")
Back to Archived Journals » Research and Reports in Biology » Volume 6
Calcium sparks in the heart: dynamics and regulation
Authors Hoang-Trong T, Ullah A, Jafri MS
Received 2 July 2015
Accepted for publication 12 August 2015
Published 16 October 2015 Volume 2015:6 Pages 203—214
DOI https://doi.org/10.2147/RRB.S61495
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Zvi Kelman
Tuan M Hoang-Trong,1 Aman Ullah,1 M Saleet Jafri1,2
1Department of Molecular Neuroscience, Krasnow Institute for Advanced Study, George Mason University, Fairfax, VA, USA; 2Biomedical Engineering and Technology, University of Maryland, Baltimore, MD, USA
Abstract: Ca2+ plays a central role in the contraction of the heart. It is the bi-directional link between electrical excitation of the heart and contraction. Electrical excitation initiates Ca2+ influx across the sarcolemma and T-tubular membrane that triggered calcium release from the sarcoplasmic reticulum (SR). Ca2+ sparks are the elementary events of calcium release from the SR. Therefore, understanding the dynamics of Ca2+ sparks is essential for understanding the function of the heart. To this end, numerous experimental and computational studies have focused on this topic, exploring the mechanisms of calcium spark initiation, termination, and regulation and what role these play in normal and patho-physiology. The proper understanding of Ca2+ spark regulation and dynamics serves as the foundation for our insights into a multitude of pathological conditions that may develop and that can be the result of structural and/or functional changes at the cellular or subcellular level. Computational modeling of Ca2+ spark dynamics has proven to be a useful tool to understand Ca2+ spark dynamics. This review addresses our current understanding of Ca2+ sparks and how synchronized SR Ca2+ release, in which Ca2+ sparks is a major pathway, is linked to the different cardiac diseases, especially arrhythmias.
Keywords: leak, arrhythmia, excitation-contraction coupling, phosphorylation
Introduction
The primary function of the heart is to pump blood to carry oxygen and nutrients to the tissue and carbon dioxide and waste products away from the tissue for removal from the body. The well-regulated, rhythmic contraction of the heart accomplishes this goal through a wide range of metabolic demands. Excitation–contraction coupling (ECC) is the process starting with the electrical excitation of the heart and ending with the contraction of the heart muscle. The intermediate step is the mobilization of calcium which not only binds to myofilaments to cause contraction but it also provides feedback on the electrical signal.1 Calcium is also involved a myriad of intracellular signaling processes and regulates several signaling cascades.2–6
Ca2+ sparks are the elementary Ca2+ release events in heart.7 In this review several scientific questions about Ca2+ sparks will be addressed. These include a discussion of the mechanisms of Ca2+ spark initiation and termination. These areas have been a topic of much scientific inquiry with competing hypotheses. The review will also discuss the role of Ca2+ spark dynamics in normal cardiac myocyte function and disease. To this end, the topics of cellular Ca2+ homeostasis, Ca2+ signaling, heart failure (HF), and arrhythmia will be discussed.
Excitation-contraction coupling and Ca2+ sparks
ECC is tightly controlled by the action of different ions. Central to ECC is Ca2+ signaling which is described in detail in the review by Fearnley et al.8 Here we give a brief overview.
The transmembrane potential depolarization triggers the voltage-gated ionic channels (primarily Na+, K+, and Ca2+ selective permeable channels) with different activation and deactivation time constants. These opening channels generate different ionic currents (INa, IK [IKtof, IKtos, IK1, IKss], and ICa). The translocation of ions across the membrane via these opening, ion channels causes a shift in ionic gradients (Δ[Na+], Δ[K+], Δ[Ca2+]) and subsequently affects the transmembrane potential (Vm). In a cycle of the ECC, at the cellular level, the transmembrane potential transiently depolarizes forming the so-called action potential (AP). Cardiac cells, at different regions of the heart, have different expression levels of ionic channels and thus, the AP can have different waveforms.9
The aforementioned voltage-gated ion channels are located in the sarcolemma as well as in the t-tubule system which penetrate throughout the ventricular myocyte. Atrial myocytes have varying amounts of t-tubules. The t-tubules are located at the ends of the sarcomeres. Closely apposed (12–15 nm distance) to the T-tubular membrane at select locations is the junctional sarcoplasmic reticulum (JSR) to define the dyad. This is the main site of calcium release in myocyte. A majority of the L-type Ca2+ channels (LCCs) (~80%) are found in the T-tubular membrane of the dyad. The remainder is considered to be nonjunctional, located elsewhere in the t-tubule and sarcolemma. Figure 1A shows the repeating pattern of the sarcomere in the myocyte with the dyad circled. Figure 1B shows the details of the dyad and calcium cycling in the myocyte.
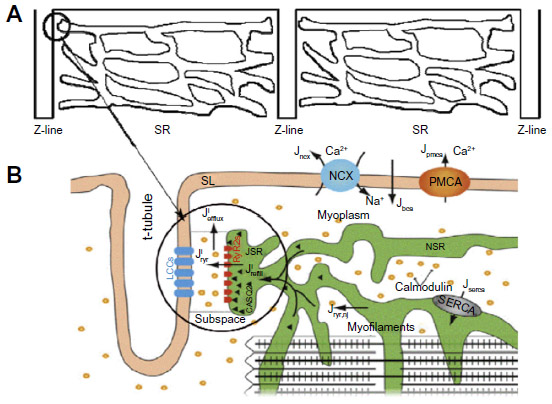 | Figure 1 A schematic diagram of calcium-induced calcium-release mechanism in a cardiac myocyte. |
In cardiac myocytes, upon membrane depolarization, the opening of voltage-gated LCC can bring the local [Ca2+] from the resting level (~0.1 μM) to a level that is high enough to trigger the opening of an array of 30–300 ryanodine receptors (RyRs) in the JSR membrane.10–13 This process is termed Ca2+-induced Ca2+ release (CICR). This is known as evoked Ca2+ sparks or triggered Ca2+ sparks. A similar phenomenon has also been found in other cell types, eg, skeletal cells and smooth muscle cells.10,14,15 On the other hand, under resting condition, without any electrical stimulus, spontaneous calcium sparks resulting from the stochastic opening of one or more RyR2 are observed.16,17
The narrow space of dyad allows the small influx of calcium via LCC or the calcium elevation due to the opening of one or a few RyR2 channel to bring the subspace calcium concentration above the half-maximal activation level (Km ~12 μM).18 Due to the restricted subspace in the dyad, the released calcium ions from the stochastic opening of a single RyR2 channel quickly diffuse to the neighboring RyR2 channel in the dyad and bind to the activation site and trigger additional channel opening. This CICR (strong positive feedback) causes a fast amplification of the Ca2+ signal.
Both the triggered and spontaneous elementary Ca2+ release events are called Ca2+ sparks and were discovered by Cheng et al.7 In rat ventricular myocytes there are ~20,000 of such Ca2+ release sites in a single cardiac cell of volume ~20 pL.7 The summation of sparks from a fraction of calcium release sites results in the Ca2+ transient which binds to troponin-C in the myofilaments. The mechanism of Ca2+ spark activation and termination is the topic of active research in the last decade.19,20
In cardiac ventricular myocytes, the microdomain of calcium release is a dyad subspace formed by a cluster of RyR2 (30–100 channels) on the terminal cisternae side and a smaller cluster of LCC on the T-tubular wall, in the case of cardiac myocyte, as shown in Figure 2. The dimension of this subspace is at the range of 10–15 nm height and 200–400 nm width. The physiological role of the variation in the cluster size and the spatial locations of these clusters, however, are not clearly understood.
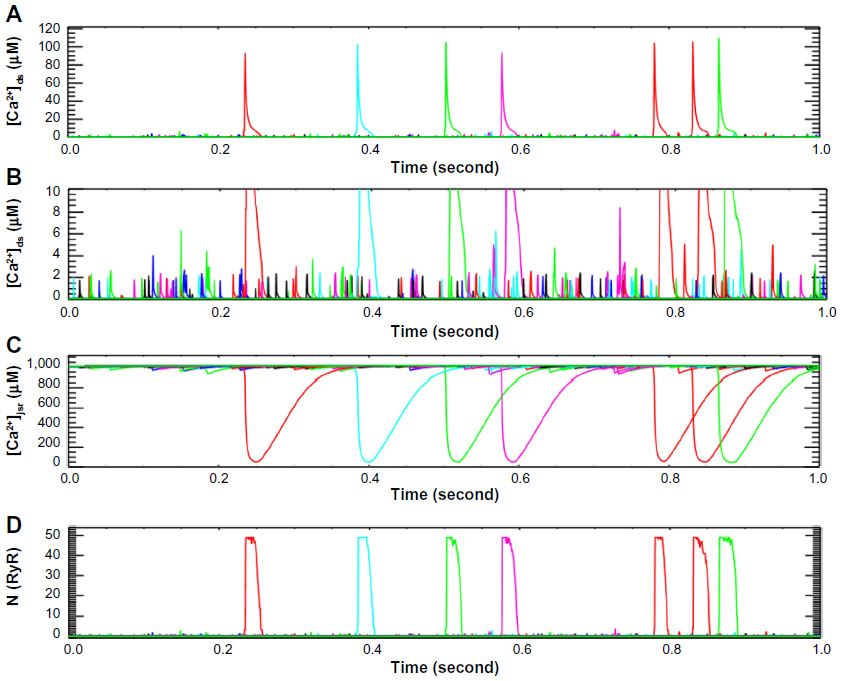 | Figure 2 Simulation with sticky cluster model incorporated into a model for excitation–contraction coupling in the rat ventricular myocyte. |
T-tubules have been found in the cardiac tissues of all mammalian species (eg, rat, mice, guinea pigs, dogs, rabbits, and humans), but appear to be absent in avian, reptile, and amphibian cardiac tissue.21 Interestingly, t-tubules are far less developed in atrial, pacemaker cells, with approximately 50% of atrial myocytes having a sparse irregular T-tubular system.22,23 Although approximately one-third of the entire cell membrane area form the t-tubule networks, the total volume density is very small, eg, approximately 1%–3%.24,25
The name T-tubular system was given due to the transversal direction of the invagination. However, subsequent studies have shown that a considerable amount of tubules run in the axial direction.26,27 A better descriptive name was suggested as “transverse-axial tubular system” (TATS or T-Ax).27 Another name, sarcolemma Z rete, was also proposed due to the fact that there are a large number of tubules that run neither in axial nor in transversal directions.24 Electron microscopy has also suggested that approximately 51% of tubules are between 180 and 280 nm wide in rat and the mean width is ~400 nm in rabbit and in human.28,29 This complex structure of the T-Ax aids in uniform ECC throughout the cell. In fact the remodeling of T-Ax and the microdomains have been suggested to play an important role in the changes to ECC in HF.30–32
Spark initiation
At rest, the open probability of a single RyR2 channel is very small (<0.001).33 Calcium release from the sarcoplasmic reticulum (SR) has long been considered to be a result of CICR. In the process, elevation of [Ca2+] in the dyad caused RyRs to open releasing Ca2+ which further elevates [Ca2+] resulting in the opening of more RyRs. This strong positive feedback increases RyR2 open probability to ~0.5. Calcium sparks are considered to be activated by the CICR mechanism as well.
More recently, experiments observed that the open probability was regulated by the [Ca2+] inside the JSR.34 When SR calcium is elevated, there is an increased RyR2 open probability. Lipid bi-layer experiments have shown that this regulation is not due to feed-forward regulation in which Ca2+ exiting the RyR2 channel opens additional RyRs on the cytosolic side. Instead, Ca2+ binding to calsequestrin, triadin, and junctin-2 complex or directly to the channel in the JSR lumen confers the probability.35–37 This led to the idea that increased cytosolic [Ca2+] during a wave could enter the SR and sensitize the RyR2 for Ca2+ release.38 MacLennan, and Chen and Ramay et al suggested the mechanism of store overload-induced Ca2+ release in which increased SR [Ca2+] can trigger opening of RyRs to cause Ca2+ release.39,40 This is based upon the experimental observations that SR [Ca2+] modulates RyR2 open probability.34 A computational model by Ramay et al explored the role of SR [Ca2+] on calcium wave dynamics.40 The study suggested that SR [Ca2+] could sensitize RyRs to aid Ca2+ wave propagation only when SR Ca2+ diffusion is slow.
Computational models have allowed us to look at the initiating Ca2+ signal in detail.19 The Williams et al model for calcium dynamics in the rat ventricular myocyte includes 20,000 calcium release units consisting of clusters of stochastically gated RyR2s. The model allows the resolution of the many single RyR2 opening events that do not result in a Ca2+ spark (~3,000 per cell per second) (Figure 1). It also shows that one RyR2 opening can trigger the opening of others through CICR and that once six RyR2s in a cluster of 49 channels are open a spark usually follows. The model demonstrates the importance of CICR for initiation of a Ca2+ spark. While it is mathematically possible that a step change in JSR [Ca2+] can increase the open probability of the RyR2 at a given dyadic [Ca2+], it is not likely to happen. The changes in JSR calcium usually follow the changes in dyadic subspace calcium arguing against store overload-induced Ca2+ release as a mechanism for spark initiation.19
Spark termination
Calcium sparks activate utilizing the strong positive feedback that results from CICR phenomenon. The mechanism by which this release ends, ie, spark termination has been an active research topic for many years with several hypotheses proposed.41–43 The hypotheses can be classified as follows: 1) depletion of SR luminal Ca2+,44–46 2) inactivation of the RyR2,47,48 3) stochastic attrition,33 or possibly a combination of these mechanisms as suggested by 4) the sticky cluster spark model.42 More recently, the ideas of 5) pernicious attrition and induction decay have been presented as a mechanism of the termination of Ca2+ release.46
The first possible mechanism the Ca2+ release from the SR could terminate is that there is no Ca2+ left in the SR to release. This however, is not the case. Experimental studies, using caffeine to open RyRs, have shown that during and after a Ca2+ transient, there is still releasable Ca2+ in the SR.45 Other studies have shown that while most normal Ca2+ sparks are less than 40 ms in duration, low concentrations of ryanodine can result in sparks that last more than a second.7 This argues that there is still releasable Ca2+ in the SR. Furthermore, experimental results show there is a significant amount, 40% of free calcium in the JSR, as the nadir of JSR calcium level recorded in the form of Ca2+ blinks.49,50
The second mechanism suggests that the RyR2 inactivate, ie, they close and hence cannot release Ca2+. However, experiments in lipid bilayers have failed to show the existence of inactivation of individual RyRs. Such studies have observed a phenomenon called adaptation in which the RyR2 open probability declines after an initial increase when Ca2+ levels are increased.34,41,51,52 A certain fraction of the channels remain in the adapted state so the “adapted” channels remain closed to the subsequent higher level of calcium. However, the time course for adaptation is slow with a half-time of ~150 ms. Because the duration of the spark is smaller (~40 ms) adaptation does not seem to play a major role in spark termination.
The third suggested mechanism is stochastic attrition. In this mechanism, the RyR2s are stochastically close to terminate release. However, stochastic attrition can only work for an RyR2 cluster consisting of a few activated channels (<6). Scanning electron microscopy imaging using freeze fracture techniques have indicated that there are 30–300 RyR2 in a cluster.53 More recent optical super-resolution methods have also shown that the RyRs are clustered in the dyad.54
As these previous hypotheses could not explain spark termination, the sticky cluster hypothesis was proposed. The sticky cluster mechanism, combines strong positive feedback of CICR, large clusters of RyR2s that display coupled gating conferred by the FKBP12.6 protein,55–58 local SR depletion (JSR), and an RyR2 open probability that decreases with decreasing SR luminal Ca2+.34 The model demonstrates a mechanism of termination that can be understood from Figure 2. Opening of a single RyR2 triggers opening of additional RyR2s in the cluster (Figure 2A). This quickly increases the open probability until most of the channels in the cluster are open (Figure 2D). With this opening, the RyR2 Ca2+ release flux quickly increases which leads to a jump in the dyadic subspace [Ca2+] and emptying of the JSR (Figure 2C). The reduction in the JSR Ca2+ concentration leads to a decrease in the RyR2 Ca2+ release flux. The reduced flux leads to decreased subspace [Ca2+] which results in decreased opening of the RyRs. Furthermore, the decreased subspace JSR [Ca2+] further decreases the opening probability. As more and more channels stochastically close and fail to reopen, the number of closed channels increases and the subspace [Ca2+] decreases until all the channels close terminating release. The coordinated closing of the channels is facilitated by coupled gating but is not a strict requirement.
More recently, the concept of pernicious attrition and induction decay were proposed as the mechanism for spark termination.46 Both of these mechanisms rely on a decreasing Ca2+ release flux during the course of a Ca2+ spark being unable to maintain CICR, resulting in closure of the RyR2s in the cluster. These proposed mechanisms do not require coupled gating of the RyR2s for closure. This idea of decreased flux being unable to maintain CICR seems to be a pervasive theme in all the proposed models of release. In fact, the decreased flux mechanism was proposed as the mechanism of release in the deterministic Jafri–Rice–Winslow model of ECC in the guinea pig ventricular myocyte many years earlier.59
Calcium spark dynamics – physiological role of sparks
In cardiac cells, under physiological conditions, to avoid calcium toxicity, the basal level of cytosolic calcium concentration is kept at a low level ([Ca2+]i ~0.1 μM). This concentration can increase up to tenfold within tens to a few hundreds of milliseconds, depending upon species to facilitate the contraction of the myofilaments.1 During a cardiac cycle, the calcium homeostasis is regulated via ion channels such as the ryanodine-receptor (primarily RyR2 in cardiac myocytes) and inositol 1,4,5-trisphosphate (IP3R), and a number of special transmembrane protein complexes known as pumps and exchangers, eg, plasma membrane Ca2+ ATPase pumps (PMCA) and Na+–Ca2+ exchanger (NCX), sarcoplasmic/endoplasmic reticulum (SR/ER) Ca2+-ATPase (SERCA) pumps, and mitochondrial calcium uniporter.60 For example, in rat ventricular myocyte, the contribution of the above proteins to bringing cytosolic calcium back to the normal diastolic level is SERCA uptake (nearly 90% uptake), NCX extrusion (nearly 7%),61 by mitochondria (1%–2%),62 and sarcolemma Ca2+-ATPase (1%).60
Ca2+ sparks play a significant role in maintaining Ca2+ homeostasis in addition to their role in ECC. However, aberrant Ca2+ spark dynamics have been implicated in pathology such as HF and cardiac arrhythmia. Below we discuss the role of Ca2+ sparks in normal and pathophysiology.
Sarcoplasmic reticulum calcium leak
As mentioned above, the SERCA actively sequesters Ca2+ ions into the SR. To balance the resting SERCA activity, an SR Ca2+ leak or Ca2+ efflux from the SR is present. This leak has been proposed to take the form of spontaneous Ca2+ spark activity. However, when the leak in the form of the Ca2+ spark is calculated, it is insufficient to account for the leak. Furthermore, the SR Ca2+ leak can be measured experimentally by blocking SERCA with cyclopiazonic acid or thapsigargin and measure SR Ca2+ content with caffeine trigger Ca2+ release through the RyR2s.63 In such studies, the SR Ca2+ content declines as does the spark rate. However, after Ca2+ sparks are no longer visible, the SR Ca2+ content continues to decline also suggesting that the Ca2+ sparks do not completely account for the SR Ca2+ leak. The missing component of leak has been termed invisible leak as it is not directly measurable.
Several hypotheses have been proposed to explain the invisible leak: 1) back flux through the SERCA, 2) other ion channels, and 3) nonjunctional RyR2s. Shannon et al64 and Bers proposed that the SERCA was a reversible pump with a forward flux and a back flux.60,61 They suggested that the back flux is a source of Ca2+ leak. However, while experiments have observed that SERCA can work in reverse, this occurs under extreme nonphysiological conditions. A thermodynamically derived model by Tran et al better represents SERCA function.65
The existence of other ion channels has also been suggested. Early models used a passive Ca2+ leak to explain the entire leak. However, experiments have indicated that Ca2+ sparks form the visible part of the leak. For the invisible part of the leak, no other suitable ion channels have been found. The IP3 receptor has been suggested as a candidate for the Ca2+ leak however, the number of IP3 receptors is small (<5% the amount of RyR2s) and their physiological role in adult ventricular myocytes has not been directly observed. The nonjunctional RyR2 located away from the release sites are few in number being perhaps less than 2%–5% of the total RyR2 population and insufficient to account for the leak.
The sticky cluster model and its updated version, helps to explain mechanism of the SR Ca2+ leak. The model simulates Ca2+ sparks with realistic rate as well as openings of one or a few RyR2s that do not result in Ca2+ sparks (Figure 2B).19,42 The model also includes the opening of nonjunctional RyR2s. The model suggests that total SR Ca2+ leak flux consisting of the Ca2+ sparks, nonspark leak, and nonjunctional RyR2 openings sufficiently balances the SERCA pump. The model therefore suggests that the “invisible” SR Ca2+ leak release results from nonspark activation of RyR2 channels in the dyad and the nonjunctional RyR2s. The nonspark release of Ca2+ via RyR2 channel opening has been experimentally confirmed and coined Ca2+ quarks.66,67 Thus, the model suggests a mechanism for the SR Ca2+ leak that relies only upon components known to exist in the myocyte.
Spatial spread of sparks – calcium quarks, sparks, and waves
The Ca2+ spark is the spatial spread of the Ca2+ release in a dyad. A three-dimensional spatial model ECC is used to simulate the linescan of Ca2+ spark (Figure 3A). With activation of LCCs during an AP, the influx of calcium via LCC is sensed by the closely apposed cluster of RyR2s, triggering the release of calcium from the SR and yielding a local rise in [Ca2+], ie, [Ca2+]ds (Figure 3B). These spatially localized triggered Ca2+ sparks occurring at the “calcium release unit” (CRU) are termed local control. However, as mentioned previously, Ca2+ sparks can occur spontaneously. Smaller events of a single or a few RyR2 channels called Ca2+ quarks also occur spontaneously.
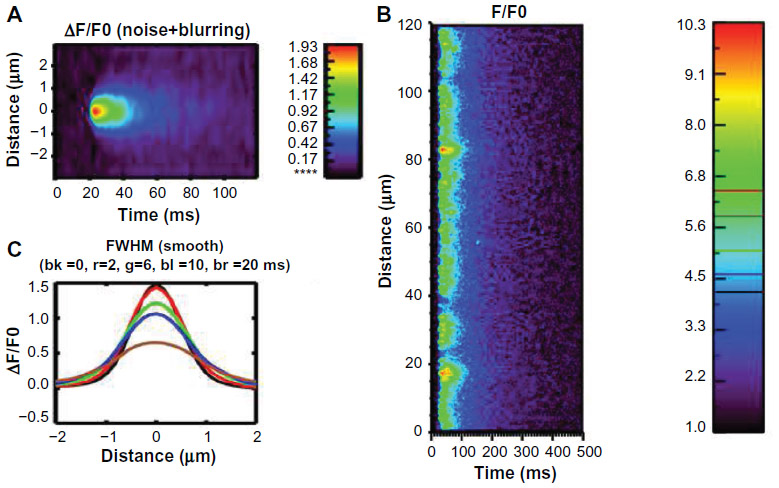 | Figure 3 Simulated Ca2+ spark. |
Ca2+ sparks can be detected using fluorescent dyes which then can be quantified using the full-width half-max (FWHM), time-to-peak, half-decay time and full-duration half-max (Figure 3C).7 The line-scan image is a 2D picture with x-axis as time, and y-axis as the spatial information. At the time point where the Ca2+ spark reaches the peak, FWHM is the spatial distance between two locations at which the amplitude is half of the peak. If we assume the line-scan going through the center of the release site, full-duration half-max is the time period from the two time points at which the value is half peak. In cardiac cells, FWHM of a Ca2+ spark is wide (~2 μm), yet does not trigger neighboring sites under normal physiological conditions. Computational models have been used to study the spatial spread of Ca2+ during a spark, yet none can produce an FWHM of 2 μm suggesting that our understanding of the physiological processes or dye properties is missing some detail. Attempts to increase the spatial dimensions of the Ca2+ release site, increase the unitary RyR2 Ca2+ flux, and increase optical blurring caused by the confocal microscope model have increased the FWHM observed in models but have not provided adequate explanation of what are the processes underlying the spread for a Ca2+ spark.
Ca2+ sparks can lead to Ca2+ waves, especially under Ca2+ overload condition.68–70 At the cellular level, the wave needs synchronized activation from multiple CRUs. For this Ca2+ release must propagate from one Ca2+ release site to the next. In ventricular cardiac cells, the mean inter-release site distance is approximately 1.6 μm longitudinal and 0.8 μm in the axial direction.61 Chen-izu et al were first to quantify the distribution of the nearest neighbor distance between the CRUs along the transversal and longitudinal directions.71 Hoang-Trong developed a three-dimensional spatio-temporal model of the rat ventricular myocyte that shows the importance of release distances on the same Z-discs, especially those near the end of the cell where the diffusion is stronger on the other end and the role of intermediate “rogue” RyR2 cluster size during the wave initiation and wave velocity.72 This is in agreement with the study by Izu et al that suggests that a Ca2+ wave requires multiple Ca2+ release units in the same Z-disk to activate to release enough Ca2+ to propagate to the next Z-disk.73 In another study, Nivala et al proposed a theoretical framework using power-law distribution as an indicator of critical state for wave initiation.74 They hypothesized that the size of the RyR2 cluster follows exponential distribution when the coupling is weak and power-law distribution if the coupling is strong. However, they did not explain the role of cluster distances and the role of intermediate cluster.
RyR2 phosphorylation and Ca2+ sparks
Phosphorylation plays a significant role in the regulation of RyR2 activity. Each of the four pore-forming subunits of the RyR2 have several phosphorylation sites. In addition to these sites, accessory proteins such as FKBP12.6 can also be phosphorylated. The state of phosphorylation depends on the activity of the numerous kinases and phosphatases in the myocyte. Of these, the most widely studied are protein kinase A (PKA) and Ca2+/calmodulin-dependent protein kinase II (CaMKII). This does not rule out the potential role of other kinases that phosphorylate serine/threonine residues of proteins.75 Of these many phosphorylation sites on the RyR2, it is difficult to assess which particular ones are phosphorylated at any given time. However, experimental observations indicate that with phosphorylation by either PKA or CaMKII, the open probability of the RyR2s increases.76 One physiological role of this is during exercise (fight or flight response), where PKA phosphorylation increases the size of the Ca2+ transient and the ability of the heart to pump blood.77,78 On the other hand, increasing the spontaneous spark rate and the Ca2+ leak from the SR. Increases in Ca2+ leak have been implicated in increased risk of cardiac arrhythmia. Abnormal RyR2 phosphorylation plays a role in atrial fibrillation (AF), HF, and catecholaminergic polymorphic ventricular tachycardia (CPVT).75 The role of Ca2+ sparks in arrhythmia in these conditions is discussed below.
Role of Ca2+ sparks in pathology
Ca2+ sparks and arrhythmia
Heart disease is the leading cause of death accounting for 24.6% of the total deaths in the United States.79 Of the deaths due to heart disease, cardiac arrhythmia is the largest cause of death; approximately 450,000 adults each year.80 Cardiac arrhythmia occurs when the normal pattern of depolarization and repolarization of the heart is disrupted. If severe, the heart may not pump enough blood to the body, reducing blood supply to the brain that can cause a person to lose consciousness in just a few minutes. In the extreme, sudden cardiac death occurs. In over half of the cases, however, sudden cardiac arrest occurs without prior symptoms. Until today, the underlying cellular mechanism is still unclear, with the most common life-threatening arrhythmia being ventricular fibrillation. As a result, the proper use of antiarrhythmic drugs is critical to avoid unexpected side effects that can increase the risk or mortality. For example, flecainide increases the risk of mortality in patients with reduced left ventricular function following a myocardial infarction.81 On the other hand, flecainide is useful in suppressing arrhythmia in atrial fibrillation patients with no structural defects in the heart.
Experiments have indicated that calcium release is stochastic and regulated locally suggesting the possibility of spatially heterogeneous calcium levels in the cells. This spatial heterogeneity might be important mediating different signaling pathways. In addition, the disruption in the pump/leak balance can be a precursor to many pathological conditions, especially the triggering of arrhythmogenic Ca2+ waves.82,83 Spontaneous Ca2+ release from the SR elevates intracellular Ca2+ concentration which can activate Na+–Ca2+ exchange. The activation of Na+–Ca2+ exchange can cause afterdepolarizations.76,84,85 If this occurs during the AP (systole) it is called an early afterdepolarization (EAD). If this occurs during diastole, it is called a delayed afterdepolarization (DAD).
An example of a genetic defect resulting in defective Ca2+ dynamics is CPVT. CPVT is a rare disease with an occurrence of one among 10,000 people, and a high mortality rate (30%–35%) comprising 15% of all unexplained sudden cardiac death.86,87 CPVT patients are seemingly normal, but upon physical activity can develop a fatal arrhythmia.86 CPVT results from a mutation of the RyR2 (CPVT1), calsequestrin (CPVT2), calmodulin (CPVT4), and triadin (CPVT5). For CPVT3 the gene is unknown. Experiments have isolated some of the gene variants. For example, mutations to calsequestrin result in a steeper dependence of RyR2 open probability on SR [Ca2+], that is, the spontaneous Ca2+ spark rate of a given SR [Ca2+] is higher.88 Mutations to RyR2 can also alter this luminal dependence or increase the sensitivity of the RyR2 to Ca2+ which enhances Ca2+ sensitivity with ²-adrenergic stimulation.89 This increased sensitivity results in an increased Ca2+ spark rate. Using flecainide to block the open state and tetracaine to prolong the closed state, Hilliard et al showed that blocking the open state can be a new therapeutic strategy to prevent diastolic Ca2+ wave resulting in CPVT.90 Using verapamil which is an L-type Ca2+ channel blocker, can decrease the prevalence of ventricular tachycardia in patients with CPVT during stress, but it does not completely suppress CPVT.86 Further studies are needed to find interventions that might cause normal Ca2+ spark dynamics associated with the changes in the function of Ca2+ handling proteins.
A recently discovered signaling pathway in cardiac cells, called X-ROS (reactive oxygen species) signaling, has shown the role of mechanical stress in increasing spark frequency.91 Under rapidly applied stretching of the myocyte, nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2) is activated to produce ROS near microtubules where NOX2 is located. ROS production triggers a burst of increased spontaneous Ca2+ sparks. One important discovery is that when the chemical reducing capacity of the cell is decreased, X-ROS signaling increases SR Ca2+ leak and global oxidative stress, thereby increasing the possibility of arrhythmia. Oxidative stress can thus lead to pathological conditions such as systolic dysfunction, arrhythmia and HF.92 Skeletal muscle has a similar X-ROS mechanism yet with important and distinctive differences.93 A computational model that simulates the effect of ROS on RyR2 gating has been developed to provide quantitative information that is not currently available from experimental means.94 In the mouse model of Duchenne muscular dystrophy (the mdx mouse), stretching the cardiomyocytes reveals that stretch-induced ROS production can result in arrhythmogenic Ca2+ waves.
Ca2+ sparks and heart failure
Heart failure is a condition where the heart loses its ability to function as a pump. Failing heart has an increased risk for arrhythmia. On the single myocyte level, the physiological changes include a prolonged AP and a Ca2+ transient with reduced amplitude. Another characteristic is an increased SR Ca2+ leak and propensity for spontaneous Ca2+ waves. These changes result from certain structural and gene functional changes that result from changes to gene expression and compensatory posttranslational modification of proteins. These changes include upregulation of K+ channels and NCX, and downregulation of SERCA. There is also phosphorylation of the RyR2s by PKA and CaMKII and rearrangement of the T-tubular system.30,75,95 Computational modeling has shown that the changes to Ca2+ dynamics contribute significantly to AP prolongation as well as the changes in the Ca2+ transient (Figure 4A and D).96 Other computational studies suggest that these changes result in an increased diastolic spark rate (SR Ca2+ leak) and an increased late phase of Ca2+ release during the AP (Figure 4C). This late Ca2+ release is enhanced by the remodeling of the T-tubular network which is referred to as orphaning, as RyR2 clusters are no longer associated with a t-tubule.95 It leads to an increased potential for arrhythmia by increasing depolarization late in the AP which increases the potential for reexcitation of the cardiac tissue. With the aforementioned changes to Ca2+ spark dynamics are altered levels of the SR [Ca2+] due to a resetting of the pump/leak balance (Figure 4B).
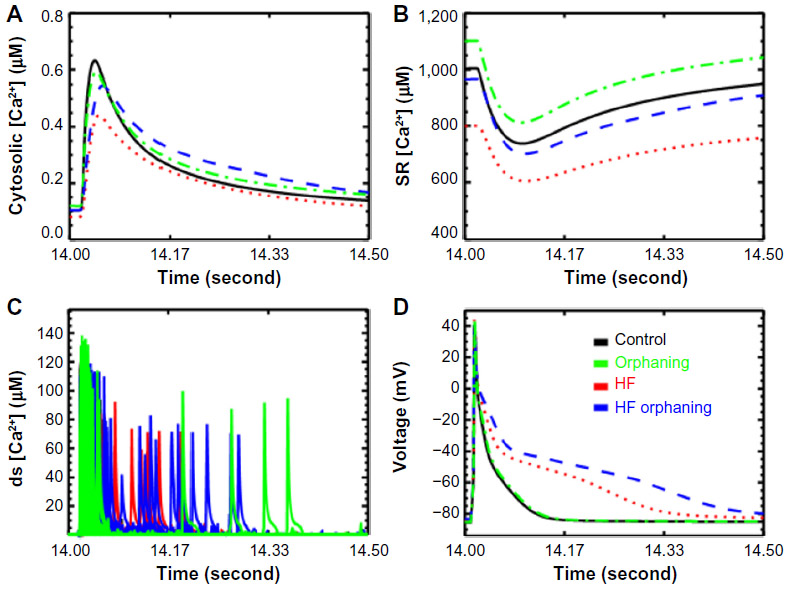 | Figure 4 Simulation with sticky cluster model incorporated into a model for excitation–contraction coupling in the rat ventricular myocyte to explore heart failure. |
Atrial fibrillation and Ca2+ sparks
AF is a pathology in which the atria undergoes rapid uncoordinated depolarization. It is classified as paroxysmal, persistent, and permanent.76 Paroxysmal AF consists of transient episodes that terminate on their own. Persistent AF requires some sort of medical intervention to ensure termination. Permanent AF persists in spite of medical intervention. While AF is not fatal, it can be uncomfortable for the patient and prolonged AF increases the risk of stroke.97,98 With time, AF results in a remodeling of the atrial tissue that involves both changes in function as well as the ultrastructure of the cells and tissue involved.
Ca2+ spark dynamics play an important role in the initiation and maintenance of AF.85 AF is accompanied by increased SR Ca2+ leak, which elevates intracellular Ca2+ levels. This leads to Ca2+-dependent inhibition of Na+ channels which causes conduction slowing. Furthermore, the elevated intracellular Ca2+ levels and increased RyR2 activity results in increased EADs and DADs.85,99 Phosphorylation of the RyR2s by CaMKII increases SR Ca2+ leak and the risk of afterdepolarization.100
Contributing to the maintenance of AF are changes in gene expression and phosphorylation state of the myocyte. The calmodulin levels increase 60%, CaMKII autophosphorylation at Thr287 increases 87%, and RyR2 phosphorylation by PKA at site Ser2808 increases 235%, and at the CaMKII site, Ser2814 increases by 77%.76,100 Furthermore, there is increased phosphorylation of phospholamban, thereby increasing SR Ca2+ load.101 These contribute further to Ca2+ spark activity further perpetuating conditions that make AF more easily initiated and sustained.
Studies suggest that during congestive heart failure, atrial SR Ca2+ load is increased and calsequestrin (CASQ) expression is reduced.76 This causes increases in Ca2+ spark rate, ie, the SR Ca2+ leak. Therefore, the risk of EADs and DADs and resulting cardiac arrhythmia increases.
Discussions/perspectives
Ca2+ spark is the elementary event of calcium release that regulates the excitation and contraction of the cardiac myocytes. The presence of Ca2+ sparks has also been found in nerve cells in which inositol 1,4,5-trisphosphate and RyR2 both control the release of Ca2+ from the endoplasmic reticulum.102,103 However, its role is less well understood than that in muscles. In cardiac cells, despite its stable regulation via the local control mechanism, under calcium overload or sensitization of RyR2 channels, the spark frequency can increase which disrupts the pump/leak balance, eventually leading to ectopic heart beat or inefficient contraction.
The extent of this SR Ca2+ leak is important as it can cause systolic dysfunction (due to less SR Ca2+ available to release) and diastolic dysfunction (due to elevated myoplasmic calcium basal level). The elevation of myoplasmic calcium level can cause calcium wave and thus increase the potential of triggered arrhythmias.104 To quantify the control of calcium leak, it is important to have a mechanistic computational model that can replicate the different pathways of Ca2+ leak.19
Due to the extensive computational high demand for developing a mechanistic whole-cell model that is able to capture the calcium dynamics at the single-channel level, developing such computational models is still a challenging problem.105,106 In such models, it is important to incorporate the spatial placement of all calcium-release sites in the heart, as well as nonuniform distribution of ion channels, and exchangers on the sarcolemma, including the external surface and the t-tubule systems. These models can provide an unprecedented tool to study calcium waves and the role of t-tubule de-tubulation in cardiac diseases. Such studies can give insight into the role of Ca2+ spark dynamics and the role they play in cardiac arrhythmia and disease.
Acknowledgments
This work was supported by the National Institutes of Health R01-HL105239 and 1U01HL116321.
Disclosure
The authors report no conflicts of interest in this work.
References
Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415: 198–205. | |
Berridge MJ, Lipp P, Bootman MD. The versatility and universality of Calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. | |
Bootman MD, Thomas D, Tovey SC, Berridge MJ, Lipp P. Nuclear calcium signalling. Cell Mol Life Sci. 2000;57:371–378. | |
Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. | |
Gaspers LD, Pierobon N, Thomas AP. Chapter 18, Calcium Signaling, in Signaling pathways in liver disease. 2005, Springer, NY. | |
Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. | |
Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262(5134):740–744. | |
Fearnley CJ, Roderick HL, Bootman MD. Calcium signaling in cardiac myocytes. Cold Spring Harb Perspect Biol. 2011;3(11):a004242. | |
Santana LF, Cheng EP, Lederer WJ. How does the shape of the cardiac action potential control calcium signaling and contraction in the heart? J Mol Cell Cardiol. 2010;49(6):901–903. | |
Klein MG, Cheng H, Santana LF, Jiang Y-H, Lederer WJJ, Schneider MF. Two mechanisms of quantized calcium release in skeletal muscle. Nature. 1996;379(6564):455–459. | |
Santana LFF, Cheng H, Gómez AMM, Cannell MBB, Lederer WJJ. Relation between the sarcolemmal Ca2+ current and Ca2+ sparks and local control theories for cardiac excitation-contraction coupling. Circ Res. 1996;78:166–171. | |
Peskoff A, Langer GA. Calcium concentration and movement in the ventricular cardiac cell during an excitation-contraction cycle. Biophys J. 1998;74:153–174. | |
Stern MD, Song L-s, Cheng H, et al. Local control models of cardiac excitation-contraction coupling. A possible role for allosteric interactions between ryanodine receptors. J Gen Physiol. 1999;113:469–489. | |
Hollingworth S, Peet J, Chandler WK, Baylor SM. Calcium sparks in intact skeletal muscle fibers of the frog. J Gen Physiol. 2001;118(6):653–678. | |
Nelson MT, Cheng H, Rubart M, et al. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270(5236):633–637. | |
López-López JR, Shacklock PS, Balke CW, Wier WG. Local, stochastic release of Ca2+ in voltage-clamped rat heart cells: visualization with confocal microscopy. J Physiol. 1994;480(Pt 1):21–29. | |
Cannell MB, Cheng H, Lederer WJ. The control of calcium release in heart muscle. Science. 1995;268:1045–1049. | |
Qin J, Valle G, Nani A, et al. Ryanodine receptor luminal Ca2+ regulation: swapping calsequestrin and channel isoforms. Biophys J. 2009;97: 1961–1970. | |
Williams GSB, Chikando AC, Tuan H-TM, Sobie Ea, Lederer WJ, Jafri MS. Dynamics of calcium sparks and calcium leak in the heart. Biophys J. 2011;101(6):1287–1296. | |
Gyorke S. Ca2+ spark termination: inactivation and adaptation may be manifestations of the same mechanism. J Gen Physiol. 1999;114: 163–166. | |
Brette F, Orchard C. T-tubule function in mammalian cardiac myocytes. Circ Res. 2003;92:1182–1192. | |
Kirk MM, Izu LT, Chen-Izu Y, et al. Role of the transverse-axial tubule system in generating calcium sparks and calcium transients in rat atrial myocytes. J Physiol. 2003;547:441–451. | |
Richards MA, Clarke JD, Saravanan P, et al. Transverse (t-) tubules are a common feature in large mammalian atrial myocytes including human. Am J Physiol. Heart Circ Physiol. 2011;301: H1996–H2005. | |
Soeller C, Cannell MB. Examination of the transverse tubular system in living cardiac rat myocytes by 2-photon microscopy and digital image processing techniques. Circ Res. 1999;84:266–275. | |
Page E, McCallister LP, Power B. Sterological measurements of cardiac ultrastructures implicated in excitation-contraction coupling. Proc Natl Acad Sci U S A. 1971;68:1465–1466. | |
Forssmann WG, Girardier L. A study of the T system in rat heart. J Cell Biol. 1970;44(1):1–19. | |
Forbes MS, Hawkey LA, Sperelakis N. The transverse-axial tubular system (TATS) of mouse myocardium: its morphology in the developing and adult animal. Am J Anat. 1984;170(2):143–162. | |
Savio-Galimberti E, Frank J, Inoue M, et al. Novel features of the rabbit transverse tubular system revealed by quantitative analysis of three-dimensional reconstructions from confocal images. Biophys J. 2008;95(4):2053–2062. | |
Cannell MB, Crossman DJ, Soeller C. Effect of changes in action potential spike configuration, junctional sarcoplasmic reticulum micro-architecture and altered t-tubule structure in human heart failure. J Muscle Res Cell Motil. 2006;27(5–7):297–306. | |
Guo A, Zhang C, Wei S, Chen B, Song LS. Emerging mechanisms of T-tubule remodelling in heart failure. Cardiovasc Res. 2013;98(2):204–215. | |
Wei S, Guo A, Chen B, et al. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107(4):520–531. | |
Sipido KR, Cheng H. T-tubules and ryanodine receptor microdomains: on the road to translation. Cardiovasc Res. 2013;98(2):159–161. | |
Stern MD. Theory of excitation-contraction coupling in cardiac muscle. Biophys J. 1992;63:497–517. | |
Gyorke I, Gyorke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J. 1998;75(6):2801–2810. | |
Zhang L, Kelley J, Schmeisser G, Kobayashi YM, Jones LR. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J Biol Chem. 1997;272(37):23389–23397. | |
Yano K, Zarain-Herzberg A. Sarcoplasmic reticulum calsequestrins: structural and functional properties. Molecular and cellular biochemistry. June 15, 1994;135(1):61–70. | |
Gyorke S, Gyorke I, Lukyanenko V, Terentyev D, Viatchenko-Karpinski S, Wiesner TF. Regulation of sarcoplasmic reticulum calcium release by luminal calcium in cardiac muscle. Front Biosci. 2002;7:d1454–d1463. | |
Keller M, Kao JP, Egger M, Niggli E. Calcium waves driven by “sensitization” wave-fronts. Cardiovasc Res. 2007;74(1):39–45. | |
MacLennan DH, Chen SR. Store overload-induced Ca2+ release as a triggering mechanism for CPVT and MH episodes caused by mutations in RYR and CASQ genes. J Physiol. 2009;587(Pt 13):3113–3115. | |
Ramay HR, Jafri MS, Lederer WJ, Sobie EA. Predicting local SR Ca(2+) dynamics during Ca(2+) wave propagation in ventricular myocytes. Biophys J. 2010;98(11):2515–2523. | |
Lukyanenko V, Wiesner TF, Gyorke S. Termination of Ca2+ release during Ca2+ sparks in rat ventricular myocytes. J physiol. 1998;507 (Pt 3):667–677. | |
Sobie EA, Dilly KW, dos Santos Cruz J, Lederer WJ, Jafri MS. Termination of cardiac Ca(2+) sparks: an investigative mathematical model of calcium-induced calcium release. Biophys J. 2002;83(1):59–78. | |
Laver DR, Kong CHT, Imtiaz MS, Cannell MB. Termination of calcium-induced calcium release by induction decay: an emergent property of stochastic channel gating and molecular scale architecture. J Mol Cell Cardiol. 2013;54:98–100. | |
Varro A, Negretti N, Hester SB, Eisner DA. An estimate of the calcium content of the sarcoplasmic reticulum in rat ventricular myocytes. Pflugers Arch. 1993;423:158–160. | |
Bassani JW, Yuan W, Bers DM. Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am J Physiol. 1995;268:C1313–C1319. | |
Gillespie D, Fill M. Pernicious attrition and inter-RyR2 CICR current control in cardiac muscle. J Mol Cell Cardiol. 2013;58:53–58. | |
Gyorke S, Fill M. Ryanodine receptor adaptation: control mechanism of Ca(2+)-induced Ca2+ release in heart. Science. 1993;260:807–809. | |
Cheng H, Fill M, Valdivia H, Lederer WJ, Sachs F, Qin F. Models of Ca2+ release channel adaptation. Science. 1995;267:2009–2011. | |
Zima AV, Picht E, Bers DM, Blatter La. Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ Res. 2008;103:e105–e115. | |
Brochet DXP, Yang D, Di Maio A, Lederer WJ, Franzini-Armstrong C, Cheng H. Ca2+ blinks: rapid nanoscopic store calcium signaling. Proc Natl Acad Sci U S A. 2005;102:3099–3104. | |
Terentyev D. Luminal Ca2+ Controls Termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circ Res. 2002;91:414–420. | |
Valdivia HH, Kaplan JH, Ellis-Davies GC, Lederer WJ. Rapid adaptation of cardiac ryanodine receptors: modulation by Mg2+ and phosphorylation. Science. 1995;267(5206):1997–2000. | |
Franzini-Armstrong C, Protasi F, Ramesh V. Comparative ultrastructure of Ca2+ release units in skeletal and cardiac muscle. Ann N Y Acad Sci. 1998;853:20–30. | |
Baddeley D, Jayasinghe ID, Lam L, Rossberger S, Cannell MB, Soeller C. Optical single-channel resolution imaging of the ryanodine receptor distribution in rat cardiac myocytes. Proc Natl Acad Sci U S A. 2009;106(52):22275–22280. | |
Marx SO, Gaburjakova J, Gaburjakova M, Henrikson C, Ondrias K, Marks AR. Coupled gating between cardiac calcium release channels (ryanodine receptors). Circ Res. 2001;88:1151–1158. | |
Wagenknecht T, Radermacher M, Grassucci R, Berkowitz J, Xin HB, Fleischer S. Locations of calmodulin and FK506-binding protein on the three-dimensional architecture of the skeletal muscle ryanodine receptor. J Biol Chem. 1997;272:32463–32471. | |
Gómez AM, Schuster I, Fauconnier J, Prestle J, Hasenfuss G, Richard S. FKBP12.6 overexpression decreases Ca2+ spark amplitude but enhances [Ca2+]i transient in rat cardiac myocytes. Am J Physiol Heart Circ Physiol. 2004;287:H1987–H1993. | |
Guo T, Cornea RL, Huke S, et al. Kinetics of FKBP12.6 binding to ryanodine receptors in permeabilized cardiac myocytes and effects on Ca sparks. Circ Res. 2010;106:1743–1752. | |
Jafri MS, Rice JJ, Winslow RL. Cardiac Ca2+ dynamics: the roles of ryanodine receptor adaptation and sarcoplasmic reticulum load. Biophys J. 1998;74(3):1149–1168. | |
Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res. 2000;87:275–281. | |
Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. 2nd ed. Dordrecht: Kluwer Academic Publishers; 2001. | |
Bers DM, Bassani JW, Bassani RA. Na-Ca exchange and Ca fluxes during contraction and relaxation in mammalian ventricular muscle. Ann N Y Acad Sci. 1996;779:430–442. | |
Zima AV, Bovo E, Bers DM, Blatter LA. Ca(2)+ spark-dependent and -independent sarcoplasmic reticulum Ca(2)+ leak in normal and failing rabbit ventricular myocytes. J Physiol. 2010;588(Pt 23):4743–4757. | |
Shannon TR, Ginsburg KS, Bers DM. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res. 2002;91(7):594–600. | |
Tran K, Smith NP, Loiselle DS, Crampin EJ. A thermodynamic model of the cardiac sarcoplasmic/endoplasmic Ca(2+) (SERCA) pump. Biophys J. 2009;96(5):2029–2042. | |
Lipp P, Niggli E. Submicroscopic calcium signals as fundamental events of excitation-contraction coupling in guinea-pig cardiac myocytes. J Physiol. 1996;492(Pt 1):31–38. | |
Brochet DXP, Xie W, Yang D, Cheng H, Lederer WJ. Quarky calcium release in the heart. Circ Res. 2011;108:210–218. | |
Cheng H, Lederer MR, Lederer WJ, Cannell MB. Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am J Physiol. 1996;270: C148–C159. | |
Wier WG, ter Keurs HE, Marban E, Gao WD, Balke CW. Ca2+ ‘sparks’ and waves in intact ventricular muscle resolved by confocal imaging. Circ Res. 1997;81(4):462–469. | |
Lukyanenko V, Gyorke S. Ca2+ sparks and Ca2+ waves in saponin-permeabilized rat ventricular myocytes. J Physiol. 1999;521 Pt 3:575–585. | |
Chen-izu Y, Mcculle SL, Ward CW, et al. Three-dimensional distribution of ryanodine receptor clusters in cardiac myocytes. Biophys J. 2006;91:1–13. | |
Hoang-Trong MT. Computational Studies of Ca2+ Dynamics in Cardiac Cells using GPGPU. Fairfax: Department of Bioinformatics and Computational Biology, George Mason University; 2014. | |
Izu LT, Wier WG, Balke CW. Evolution of cardiac calcium waves from stochastic calcium sparks. Biophys J. 2001;80(1):103–120. | |
Nivala M, de Lange E, Rovetti R, Qu Z. Computational modeling and numerical methods for spatiotemporal calcium cycling in ventricular myocytes. Front Physiol. 2012;3:114. | |
Dobrev D, Wehrens XH. Role of RyR2 phosphorylation in heart failure and arrhythmias: vontroversies around ryanodine receptor phosphorylation in cardiac disease. Circ Res. 2014;114(8):1311–1319; discussion 1319. | |
Wakili R, Voigt N, Kaab S, Dobrev D, Nattel S. Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Investig. 2011;121(8):2955–2968. | |
Eisner D. Calcium in the heart: from physiology to disease. Exp Physiol. 2014;99(10):1273–1282. | |
Fu Y, Westenbroek RE, Scheuer T, Catterall WA. Phosphorylation sites required for regulation of cardiac calcium channels in the fight-or-flight response. Proc Natl Acad Sci U S A. 2013;110(48):19621–19626. | |
Kochanek KD, Xu J, Murphy SL, Minino AM, Kung H-C. Deaths: final data for 2009. Natl Vital Stat Rep. 2011;60(3):1–116. | |
Brunello L, Slabaugh JL, Radwanski PB, et al. Decreased RyR2 refractoriness determines myocardial synchronization of aberrant Ca2+ release in a genetic model of arrhythmia. Proc Natl Acad Sci U S A. 2013;110(25):10312–10317. | |
Apostolakis S, Oeff M, Tebbe U, Fabritz L, Breithardt G, Kirchhof P. Flecainide acetate for the treatment of atrial and ventricular arrhythmias. Expert Opin Pharmacother. 2013;14(3):347–357. | |
ter Keurs HEDJ, Wakayama Y, Sugai Y, et al. Role of sarcomere mechanics and Ca2+ overload in Ca2+ waves and arrhythmias in rat cardiac muscle. Annals of the New York Academy of Sciences. 2006;1080:248–267. | |
Wakayama Y, Miura M, Stuyvers BD, Boyden PA, ter Keurs HEDJ. Spatial nonuniformity of excitation-contraction coupling causes arrhythmogenic Ca2+ waves in rat cardiac muscle. Circ Res. 2005;96:1266–1273. | |
Hove-Madsen L, Llach A, Bayes-Genis A, et al. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation. 2004;110(11):1358–1363. | |
Heijman J, Voigt N, Nattel S, Dobrev D. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res. 2014;114(9):1483–1499. | |
Liu N, Ruan Y, Priori SG. Catecholaminergic polymorphic ventricular tachycardia. Prog Cardiovasc Dis. 2008;51(1):23–30. | |
Liu N, Rizzi N, Boveri L, Priori SG. Ryanodine receptor and calsequestrin in arrhythmogenesis: what we have learnt from genetic diseases and transgenic mice. J Mol Cell Cardiol. 2009;46:149–159. | |
Lahat H, Pras E, Olender T, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69(6):1378–1384. | |
Priori SG, Napolitano C, Tiso N, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103(2):196–200. | |
Hilliard Fa, Steele DS, Laver D, et al. Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass. J Mol Cell Cardiol. 2010;48:293–301. | |
Prosser BL, Ward CW, Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. 2011;333:1440–1445. | |
Marks AR. Cardiac intracellular calcium release channels: role in heart failure. Circ Res. 2000;87(1):8–11. | |
Khairallah RJ, Shi G, Sbrana F, et al. Microtubules underlie dysfunction in duchenne muscular dystrophy. Sci Signal. 2012;5(236):ra56. | |
Limbu S, Hoang Trong MT, Prosser BL, Lederer WJ, Jafri MS. Modeling Local X-ROS and Calcium Signaling in Heart. (on submition) 2015. | |
Wagner E, Lauterbach MA, Kohl T, et al. Stimulated emission depletion live-cell super-resolution imaging shows proliferative remodeling of T-tubule membrane structures after myocardial infarction. Circ Res. 2012;111(4):402–414. | |
Winslow RL, Rice J, Jafri S. Modeling the cellular basis of altered excitation-contraction coupling in heart failure. Prog Biophys Mol Biol. 1998;69(2–3):497–514. | |
Goralnick E, Bontempo LJ. Atrial fibrillation. Emerg Med Clin North Am. 2015;33(3):597–612. | |
Miragoli M, Glukhov AV. Atrial fibrillation and fibrosis: beyond the cardiomyocyte centric view. Biomed Res Int. 2015;2015:798768. | |
Neef S, Dybkova N, Sossalla S, et al. CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ Res. 2010;106(6):1134–1144. | |
Voigt N, Li N, Wang Q, et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation. 2012;125(17):2059–2070. | |
Voigt N, Heijman J, Wang Q, et al. Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation. 2014;129(2):145–156. | |
Miyazaki K, Ross WN. Ca2+ sparks and puffs are generated and interact in rat hippocampal CA1 pyramidal neuron dendrites. J Neurosci. 2013;33(45):17777–17788. | |
Ross WN. Understanding calcium waves and sparks in central neurons. Nat Rev Neurosci. 2012;13(3):157–168. | |
Tuan HT, Williams GS, Chikando AC, Sobie EA, Lederer WJ, Jafri MS. Stochastic simulation of cardiac ventricular myocyte calcium dynamics and waves. Conf Proc IEEE Eng Med Biol Soc. 2011;2011: 4677–4680. | |
Williams GSB, Smith GD, Sobie Ea, Jafri MS. Models of cardiac excitation-contraction coupling in ventricular myocytes. Math Biosci. 2010;226:1–15. | |
Winslow RL, Cortassa S, O’Rourke B, Hashambhoy YL, Rice JJ, Greenstein JL. Integrative modeling of the cardiac ventricular myocyte. Wiley Interdiscip Rev Syst Biol Med. 2011;3:392–413. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.