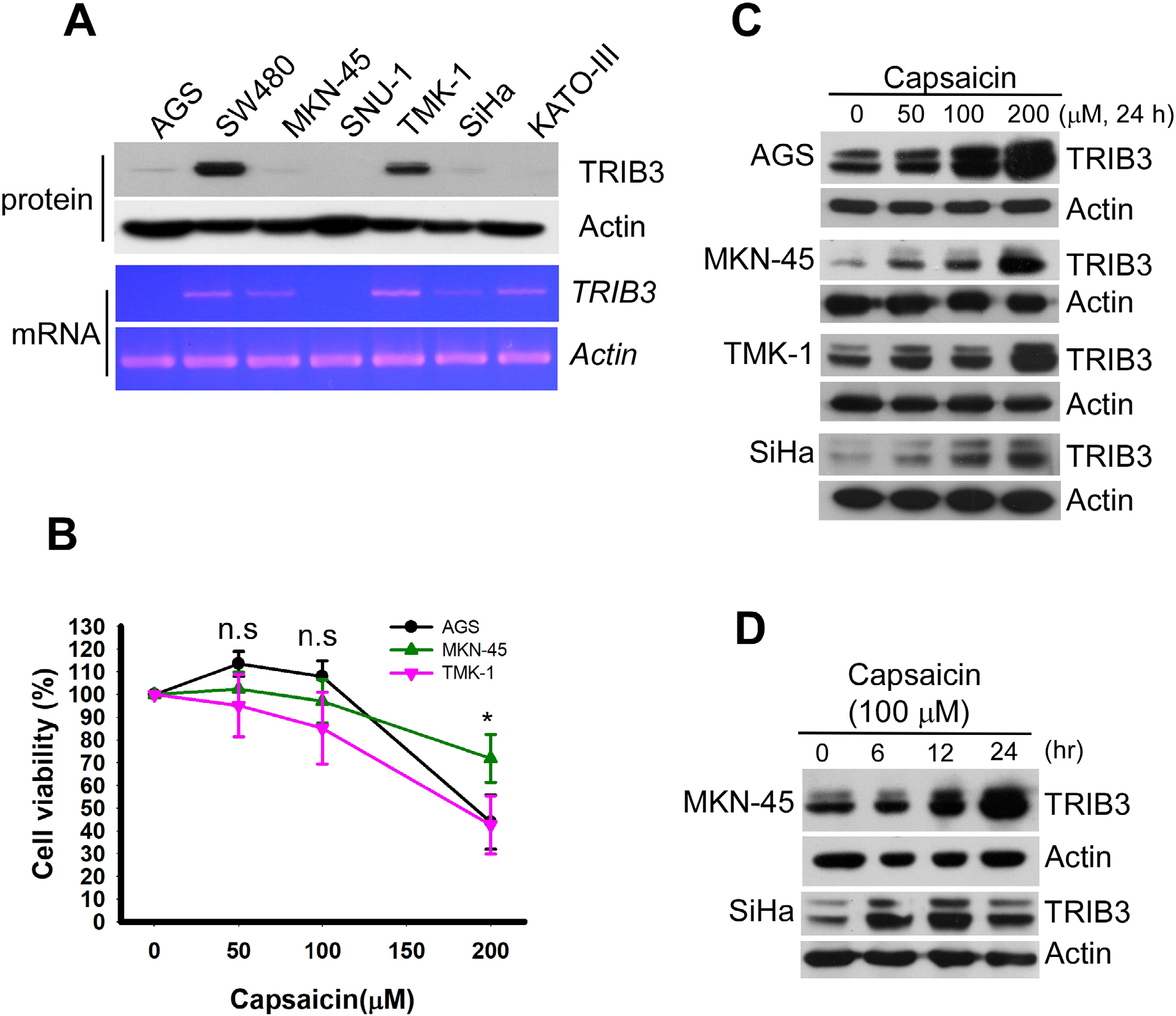 | Figure 1 Capsaicin induces the upregulation of TRIB3 in different cancer cell lines. Notes: (A) Cancer cell lines vary in their TRIB3 expression levels. The protein (upper panel) and mRNA (lower panel) expression levels of TRIB3 were measured in seven different cancer cell lines. (B) AGS, MKN-45, and TMK-1 cells were seeded in a 96-well plate for 24 hours and then exposed to 0, 50, 100, 200 µM capsaicin for 48 hours. Cell viability was determined using the WST-1 reagent. n=5. *P<0.05 as the cell viability of MKN-45 was significantly different from those of AGS and TMK-1. (C) Cells were exposed to different concentrations of capsaicin and the protein levels of TRIB3 were determined. (D) MKN-45 and SiHa cells were treated with 100 µM capsaicin for 6–24 hours and the protein levels of TRIB3 were determined. All data were obtained from at least two to four independent experiments. Abbreviation: n.s., not significant. |
Interestingly, we observed a marked increase in the cleaved forms of caspase 3 and PARP accompanied with TRIB3 upregulation in capsaicin-treated AGS (Figure 2A) and SiHa (Figure 2B), indicating that these cells underwent apoptotic cell death. Consistent with the known ability of TRIB3 to negatively regulate the activation of AKT, cells subjected to the capsaicin-induced upregulation of TRIB3 showed gradual decreases in the activation of AKT (Figure 2A and B). Dose-dependent induction of apoptosis was further confirmed in other cell lines. The increased cleaved form of PARP was also observed in capsaicin-treated TMK-1 (Figure 2C) and MKN-45 (Figure 2D) cells. Further analysis of factors involved in various apoptotic pathways revealed that capsaicin triggered significant increases in molecules involved in both intrinsic mitochondrial pathway (eg, cleaved caspase 9 and Bax) and extrinsic pathway (eg, cleaved caspase 8), as shown in Figure 2D.
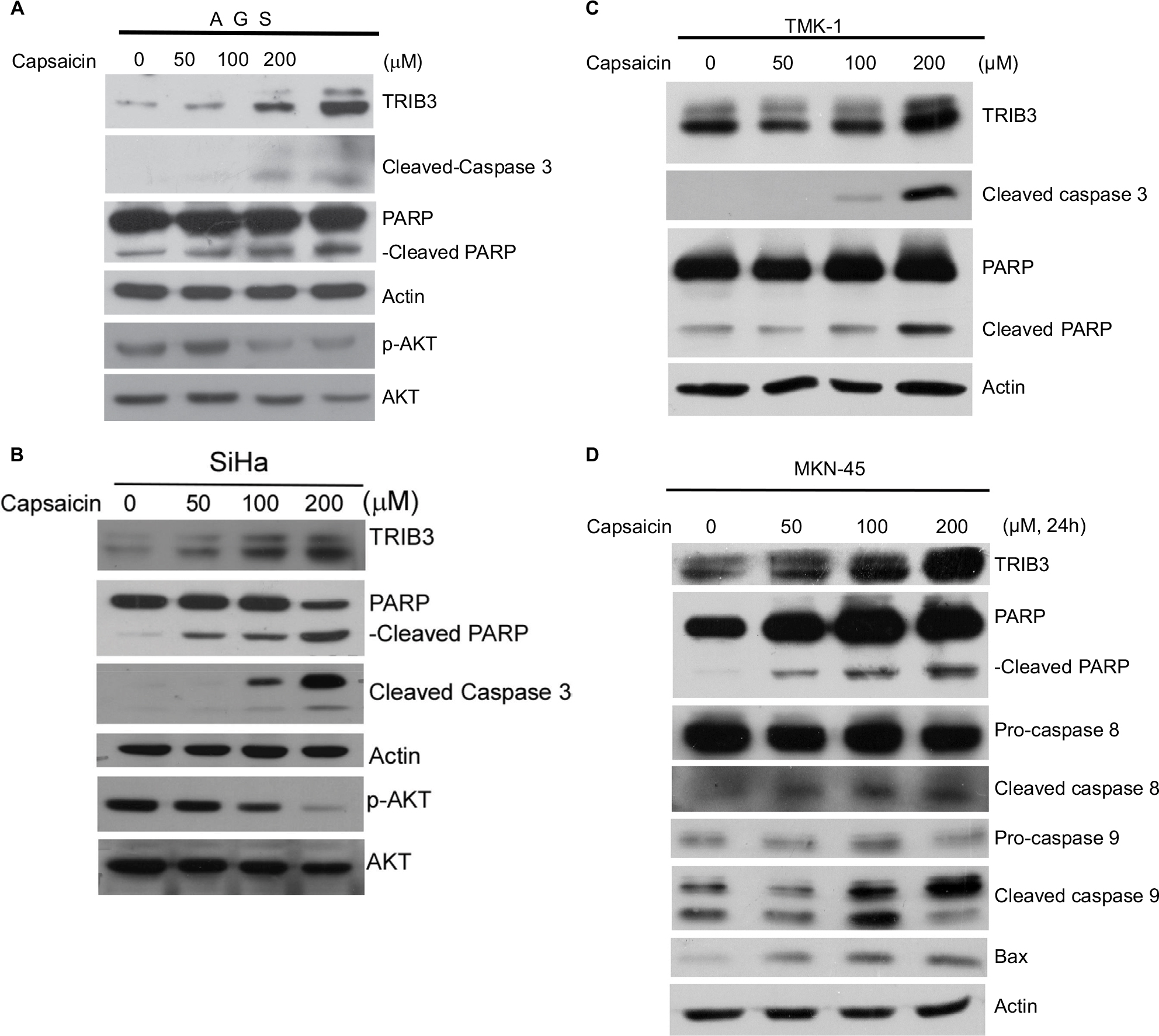 | Figure 2 Capsaicin-induced upregulation of TRIB3 is accompanied by apoptosis. Notes: (A–D) Cells were exposed to different concentrations of capsaicin and the specific protein levels were determined. All data were obtained from at least two to four independent experiments. |
Induction of TRIB3 is essential for capsaicin-induced apoptosis
Next, we investigated the functional involvement of TRIB3 in capsaicin-mediated apoptosis. TRIB3-Myc was ectopically expressed in AGS cells for 24 hours, and then the cells were treated with capsaicin. As shown in Figure 3A, ectopic expression of Myc-tagged TRIB3 augmented capsaicin-induced cleavage of caspase 3 and PARP (Figure 3A). Monitoring of cell proliferation using a RTCA system revealed that overexpression of TRIB3 attenuated cell proliferation in AGS (Figure 3B). This suggests that TRIB3 contributes to the antiproliferative and proapoptotic effects of capsaicin. Conversely, knockdown of cellular TRIB3 with specific si-RNA (si-TRIB3) decreased the levels of cleaved caspase 3 and cleaved PARP in capsaicin-treated AGS and TMK-1 cells, compared to the results from cells treated with si-control (Figure 3C). Together, these findings support the hypothesis that capsaicin elevates TRIB3 expression, and that TRIB3 contributes to cell death in capsaicin-treated cancer cells.
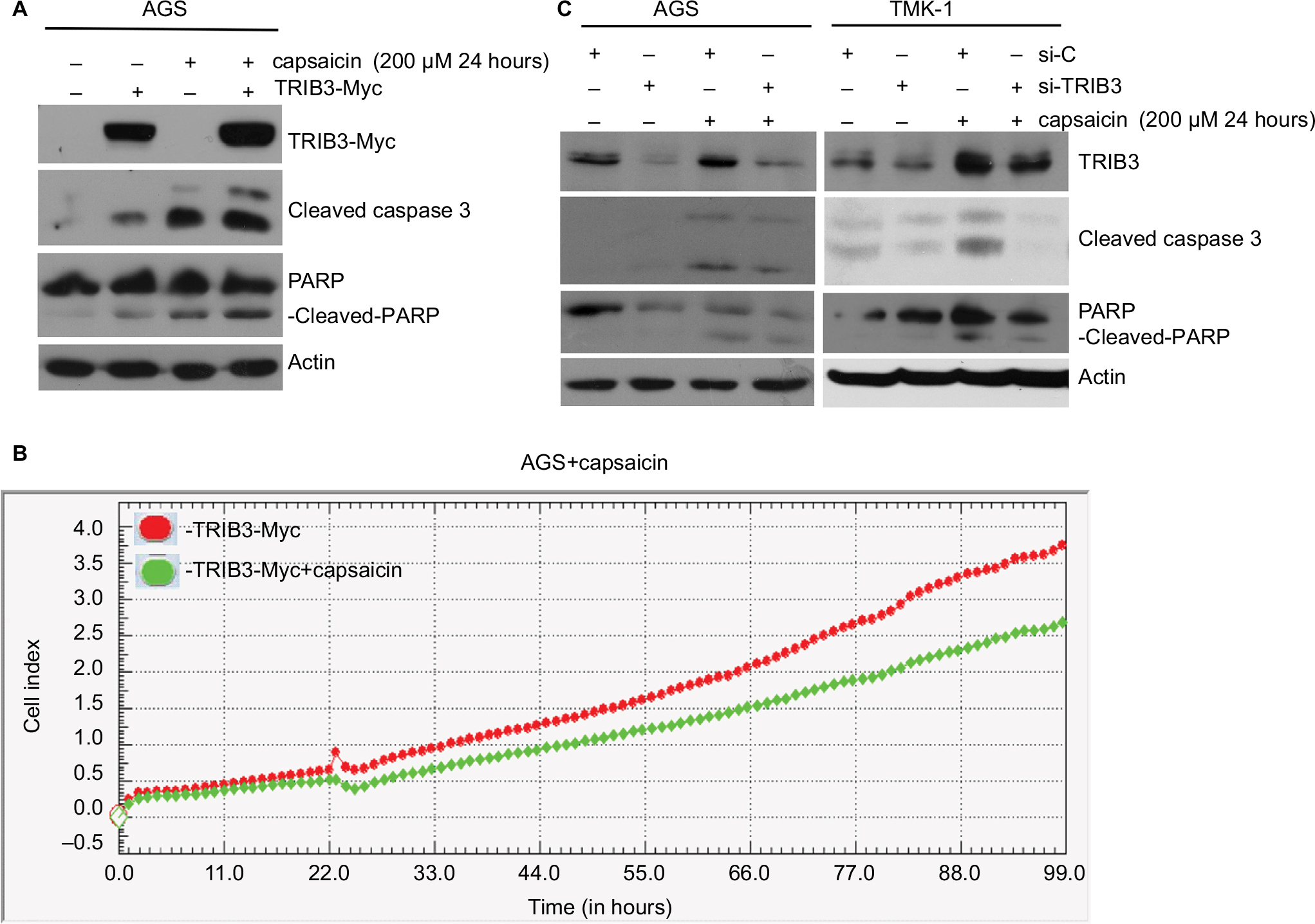 | Figure 3 Induction of TRIB3 is essential for capsaicin-induced apoptosis. Notes: (A) AGS cells were induced to express 2 µg ectopic TRIB3-Myc for 24 hours and exposed to 200 µM capsaicin for another 24 hours, and then the protein levels of TRIB3, TRIB3-Myc, cleaved caspase 3, and cleaved PARP were determined. (B) 2 µg TRIB3-Myc was expressed in AGS cells for 24 hours before exposure to 200 µM capsaicin for another 24 hours, and cell impedance was determined using an RTCA system. (C) AGS and TMK-1 were subjected to si-RNA–mediated knockdown of endogenous TRIB3 for 24 hours and then exposed to 200 µM capsaicin for another 24 hours. The protein levels of TRIB3, cleaved caspase 3, and cleaved PARP were determined. All data were obtained from at least three to five independent experiments. Abbreviation: RTCA, real-time cell analysis. |
Capsaicin enhances TRIB3 expression at the levels of mRNA expression and protein stability
Given the ability of capsaicin to induce TRIB3, we set out to test whether capsaicin could regulate the expression and function of TRIB3 in cancer cell lines. TRIB3 has been shown as a short-lived protein that is frequently degraded through the ubiquitin–proteasome pathway.44,45 To test whether the differences of TRIB3 in these cancer cell lines were due to differences in protein stability, we examined the levels of TRIB3 in four different cancer cell lines with MG132 treatment. Inhibition of proteasome pathway by MG132 further enhanced the levels of TRIB3 in a time-dependent manner (Figure 4A). This confirmed that the turnover of TRIB3 occurs through 26S proteasome-mediated proteolysis in these cancer cell lines. Furthermore, we treated cells with cycloheximide (a protein synthesis inhibitor) and tested the half-life of TRIB3 by Western blotting at different time points posttreatment (0, 0.5, 1, 2 hours). As shown in Figure 4B, the protein turnover of TRIB3 was not significantly different among the tested cancer lines. These results suggest that the differential expression of TRIB3 might reflect differences in transcriptional regulation among the cancer cell lines. To examine whether capsaicin induced TRIB3 expression by increasing gene transcription, we treated cells with capsaicin for 24 hours and then determined the TRIB3 mRNA levels. Our qPCR analyses revealed that capsaicin significantly increased the mRNA level of TRIB3 in AGS cells (Figure 4C), indicating that increased gene expression, at least in part, might be involved in the capsaicin-induced upregulation of TRIB3. Since protein turnover also plays a critical role in regulating the protein level and function of TRIB3,44–46 the cycloheximide experiment was performed, which revealed that TRIB3 protein stability was moderately, but significantly enhanced by capsaicin in AGS (Figure 4D) and MKN-45 (Figure 4E) cells. Thus, stabilization of the TRIB3 protein also appears to be an important underlying mechanism of capsaicin-mediated TRIB3 upregulation. Taken together, our data suggest that both transcriptional upregulation and increased protein stability might contribute to capsaicin-mediated TRIB3 expression.
 | Figure 4 Capsaicin enhances TRIB3 mRNA expression and protein stability. Notes: (A) AGS, MKN-45, and SiHa cells were treated with 10 µM MG132 for the indicated time periods, cell lysates were prepared, and the protein levels of TRIB3 were detected by immunoblotting. (B) Cells were treated with 50 µg/mL CHX for the indicated time periods, cell lysates were prepared, and the protein levels of TRIB3 were measured. (C) AGS cells were exposed to capsaicin for 24 hours, total mRNA was isolated, and real-time PCR was used to measure the mRNA expression of TRIB3. n=5. *P<0.05. The results of each experiment were normalized with respect to the expression of the internal control gene, TBP. (D) AGS and (E) MKN-45 cells were pretreated with or without capsaicin for 22 hours and then co-treated with 50 µg/mL CHX for 0–2 hours. The protein levels of TRIB3 were determined. All data were obtained from at least three to six independent experiments. n=3 for AGS, n=6 for MKN-45. Abbreviations: CHX, cycloheximide; PCR, polymerase chain reaction; TBP, TATA- box binding protein. |
Molecular regulation of TRIB3 expression by capsaicin
Since capsaicin has been reported to modulate various signaling pathways and alter cell growth, we investigated the potential roles of several signaling pathways in controlling capsaicin-enhanced TRIB3 expression. Indeed, capsaicin was found to dose-dependently enhance the activation of ERK, JNK, and p38 in AGS cells (Figure 5A). However, inhibitor experiments revealed that JNK and p38 MAPK signaling did not involve capsaicin-mediated upregulation of TRIB3 (Figure 5B). Inhibition of MEK-ERK1/2 signaling significantly suppressed the protein level of TRIB3 induced by capsaicin (Figure 5C). To further study whether MEK-ERK1/2 pathway participated in capsaicin-induced TRIB3 gene expression, cells were co-treated with U0126 and capsaicin for 24 hours and then the TRIB3 mRNA levels were determined. The result indicated that MEK1/2 inhibitor U0126 did not interfere with the mRNA expression of TRIB3 in capsaicin-treated AGS cells (Figure 5D), implying that MEK-ERK1/2 signaling may regulate the protein expression of TRIB3 induced by capsaicin at the posttranscriptional level. In addition, knockdown of TRIB3 did not suppress the capsaicin-mediated activation of MAPK signaling in AGS (data not shown). Collectively, our results suggest that capsaicin upregulates TRIB3 expression by increasing its gene transcription and protein stability. Mechanistically, although capsaicin activates three MAPK signaling pathways, JNK and p38 activation does not appear to participate in the capsaicin-induced upregulation of TRIB3, nor does TRIB3 appear to contribute to the capsaicin-mediated activation of MAPK signaling.
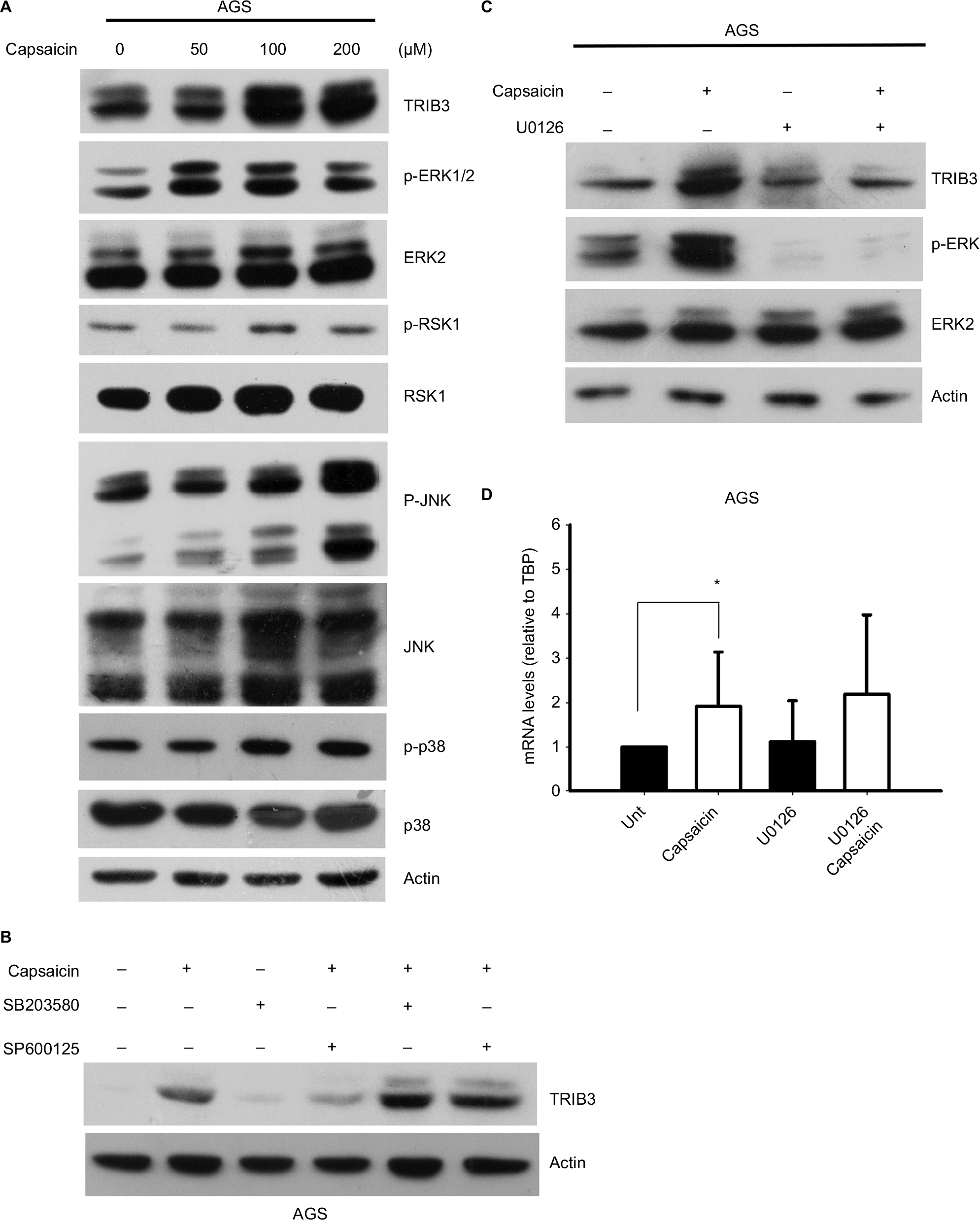 | Figure 5 JNK and p38 MAPK signaling pathways do not appear to participate in the capsaicin-triggered upregulation of TRIB3. Notes: (A) AGS cells were exposed to different doses of capsaicin for 24 hours, cell lysates were prepared, and the levels of various signaling proteins were determined. (B) AGS cells were pretreated with 10 µM SB203580 or SP600125 for 1 hour and then with 100 µM capsaicin for another 24 hours. The levels of TRIB3 were determined. (C, D) AGS cells were pretreated with 10 µM U0126 for 1 hour and then with 100 µM capsaicin for another 24 hours. The protein (C) and mRNA (D) levels of TRIB3 were determined. n=5. *P<0.05. The mRNA levels of TRIB3 were normalized with respect to the expression of the internal control gene, TBP. Abbreviation: TBP, TATA-box binding protein. |
Discussion
Increasing evidence suggests that capsaicin has anticancer effects, but we have not yet fully elucidated the molecular mechanism underlying capsaicin-induced growth inhibition and apoptosis of human cancer cells. In this study, we report that capsaicin dose- and time-dependently induces TRIB3 expression and promotes apoptosis in different cancer cells. Moreover, the antiproliferative and proapoptotic effects of capsaicin were found to correlate with its induction of TRIB3 expression, which occurs via both transcriptional and posttranslational mechanisms (Figure 6). Although the detailed mechanism underlying the capsaicin-induced gene upregulation and protein stabilization of TRIB3 is still poor understood, our findings clearly indicate that capsaicin-induced alterations in TRIB3 might be a major event leading to apoptosis in capsaicin-treated cancer cells.
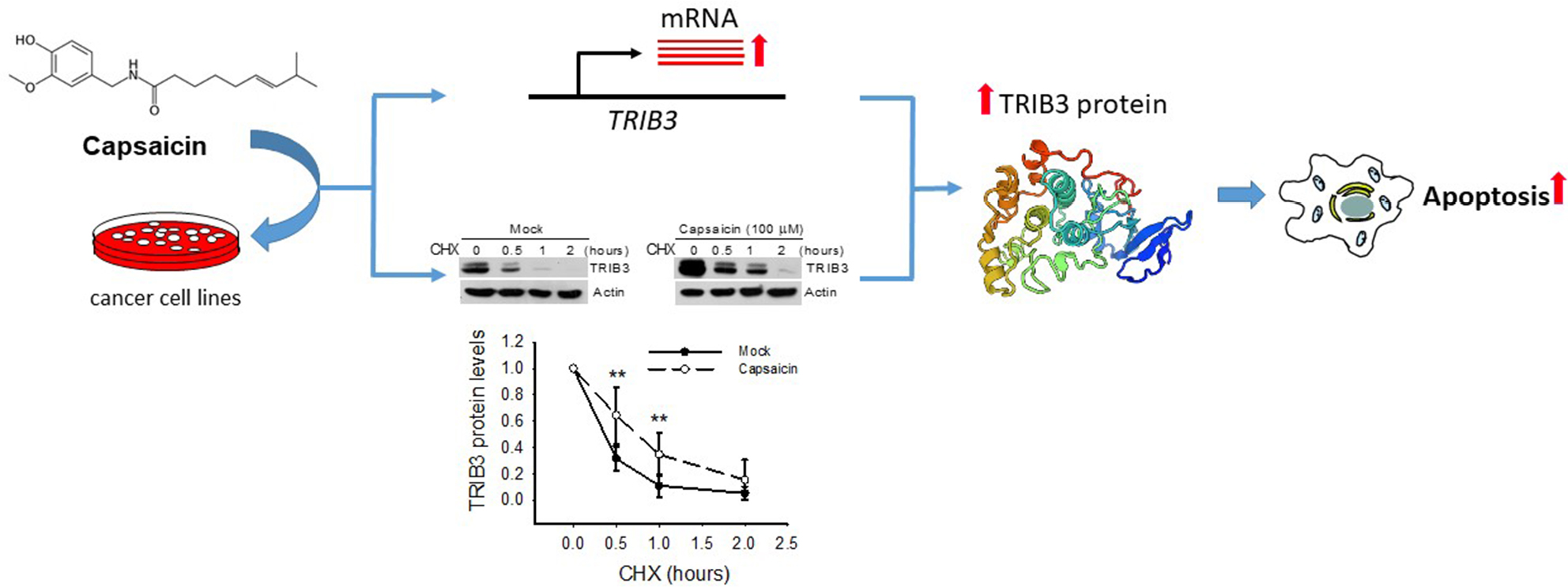 | Figure 6 Capsaicin induced apoptosis through upregulating TRIB3 expression in cancer cells. Notes: The proposed model is consistent with the published studies and the results described here. Capsaicin dose- and time-dependently induces TRIB3 expression via both stimulating gene transcription and stabilizing protein stability mechanisms to promote apoptosis in cancer cells. Abbreviation: CHX, cycloheximide. |
Capsaicin has been shown to exert profound antiproliferative activity in animal/cell models of cancer, mostly by inducing apoptosis. The modulations of signaling pathways and transcription factor functions are major mechanisms contributing to the antiproliferative effect of capsaicin. In addition, capsaicin can induce apoptosis by indirectly inhibiting the ubiquitin–proteasome system, leading to the accumulation of target substrates normally targeted by the proteasome, such as p53, Bax, and p27.47 By inhibiting the interaction between p53 and MDM2, capsaicin has been reported to stabilize the p53 protein and enhance the transcriptional activity of its encoding gene in human colon cancer cells.22 In the present study, we report that TRIB3 is a potent target of capsaicin and its upregulation might play a critical role in the capsaicin-mediated apoptosis of different human cancer cells.
Studies have shown that the protein level of TRIB3 is tightly regulated at several levels, including both transcriptional and translational mechanisms. For example, the TRIB3 mRNA is induced by the unfolded protein response via the transcription factor, ATF4, as well as by the NFκB and PI3K pathways.48 Moreover, the TRIB3 mRNA may be stabilized by the hypoxia-regulated protein, HuR.48 At the protein level, TRIB3 is modulated by ubiquitin-mediated proteolysis. Siah1 interacts with and ubiquitinates TRIB3 in vivo to regulate its turnover via the ubiquitin–proteasome pathway.44 A typical D-box motif localized in the carboxyl-terminal domain of TRIB3 is required for its protein stability, and the APC/CCdh1 protein is involved in ubiquitinating and destabilizing the TRIB3 protein.45 In the present study, we reveal that capsaicin enhances TRIB3 gene expression and protein stability, thereby augmenting apoptotic cell death in cancer cells. Although JNK and p38 MAPK were not involved in the induction of TRIB3 by capsaicin, MEK-ERK1/2 signaling may regulate capsaicin-induced TRIB3 at the posttranscriptional level. The detailed mechanism should be further investigated in the future.
Using a functional genetic analysis, we identified the –1561 to +77 bp region upstream of the TRIB3 gene as being relevant to the construction of a luciferase reporter system. However, our results revealed that this upstream region was not responsible for the induction of TRIB3 by capsaicin (data not shown). Future studies are needed to elucidate the detailed mechanisms (eg, TRIB3 phosphorylation, ubiquitin ligase activity, and/or proteasomal function) responsible for the capsaicin-mediated changes in the gene expression and stability of TRIB3.
TRIB3 is often activated by a wide variety of stress signals, including ER stress, hypoxia, and starvation. We previously reported that TRIB3 gene expression is dose-dependently upregulated by gold nanoparticle exposure in a cell model,49 supporting the notion that TRIB3 may act as a marker of stress and increased metabolism. However, the existing results are rather contradictory regarding the potential pro- or antiapoptotic roles of TRIB3 in response to diverse stresses. This suggests that there may be fundamentally different correlations between TRIB3 mRNA/protein changes and the resulting cellular phenotype. Alternatively, the contradictory results may reflect that TRIB3 is responsible for the set points of several regulatory feedback loops.
Interestingly, we have reported that TRIB3 downregulation enhances doxorubicin-induced cytotoxicity in gastric cancer cells,50 indicating TRIB3 plays a prosurvival role in doxorubicin-induced cytotoxicity in gastric cancer cells. Furthermore, we also suggest that ER stress and extrinsic apoptotic pathways may not be involved in doxorubicin-induced cytotoxicity. Interestingly, in this study, we found that capsaicin-enhanced TRIB3 expression is accompanied by increased levels of Bip (GRP78) and cleaved caspase 12 (data not shown), two well-known markers for ER stress. We anticipate that increased TRIB3 expression plays an important role in capsaicin-induced apoptosis through the induction of ER stress. Consistent with previous reports, it seems like that TRIB3 has the opposite role in apoptosis in gastric cancer cell lines, dependent on apoptotic inducers such as capsaicin or doxorubicin. More specifically, ER stress might be an important switch to determine the pro- or antiapoptotic role of TRIB3. In conclusion, the apparent dual function of TRIB3 may act as a key cellular context-dependent switch between cell survival and apoptosis, and it is not a bona fide member of apoptotic machinery. Therefore, we need to make very careful conclusion for the function of this protein in apoptosis.
Gastric cancer is the fourth most common cancer worldwide, but conventional chemotherapy and radiation are not effective in prolonging the long-term survival of gastric cancer patients. The existing evidence suggests that combinatorial anticancer therapeutic interventions, such as the addition of natural compounds to the conventional strategies, are needed to completely eradicate neoplastic lesions and minimize the side effects. Capsaicin has been demonstrated to negatively regulate cell survival, growth, inflammation, and differentiation. In this study using cell culture model, we presented a novel mechanism whereby capsaicin upregulates TRIB3 to promote apoptosis in gastric cancer cell lines. This suggests that the combination of capsaicin with effectively targeted chemotherapeutic agents might provide an efficient strategy for the treatment of human tumors, such as gastric cancer. However, the effect of capsaicin in cell culture may not be well correlated with the responses and mechanisms in vivo and the concentrations of capsaicin used in this study were not physiologically relevant. The significance of TRIB3 in capsaicin-mediated apoptosis in vivo should be further investigated.
Acknowledgments
We appreciate the help of Dr Chun-Ying Wu (Department of Education and Research and Division of Gastroenterology, Taichung Veterans General Hospital, Taichung, Taiwan; current address is Taipei Veterans General Hospital and Faculty of Medicine, School of Medicine, National Yang-Ming University, Taipei, Taiwan) who kindly provided us these cancer cell lines for this study. We thank the cell bank at Bioresource Collection and Research Center (Taiwan) for the providing the STR profiling data. Financial support was provided by a grant from the Ministry of Health and Welfare (Feng Yuan Hospital Research Project 103-003), Taiwan.
Disclosure
The authors report no conflicts of interest in this work.
References
Jung MY, Kang HJ, Moon A. Capsaicin-induced apoptosis in SK-Hep-1 hepatocarcinoma cells involves Bcl-2 downregulation and caspase-3 activation. Cancer Lett. 2001;165(2):139–145. | ||
Tanaka T, Kohno H, Sakata K, et al. Modifying effects of dietary capsaicin and rotenone on 4-nitroquinoline 1-oxide-induced rat tongue carcinogenesis. Carcinogenesis. 2002;23(8):1361–1367. | ||
Ito K, Nakazato T, Yamato K, et al. Induction of apoptosis in leukemic cells by homovanillic acid derivative, capsaicin, through oxidative stress: implication of phosphorylation of p53 at Ser-15 residue by reactive oxygen species. Cancer Res. 2004;64(3):1071–1078. | ||
Kim CS, Park WH, Park JY, et al. Capsaicin, a spicy component of hot pepper, induces apoptosis by activation of the peroxisome proliferator-activated receptor gamma in HT-29 human colon cancer cells. J Med Food. 2004;7(3):267–273. | ||
Lo YC, Yang YC, Wu IC, et al. Capsaicin-induced cell death in a human gastric adenocarcinoma cell line. World J Gastroenterol. 2005;11(40):6254–6257. | ||
Mori A, Lehmann S, O’Kelly J, et al. Capsaicin, a component of red peppers, inhibits the growth of androgen-independent, p53 mutant prostate cancer cells. Cancer Res. 2006;66(6):3222–3229. | ||
Wu CC, Lin JP, Yang JS, et al. Capsaicin induced cell cycle arrest and apoptosis in human esophagus epidermoid carcinoma CE 81T/VGH cells through the elevation of intracellular reactive oxygen species and Ca2+ productions and caspase-3 activation. Mutat Res. 2006;601(1–2):71–82. | ||
Sánchez AM, Malagarie-Cazenave S, Olea N, Vara D, Chiloeches A, Díaz-Laviada I. Apoptosis induced by capsaicin in prostate PC-3 cells involves ceramide accumulation, neutral sphingomyelinase, and JNK activation. Apoptosis. 2007;12(11):2013–2024. | ||
Zhang R, Humphreys I, Sahu RP, Shi Y, Srivastava SK. In vitro and in vivo induction of apoptosis by capsaicin in pancreatic cancer cells is mediated through ROS generation and mitochondrial death pathway. Apoptosis. 2008;13(12):1465–1478. | ||
Chou CC, Wu YC, Wang YF, Chou MJ, Kuo SJ, Chen DR. Capsaicin-induced apoptosis in human breast cancer MCF-7 cells through caspase-independent pathway. Oncol Rep. 2009;21(3):665–671. | ||
Brown KC, Witte TR, Hardman WE, et al. Capsaicin displays anti-proliferative activity against human small cell lung cancer in cell culture and nude mice models via the E2F pathway. PLoS One. 2010;5(4):e10243. | ||
Lu HF, Chen YL, Yang JS, et al. Antitumor activity of capsaicin on human colon cancer cells in vitro and colo 205 tumor xenografts in vivo. J Agric Food Chem. 2010;58(24):12999–13005. | ||
Meral O, Alpay M, Kismali G, et al. Capsaicin inhibits cell proliferation by cytochrome c release in gastric cancer cells. Tumour Biol. 2014;35(7):6485–6492. | ||
Huh HC, Lee SY, Lee SK, Park NH, Han IS. Capsaicin induces apoptosis of cisplatin-resistant stomach cancer cells by causing degradation of cisplatin-inducible Aurora-A protein. Nutr Cancer. 2011;63(7):1095–1103. | ||
Ying H, Wang Z, Zhang Y, et al. Capsaicin induces apoptosis in human osteosarcoma cells through AMPK-dependent and AMPK-independent signaling pathways. Mol Cell Biochem. 2013;384(1–2):229–237. | ||
Lee GR, Jang SH, Kim CJ, et al. Capsaicin suppresses the migration of cholangiocarcinoma cells by down-regulating matrix metalloproteinase-9 expression via the AMPK-NF-κB signaling pathway. Clin Exp Metastasis. 2014;31(8):897–907. | ||
Bu HQ, Cai K, Shen F, et al. Induction of apoptosis by capsaicin in hepatocellular cancer cell line SMMC-7721 is mediated through ROS generation and activation of JNK and p38 MAPK pathways. Neoplasma. 2015;62(4):582–591. | ||
Park SY, Kim JY, Lee SM, et al. Capsaicin induces apoptosis and modulates MAPK signaling in human gastric cancer cells. Mol Med Rep. 2014;9(2):499–502. | ||
Zhang JH, Lai FJ, Chen H, et al. Involvement of the phosphoinositide 3-kinase/Akt pathway in apoptosis induced by capsaicin in the human pancreatic cancer cell line PANC-1. Oncol Lett. 2013;5(1):43–48. | ||
Malagarie-Cazenave S, Olea-Herrero N, Vara D, Morell C, Díaz-Laviada I. The vanilloid capsaicin induces IL-6 secretion in prostate PC-3 cancer cells. Cytokine. 2011;54(3):330–337. | ||
Kim JD, Kim JM, Pyo JO, et al. Capsaicin can alter the expression of tumor forming-related genes which might be followed by induction of apoptosis of a Korean stomach cancer cell line, SNU-1. Cancer Lett. 1997;120(2):235–241. | ||
Jin J, Lin G, Huang H, et al. Capsaicin mediates cell cycle arrest and apoptosis in human colon cancer cells via stabilizing and activating p53. Int J Biol Sci. 2014;10(3):285–295. | ||
Chakraborty S, Adhikary A, Mazumdar M, et al. Capsaicin-induced activation of p53-SMAR1 auto-regulatory loop down-regulates VEGF in non-small cell lung cancer to restrain angiogenesis. PLoS One. 2014;9(6):e99743. | ||
Singh S, Natarajan K, Aggarwal BB. Capsaicin (8-methyl-N-vanillyl-6-nonenamide) is a potent inhibitor of nuclear transcription factor-kappa B activation by diverse agents. J Immunol. 1996;157(10):4412–4420. | ||
Surh YJ, Han SS, Keum YS, Seo HJ, Lee SS. Inhibitory effects of curcumin and capsaicin on phorbol ester-induced activation of eukaryotic transcription factors, NF-kappaB and AP-1. Biofactors. 2000;12(1–4):107–112. | ||
Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3(10):768–780. | ||
Hegedus Z, Czibula A, Kiss-Toth E. Tribbles: a family of kinase-like proteins with potent signalling regulatory function. Cell Signal. 2007;19(2):238–250. | ||
Ord D, Meerits K, Ord T. TRB3 protects cells against the growth inhibitory and cytotoxic effect of ATF4. Exp Cell Res. 2007;313(16):3556–3567. | ||
Takahashi Y, Ohoka N, Hayashi H, Sato R. TRB3 suppresses adipocyte differentiation by negatively regulating PPARgamma transcriptional activity. J Lipid Res. 2008;49(4):880–892. | ||
Hua F, Mu R, Liu J, et al. TRB3 interacts with SMAD3 promoting tumor cell migration and invasion. J Cell Sci. 2011;124(Pt 19):3235–3246. | ||
Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300(5625):1574–1577. | ||
Kiss-Toth E, Bagstaff SM, Sung HY, et al. Human tribbles, a protein family controlling mitogen-activated protein kinase cascades. J Biol Chem. 2004;279(41):42703–42708. | ||
Wu M, Xu LG, Zhai Z, Shu HB. SINK is a p65-interacting negative regulator of NF-kappaB-dependent transcription. J Biol Chem. 2003;278(29):27072–27079. | ||
Schwarzer R, Dames S, Tondera D, Klippel A, Kaufmann J. TRB3 is a PI 3-kinase dependent indicator for nutrient starvation. Cell Signal. 2006;18(6):899–909. | ||
Bowers AJ, Scully S, Boylan JF. SKIP3, a novel Drosophila tribbles ortholog, is overexpressed in human tumors and is regulated by hypoxia. Oncogene. 2003;22(18):2823–2835. | ||
Corcoran CA, Luo X, He Q, Jiang C, Huang Y, Sheikh MS. Genotoxic and endoplasmic reticulum stresses differentially regulate TRB3 expression. Cancer Biol Ther. 2005;4(10):1063–1067. | ||
Zareen N, Biswas SC, Greene LA. A feed-forward loop involving Trib3, Akt and FoxO mediates death of NGF-deprived neurons. Cell Death Differ. 2013;20(12):1719–1730. | ||
Li T, Su L, Zhong N, et al. Salinomycin induces cell death with autophagy through activation of endoplasmic reticulum stress in human cancer cells. Autophagy. 2013;9(7):1057–1068. | ||
Salazar M, Lorente M, Garcia-Taboada E, et al. The pseudokinase tribbles homologue-3 plays a crucial role in cannabinoid anticancer action. Biochim Biophys Acta. 1831;2013(10):1573–1578. | ||
Erazo T, Lorente M, López-Plana A, et al. The new antitumor drug ABTL0812 inhibits the Akt/mTORC1 axis by upregulating tribbles-3 pseudokinase. Clin Cancer Res. 2016;22(10):2508–2519. | ||
Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005;24(6):1243–1255. | ||
Shimizu K, Takahama S, Endo Y, Sawasaki T. Stress-inducible caspase substrate TRB3 promotes nuclear translocation of procaspase-3. PLoS One. 2012;7(8):e42721. | ||
Xu M, Lu N, Sun Z, et al. Activation of the unfolded protein response contributed to the selective cytotoxicity of oroxylin A in human hepatocellular carcinoma HepG2 cells. Toxicol Lett. 2012;212(2):113–125. | ||
Zhou Y, Li L, Liu Q, et al. E3 ubiquitin ligase SIAH1 mediates ubiquitination and degradation of TRB3. Cell Signal. 2008;20(5):942–948. | ||
Ohoka N, Sakai S, Onozaki K, Nakanishi M, Hayashi H. Anaphase-promoting complex/cyclosome-cdh1 mediates the ubiquitination and degradation of TRB3. Biochem Biophys Res Commun. 2010;392(3):289–294. | ||
Wennemers M, Bussink J, van den Beucken T, Sweep FC, Span PN. Regulation of TRIB3 mRNA and protein in breast cancer. PLoS One. 2012;7(11):e49439. | ||
Maity R, Sharma J, Jana NR. Capsaicin induces apoptosis through ubiquitin-proteasome system dysfunction. J Cell Biochem. 2010;109(5):933–942. | ||
Rzymski T, Paantjens A, Bod J, Harris AL. Multiple pathways are involved in the anoxia response of SKIP3 including HuR-regulated RNA stability, NF-kappaB and ATF4. Oncogene. 2008;27(33):4532–4543. | ||
Liang R-Y, Tu H-F, Tan X, Yeh Y-S, Chueh PJ, Chuang S-M. A gene signature for gold nanoparticle-exposed human cell lines. Toxicol Res. 2015;4(2):365–375. | ||
Wu IJ, Lin RJ, Wang HC, Yuan TM, Chuang SM. TRIB3 downregulation enhances doxorubicin-induced cytotoxicity in gastric cancer cells. Arch Biochem Biophys. 2017;622:26–35. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.