")
Back to Journals » Adolescent Health, Medicine and Therapeutics » Volume 8
Burkitt lymphoma in adolescents and young adults: management challenges
Authors Dozzo M, Carobolante F, Donisi PM, Scattolin A, Maino E, Sancetta R, Viero P, Bassan R
Received 19 August 2016
Accepted for publication 8 October 2016
Published 23 December 2016 Volume 2017:8 Pages 11—29
DOI https://doi.org/10.2147/AHMT.S94170
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Alastair Sutcliffe
Massimo Dozzo,1 Francesca Carobolante,1 Pietro Maria Donisi,2 Annamaria Scattolin,1 Elena Maino,1 Rosaria Sancetta,1 Piera Viero,1 Renato Bassan1
1Complex Operative Unit of Hematology, Ospedale dell’Angelo, 2Simple Departmental Operative Unit of Anatomic Pathology, Ospedale Ss. Giovanni e Paolo, Venice, Italy
Abstract: About one-half of all Burkitt lymphoma (BL) patients are younger than 40 years, and one-third belong to the adolescent and young adult (AYA) subset, defined by an age between 15 and 25–40 years, based on selection criteria used in different reports. BL is an aggressive B-cell neoplasm displaying highly characteristic clinico-diagnostic features, the biologic hallmark of which is a translocation involving immunoglobulin and c-MYC genes. It presents as sporadic, endemic, or epidemic disease. Endemicity is pathogenetically linked to an imbalance of the immune system which occurs in African children infected by malaria parasites and Epstein–Barr virus, while the epidemic form strictly follows the pattern of infection by HIV. BL shows propensity to extranodal involvement of abdominal organs, bone marrow, and central nervous system, and can cause severe metabolic and renal impairment. Nevertheless, BL is highly responsive to specifically designed short-intensive, rotational multiagent chemotherapy programs, empowered by the anti-CD20 monoclonal antibody rituximab. When carefully applied with appropriate supportive measures, these modern programs achieve a cure rate of approximately 90% in the average AYA patient, irrespective of clinical stage, which is the best result achievable in any aggressive lymphoid malignancy to date. The challenges ahead concern the following: optimization of management in underdeveloped countries, with reduction of diagnostic and referral-for-care intervals, and the applicability of currently curative regimens; the development of lower intensity but equally effective treatments for frail or immunocompromised patients at risk of death by complications; the identification of very high-risk patients through positron-emission tomography and minimal residual disease assays; and the assessment in these and the few refractory/relapsed ones of new monoclonals (ofatumumab, blinatumomab, inotuzumab ozogamicin) and new molecules targeting c-MYC and key proliferative steps of B-cell malignancies.
Keywords: Burkitt lymphoma, adolescents, young adults, treatment, outcome
Introduction
In 1958, Denis Burkitt reported the first cases of a “sarcoma” of the jaws in children from equatorial Africa,1 discovering a disease which a few years later would be recognized as a very aggressive form of lymphoma,2 universally known as Burkitt lymphoma (BL) (Figure 1).3–5 During the following half century, massive advancement occurred in the understanding of BL pathogenesis, epidemiology (which includes both environmental and behavioral risk factors), and treatment, although with significant disparities across countries at different socioeconomic development. At present, though it is a highly malignant disease with demonstrated propensity to rapid growth and dissemination, provided it is correctly identified and treated, BL is the aggressive lymphoma subset associated with the highest cure rate in both adults and children. The aim of this review is to highlight the factors contributing to this outstanding progress in younger BL patients, from childhood to adolescence and young adulthood, and to identify and discuss the obstacles precluding a generalized cure rate. Adolescents and young adults (AYAs) may be defined by an age range between 15 and 25–40 years, according to the upper age limit adopted in the studies reporting age-related outcomes. In a recent large North-American analysis on 3961 cases of BL diagnosed between 2002 and 2008, 24% of the patients were AYAs aged 20–39 years, and 26% were aged 0–19 years, a figure which includes a proportion of adolescents between 15 and 19 years. Although an exact estimate is lacking, it is reasonable to assume that AYAs represent about one-third of all new BL cases.6
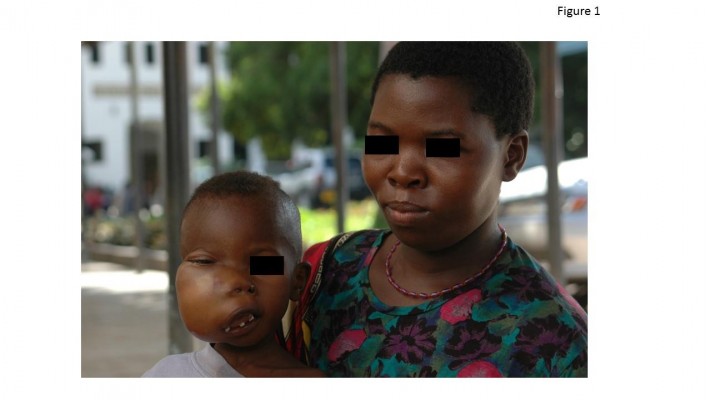
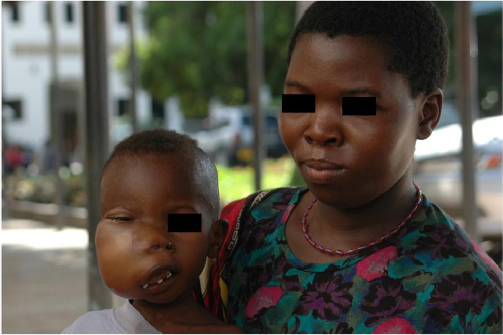 | Figure 1 Typical case of endemic BL involving the facial bones in an African child (picture courtesy of RB, taken at Ocean Road Cancer Institute, Dar-es-Salaam, Tanzania, 2007). |
Epidemiology and survival data from registries
Epidemiology and risk factors
The exact worldwide incidence of BL is difficult to assess, as collection of exact epidemiologic data is not possible in developing countries with the highest apparent incidence (equatorial Africa).7 BL is however subdivided into 3 distinct epidemiological subtypes: endemic (African), sporadic (non-endemic), and immunodeficiency-related.
Endemic BL is mainly confined to equatorial Africa (it is also present in Papua New Guinea), where it accounts for 30%–50% of all childhood cancers diagnosed each year, with an estimated incidence of 3–6 cases per 100,000 children per year.7 This incidence is approximately 50-fold higher than in the US, with a peak in children aged 4–7 years and a male-to-female ratio of approximately 2:1.8 The sporadic variant is mostly seen in the US and Western Europe. In the US, BL comprises 30% of pediatric lymphomas and <1% of adult non-Hodgkin lymphomas (NHLs).9 This translates into an estimated incidence of approximately 3 cases per million in both children and adults. In Europe, the incidence is approximately 2.2 cases per million.10 A large epidemiological survey on 3403 cases diagnosed during 1978–2002 however documents a rising incidence over the years with 3 distinct peaks at around age 10, age 40 since 1998–2002, and in the elderly.11 In adults, sporadic BL is typically seen in patients younger than 35 years, with a median age of 30 years.12,13 Sporadic BL is more common among Caucasians than Africans or Asian-Americans,9 and may be more common in some areas of Central America (Guatemala).14 In all age groups, most patients are male with a 3:1 or 4:1 male-to-female ratio.9,15,16
The immunodeficiency-associated variant is primarily seen in subjects infected by HIV who develop the AIDS and less commonly in patients with other immunodeficiencies (recipients of organ transplants). About 20% of all cases of BL are HIV+ individuals, and HIV+ individuals are 57 times more likely to develop BL than HIV− ones.17 A steep incidence was noted since the late 1980s, due to the growing epidemics of HIV infection, especially in males, mirroring the early epidemics of HIV infection among homosexual males in the US.6 In HIV+ patients, BL typically affects those with a relatively high CD4 T-cell count (>200/µl) and without opportunistic infections. In contrast to other HIV-associated lymphomas, the rate of BL in the HIV+ population has not decreased with the advent of powerful antiretroviral therapy.18
With regard to the risk factors for sporadic BL, a case–control study on 295 BL patients and 21,818 healthy subjects found the following factors to be significantly associated with a diagnosis of BL in patients younger than 50 years (n=133): history of eczema, taller height, and employment as a cleaner; instead, a history of allergy seemed to provide a protective effect.19
Survival
A 2002–2008 Surveillance, Epidemiology, and End Results (SEER) US study, including 3691 BL patients with a median age of 43 years, reported a 3-year survival of only 56%.6 Possible explanations for this suboptimal outcome were a selection bias with unequal access to tertiary medical centers delivering effective lymphoma treatments. “Real-world” results may be inferior to trial results because patient selection is operated by inclusion/exclusion criteria of prospective clinical studies. These normally include an adequate performance status and the lack of severe organ dysfunction and comorbidity, which increase the hazards related to intensive chemotherapy. Such very high-risk patients should nevertheless be treated, though their outcome can be inferior to that of trial patients. The worst setting is that of late diagnosis and referral, variously combined with disseminated disease, effusions, extensive gastrointestinal involvement, and severe impairment of metabolism and liver and kidney function. The SEER analysis emphasized how the gap between clinical trials and “real-world” outcomes was not the same across age groups. Outcome for patients aged 0–19 years was comparable to that reported by the literature, owing to the very high rate of participation of this patient population in therapeutic clinical trials.20 Reported outcomes for adult patients were significantly inferior, even in those aged 20–39 years diagnosed between 2002 and 2008, 60% of whom were alive at 5 years. Notably, the majority of BL-related deaths occurred within the first year from diagnosis, and the risk of death correlated with age. The analysis showed a cumulative incidence of death at 3 years of 37.4% for patients aged 20–39 years. Moreover, the conditional 5-year relative survival rate in patients alive for >12 months from diagnosis was 83.4%, without significant differences in age subgroups.21
Etiology, pathogenesis, and molecular pathogenesis
BL originates from germinal or post-germinal center B cells. The different clinical subtypes (endemic, sporadic, and immunodeficiency-related) likely arise from B cells at different stages of development. Epstein–Barr virus (EBV)-negative BL shows a lower degree of somatic hypermutation of variable region heavy chain immunoglobulin (Ig) genes and no signs of antigen selection, whereas EBV+ (generally endemic or AIDS-associated) BL displays significantly higher levels of somatic Ig hypermutation and antigen selection.22 These findings suggest that EBV− BL may arise from early centroblasts, while EBV+ BL may arise from memory B cells or late germinal center B cells. This difference in cell origin may also relate to a difference in c-MYC translocation breakpoints. The development of BL is in fact dependent upon the constitutive activation of the c-MYC proto-oncogene located at 8q24 and encoding for the MYC protein. This acts as a transcription factor modulating several target genes involved in cell cycle regulation, cellular differentiation, apoptosis, cellular adhesion, and metabolism.23,24 Additional factors must be present because a small percentage of HIV+ persons and healthy subjects have c-MYC translocations in B lymphocytes of enlarged lymph nodes without having BL.25
The overexpression of c-MYC is the result of translocation t(8;14), by which MYC is placed in close proximity to the promoter sequences of Ig genes, more frequently heavy chain genes mapping on 14(q32), or in 10%–15% of cases in chromosome 2 (at p12, promoter sequences of kappa light chain), or chromosome 22 (at q11, lambda light chain genes).26,27 In endemic (African) cases, the breakpoint on chromosome 14 involves the heavy chain joining region, while in non-endemic cases, the translocation involves the heavy chain class-switch region.28,29 In endemic cases, the breakpoint in chromosome 8 usually lies adjacent to c-MYC, while in sporadic cases, it often lies in intron 1 of the c-MYC gene.
Work in mouse models has shed light on the possible mechanisms leading to c-MYC translocations. Translocations involving the class-switch region of Ig heavy chain genes and c-MYC can occur with surprisingly high frequency in activated B cells undergoing class-switch recombination. These apparent “mistakes” are observed during the recombination events that allow B cells to switch from expression of IgM to other Ig types, which requires the enzyme activation-induced cytidine deaminase (AID),30 an essential cofactor for normal class-switch recombination. In further support, infection of mice with malaria (a known risk factor for human endemic BL) provokes sustained expansion of AID-expressing germinal center B cells, increasing the frequency of aggressive B-cell lymphomas bearing the molecular signatures of an AID-mediated DNA damage.31
The EBV infection is present in virtually all cases of endemic BL, approximately 30% of sporadic BL, and 40% of immunodeficiency-associated BL. One hypothesis is that EBV infection stimulates B-cell expansion, a process during which gene translocations may occur leading to activation and overexpression of c-MYC. Once BL emerges, the EBV infection is thought to have little effect on tumor progression. Of interest, EBV infection in BL displays a peculiar latent infection phenotype, characterized by lack of expression of the EBV transforming proteins LMP-1 and EBNA-2.32
Although chronic EBV infection has long been recognized to play a role in the etiology of virtually all cases of endemic (African) BL and a minority of sporadic and immunodeficiency-associated BL, this is more likely a polymicrobial process in which patients with other infections suffer from a persistent, acute phase of EBV infection.12,33 This prolonged acute phase, associated with a polyclonal B-cell expansion, could increase the likelihood of translocations involving c-MYC which in turn favor oligoclonal/clonal proliferations. Proposed diseases associated with persistent EBV infection and BL development include HIV, malaria, and arboviruses.12 Early epidemiologic data documented a high incidence of both Plasmodium falciparum malaria and endemic BL in equatorial Africa and Papua New Guinea.34,35 A subsequent study demonstrated that, when compared with age-, sex-, and location-matched controls, children with endemic BL were more likely to have had recent malaria infection (anti-HRP-II antibodies) and less likely to have had chronic malaria (anti-SE36 antibodies).36
Diagnosis and differential diagnosis
Histology and immunohistochemistry
The diagnosis of BL is based upon the evaluation of a biopsy specimen by an expert pathologist. The diagnostic hallmark of BL is the expression of markers typical of germinal center B cells. Histologically, BL is characterized by a diffuse growth pattern without any nodularity. Of interest, all the 3 clinico-epidemiologic subtypes have similar features. At low magnification, the characteristic “starry sky” pattern may be appreciated in standard hematoxylin/eosin preparations (Figure 2A and B). This is composed of a “blue” background of tightly packed round basophilic cells, without intercellular stroma, forming the “sky”, on which the “stars” of interspersed tangible-body macrophages are scattered. This is reflective of the rapid rate of cell doubling with individual cell apoptosis and tissue necrosis. At high magnification, the lymphoma cells in the classic type are intermediate-sized, monomorphic lymphocytes with scant blue cytoplasm, and lipid vacuoles. The nuclei are round with lacy chromatin and multiple, small nucleoli. Morphologic variants are designed as plasmacytoid or pleomorphic BL. On immunohistochemical analysis (Figure 2C–H), BL cells are mature B cells more similar to germinal center cells than activated B cells.22 They express monotypic surface IgM, CD19, CD20, CD79a, PAX5, and CD43, plus the plasma cell antigen CD38 and germinal center antigens CD10 and BCL6, with a Ki-67 proliferative fraction >95%. They typically do not express nuclear TdT, CD5, and BCL2. All cases with some expression of BCL2 protein should be tested for MYC, BCL2, and BCL6 breakpoints to exclude a double-/triple-hit lymphoma. In situ hybridization for EBV-encoded RNA is positive in >95% of all cases of endemic BL, in 30%–40% of AIDS-associated BL, and 20% of sporadic cases arising in Western countries.
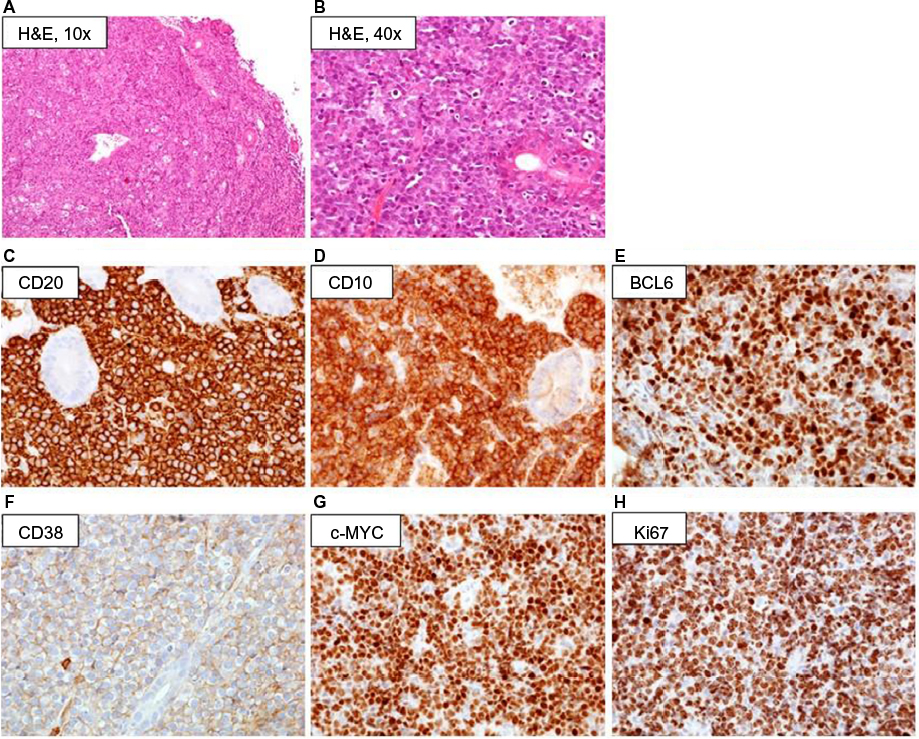 | Figure 2 Diagnostic pathology of BL: low magnification (×10, ×40) hematoxylin/eosin staining of BL sample involving the gastrointestinal tract, showing monotonous proliferation of medium-sized basophilic lymphoid cells punctuated by lightly colored macrophages (“starry sky” pattern) (A and B); immunohistochemistry, demonstrating staining for B-cell antigens including CD20 and the early CD10 antigen, with concurrent c-MYC and BCL6 expression and high proliferative rate (Ki67) (C–H). |
Genetics and genomics
The molecular hallmark of BL is the translocation of the MYC proto-oncogene to the Ig heavy or 1 light chain genes, leading to constitutive MYC activation.37 The most frequent genomic alterations are point mutations/indels in MYC, and TP53 mutations, confirming that these mutations may functionally cooperate to promote MYC-mediated oncogenesis.38 In classic BL, either endemic type or sporadic type, 90%–95% of Ig loci are involved, 85% for t(8;14)/IgH-MYC, 10% for t(8;22)/Ig-lambda-MYC, and £5% for t(2;8)/Ig-kappa-MYC. Any of the 3 translocations found in BL can be demonstrated by fluorescent in situ hybridization using break-apart fusion probes to the flanking regions of the MYC locus. MYC translocation is sensitive but not specific for BL, and some BL do not have a typical translocation, including a recently identified entity called Burkitt-like lymphoma with 11q aberration.39 Additional gene alterations include the following: truncating mutations of ARID1A and amplification of MCL1; point mutations of LRP6; truncating alterations of LRP1B, PTPRD, PTEN, NOTCH, and ATM; amplifications of RAF1, MDM4, MDM2, KRAS, IKBKE, and CDK6; deletion of CDKN2A;37 overexpression of MIR17HG; activating mutations of TCF3 and/or inactivating mutations of its negative regulator ID3; and CCND3 activating mutations.37 Generally, EBV-negative cases are more likely to express multiple genetic alterations than EBV+ cases, and generally, there are biological differences between adults and young: young patients display more frequent 13q31.3q32.1 amplification, 7q32q36 gain, and 5q23.3 copy-neutral loss of heterozygosity (CN-LOH); 17p13 and 18q21.3 CN-LOH are only detected in adults BL. ID3 mutations are present in adult but only in 42% of childhood patients; ID3 double-hit mutations, as well as 18q21 CN-LOH, seem to be associated with poorer outcome.37,38 Moreover, all BL subtypes share a very similar gene expression profile.37
Differential diagnosis
BL needs to be clearly distinguished from diffuse large B-cell lymphoma (DLBCL), unclassifiable BL/DLBCL, which has an extremely poor prognosis, and other tumors with histomorphological and immunohistochemical features intermediate between BL and DLBCL, called “gray-zone lymphoma”.40 BL and DLBCL are recognized by the World Health Organization (WHO) as separate entities having distinct genetic alterations, tumor morphology, and immunophenotype.37,38 Dysregulation of MYC through chromosomal translocation is the genetic hallmark of BL, but it can be encountered in several other lymphomas, especially DLBCL and unclassifiable DLBCL.37,38 There have been numerous reports of lymphomas with BL morphology or Burkitt-like morphology with MYC translocation in addition to complex cytogenetics or other translocations, such as t(14;18) with IgH-BCL2 gene fusion as seen in follicular lymphoma. Therefore, the concurrent analysis for MYC and BCL2/BCL6 translocations is useful to differentiate BL from gray-zone BL/DLBCL and DLBCL confounders. In hematoxylin/eosin-stained sections, BL should be distinguished from a variety of tumors, including some non-hematologic malignancies, acute myeloid leukemia/myeloblastic sarcoma, lymphoblastic lymphoma, some cases of blastic mantle cell lymphoma, and even plasmablastic myeloma. More challenging is the differential diagnosis from DLBCL, especially of the germinal center type, due to the overlapping immunophenotypes and the occasional growth pattern of DLBCL mimicking BL, with sheets of relatively monomorphic, cohesive cells, and the “starry sky” pattern related to the macrophages. These mimickers of BL, in which no MYC translocation is usually demonstrable, were previously diagnosed as Burkitt-like lymphoma or unclassifiable B-cell lymphoma, with features intermediate between DLBCL and BL. DLBCLs with similar BL appearance but positive MYC (5%–15%) as well as a BCL2 or BCL6 translocation are called “double-hit” and “triple-hit” lymphomas. In the 2016 WHO classification, “double-” and “triple-hit” lymphomas were grouped in the new category of high-grade B-cell lymphoma (HGBL) with rearrangements of MYC and BCL2 and/or BCL6.39 Cases of blastic appearance or intermediate between DLBCL and BL without MYC and/or BCL2/BCL6 rearrangements are placed in the HGBL category with no further specification. A simplified diagnostic algorithm was developed to confirm the diagnosis of BL vs DLBCL/BL, DLBCL, B-lymphoblastic lymphoma, and blastoid mantle cell lymphoma.7
Clinical features
Because the tumor-doubling time is very short (about 25 hours), patients present rapidly growing tumor masses and often have signs of rapid tumor turnover with high serum lactate dehydrogenase (LDH) concentration and elevated uric acid. Endemic or African BL usually develops in children with a jaw or facial bone tumor (50%–60% of cases), while an initial involvement of the abdomen is less common. The primary tumor can spread to mesentery, ovary, testis, kidney, breast, and meninges, spreading to lymph nodes, mediastinum, and spleen less frequently. Bone marrow involvement occurs in <10% of cases at presentation but is common at recurrence or with treatment resistance. Non-endemic or sporadic BL usually has an abdominal presentation (91%) with massive disease and ascites, involving distal ileum, stomach, cecum and/or mesentery, kidney, testis/ovary (6%), and breast, plus bone marrow (20%) and/or central nervous system (CNS; 14%).41 Presenting symptoms can be related to bowel obstruction or gastrointestinal bleeding, mimicking acute appendicitis or intussusception. Lymphadenopathy, if present, is generally localized. Bone marrow and CNS involvement is more common with recurrent or resistant disease.42 Immunodeficiency-related BL is more likely to involve lymph nodes, bone marrow, and CNS, accompanied by signs or symptoms related to the underlying immunodeficiency. A subset of patients display extensive bone marrow and blood involvement. Such cases are termed Burkitt leukemia. However, this is a different manifestation of the same disease commonly treated as advanced-stage BL.
Clinical evaluation, staging, and risk stratification
At diagnosis, the clinical evaluation aims to define the extent of the disease, primarily through an accurate physical examination. Besides enlarged lymph nodes and abdominal masses or hepatosplenomegaly, any cranial nerve palsy or a mental neuropathy should raise the suspect of CNS involvement and prompt further investigations (cerebrospinal fluid [CSF] examination, magnetic resonance imaging, and/or computed tomography [CT] of brain and spine, depending on clinical symptoms). Laboratory tests include complete blood counts, renal and liver function tests including creatinine clearance, calcium and urate level, serum LDH level, and coagulation. Checking the kidney function and urinary output is of the utmost importance. Testing for HIV and hepatitis B and C virus is recommended. Instrumental exams must be carried out expeditiously in order not to delay the start of chemotherapy, and should include a CT scan of chest, abdomen, and pelvis plus total-body positron-emission tomography (PET) scan. A bone marrow biopsy is performed to detect any degree of marrow involvement, along with an early medicated lumbar puncture for diagnostic cytology and flow cytometry analysis of the CSF.43 In selected cases, the use of anthracyclines requires a pretreatment assessment of cardiac function.44
At completion of all these investigations, BL can be staged and classified as low or high risk based on number of involved sites, presence of bulky disease, and LDH concentration, according to the Murphy staging system developed for childhood B-NHL.45 With this system, stage 1 is defined by a single nodal/extranodal site excluding mediastinum or abdomen; stage 2 is defined by a single extranodal site with regional involvement, by two or more nodal sites with or without regional node involvement on the same side of the diaphragm, or by a completely resected primary gastrointestinal tumor with or without regional involvement; stage 3 is defined by all primary intrathoracic tumors (mediastinum, pleura), by nodal/extranodal tumors on both sides of the diaphragm, by unresectable extensive intra-abdominal disease, and by paraspinal/epidural tumors regardless of other involved areas; and stage 4 is defined by CNS or bone marrow involvement. Slightly different staging systems were developed in Europe by the French-American-British/Lymphomes Malins B group and the Berlin-Frankfurt-Muenster (BFM) group. These systems, along with the classic Ann Arbor classification which is still adopted in adult patients, are commonly used in children and AYAs to define extent of disease and the risk class, and to establish the treatment intensity accordingly in some studies.46
Minimal disseminated and residual disease
Minimal disseminated disease (MDD) and minimal residual disease (MRD) refer to subclinical, submicroscopic amounts of lymphoma or leukemia cells detectable either at diagnosis (MDD in BL) or following a successful therapy of the disease in patients achieving a complete clinical response (MRD in BL/Burkitt leukemia). Because MDD/MRD is a well-known risk factor conferring a significantly worse outcome in acute lymphoblastic leukemia (ALL) and lymphoblastic lymphoma at all ages,47–50 studies were incepted in this sense in BL patients as well. Because BL is a c-MYC+ mature B-cell neoplasm, MRD in BL could be assessed using a polymerase chain reaction (PCR) specific for the MYC/IgH fusion, with a sensitivity between 10–3 and 10–5, identifying BL cells in the bone marrow and peripheral blood of 4 of 16 and 6 of 15 patients without visible blasts at diagnosis.51 Moreover, because PCR for t(8;14) is feasible for MDD/MRD analysis in 70% of the cases only, another study demonstrated an improved feasibility, up to 95% in BL, using t(8;14) PCR in combination with PCRs for IgH and Ig-kappa light chain rearrangements.52 These and other studies showed a negative prognostic impact of MDD/MRD in patients with BL and Burkitt leukemia. Mussolin et al,53 for the pediatric Italian group, using t(8;14) PCR in 47 children with Burkitt leukemia, found a significantly superior outcome in 31 patients who were PCR-negative post-induction (3-year relapse-free survival [RFS] 84%) compared to 9 PCR+ patients (3-year RFS 38%, P=0.0005). In BL, the same authors found MDD at diagnosis in 31% of 84 patients, of whom only 18% had morphological evidence of bone marrow disease. Three-year progression-free survival (PFS) was 93% in MDD-negative patients and 68% in MDD-positive patients (P=0.03), and in multivariate analysis, MDD only was significant for a higher risk of failure.53 Finally, in a larger study including 422 patients (379 BL), in the subgroup of 128 children and adolescents who had the study done, MDD was the only risk factor in multivariate analysis. Studies from the Children’s Oncology Group yielded nearly similar results.54 A report on 14 BL subjects showed that none of 12 MRD-negative after 2 months of therapy relapsed compared to both those found MRD-positive.55 Another PCR study conducted on 32 children and adolescents with a variety of aggressive B-cell lymphomas including BL (n=22)56 confirmed the presence of MDD in all cases, although only 3 had morphological evidence of marrow involvement. With regard to outcome, treatment results were excellent; however, 2 patients relapsed, both with MDD-positive specimens. In Burkitt leukemia, a small study in 10 patients identified MRD in 7 and 5 cases at end of induction and consolidation, respectively.56 Because none of MRD-positive cases relapsed, the study conclusion was that MRD monitoring at later timepoints is warranted.
Other clinical and biological risk factors
The prognosis of BL patients is significantly related to age, with an increased curability rate in pediatric patients compared to adults, with further worsening in older adults and the elderly. It is unclear whether this occurs solely because children tolerate better intensive treatments, or because distinct pathogenetic mechanisms affect disease outcome.38 Regardless of the conventional risk definition, the recent FAB96 study by Cairo et al identified the clinical variables conferring a significantly inferior event-free survival (EFS).37 Apart from stage, these were an elevated LDH, primary mediastinal involvement, and combined bone marrow/CNS disease.37 The pediatric Italian review on 379 BL patients confirmed LDH to exert the greatest prognostic relevance (P<0.0001).54 Among biomarkers, apart from MDD/MRD which is not always available, some specific cytogenetic findings, such as deletion of 13q, gain of 7q, and complex cytogenetics, could impart a worse prognosis and/or benefit from treatment intensification.37 Among genetic aberrations, ID3 and CCND3 double-hit mutations and 18q21 CN-LOH were associated with poorer therapeutic response and adverse prognosis. The occurrence of a double ID3 and CCND3 mutation may be even more deleterious by acting simultaneously on 2 different pathways that cross talk to activate the pro-survival PI3K pathway (ID3/TCF3) and drive cell cycle progression (CCND3/CDK6). TCF3 and CCND3 mutations may prevail in older and younger patients, respectively.37 A concurrent HIV+ status is another potential risk factor that may affect a proportion of younger adults.
Response evaluation
A uniform approach to describe treatment response in malignant lymphomas was proposed by Cheson et al. This ranged from a complete response (CR) indicating the disappearance of disease, to a partial response, stable disease, and progressive disease when old lesions increase in size >50% or new lesions develop. In the updated version, including PET scan results, patients with PET-negative residual lesions are considered in CR.57,58 However, a persistent residual mass in children with NHL may require a biopsy to ascertain induction failure. In pediatric lymphoblastic lymphoma, end-of-induction biopsies are often negative, which indicates the limitations of conventional diagnostic imaging.59 Functional PET scanning may help resolve this dilemma, although interpretation of results is sometimes difficult and the use of PET to guide therapeutic decision making should be considered investigational and applied only within the confines of clinical trials.60,61 Moreover, false-positive PET results were reported in a small series of children with BL.62 This may be the result of a benign inflammatory process, a xanthomatous pseudotumor, brown fat uptake, rebound thymic hyperplasia, infection, or even an effect of granulocyte colony-stimulating factor. A diagnostic challenge is the persistence of minimal residual uptake (MRU). MRU is generally felt to represent a benign process and not likely to represent malignancy; however, there are no uniform criteria to define MRU.63–65 With the most recent criteria,64,66 MRU is graded 1–5, identifying a group (grade 5) with poorer PFS than MRU grades 1–4. This new classification may be prognostically more informative than other MRU-grading systems.67 The role of PET/CT in the management path of younger patients with NHL including BL has yet to be fully established. Although PET/CT is highly promising in assessing disease involvement,68 its impact on therapeutic stratification has yet to be clarified, and its role in remission assessment has only been investigated in small series. Some reports indicated high rates of false-positive results, requiring histologic examination before treatment modification.68 The prognostic value of early (after 1–3 cycles of chemotherapy) PET/CT response assessment has also not been established,68,69 and defining the exact relationships between treatment courses and PET response is one of the remaining tasks in the management of BL lymphoma.
Shaping an effective BL therapy
BL is now regarded as one of the greatest therapeutic successes in oncology; however, due to rapid growth kinetics and frequent presence of extranodal localizations, it requires a highly specific approach. The optimal initial therapy of BL has not been clearly defined given the paucity of randomized studies in this uncommon disease and the fact that different, modern BL regimens can all achieve excellent results in younger age groups.44 Standard chemotherapy such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) is inadequate for treating BL, and there is no role for radiation therapy, even for localized disease or paraspinal presentations, which respond very quickly to chemotherapy.22
Effective treatment strategies for BL were initially pioneered in pediatric studies and in HIV− patients, who still represent the majority of BL cases in the AYA group. The use of intensified chemotherapy protocols associated with the prophylaxis of CNS localizations determined high percentages of healing (Table 1).70–80 A Children’s Cancer Group trial demonstrated that the cyclophosphamide/methotrexate-based COMP regimen was superior for the treatment of advanced-stage BL.81 A major contribution achieving cure rates of 70%–90% in children was made by Magrath et al, who developed CODOX-M/IVAC (cyclophosphamide, doxorubicin, vincristine, methotrexate, ifosfamide, cytarabine and etoposide).74 In that study, 2-year EFS was 92% in 41 patients of median age 25 years, and AYAs fared better than older adults.74
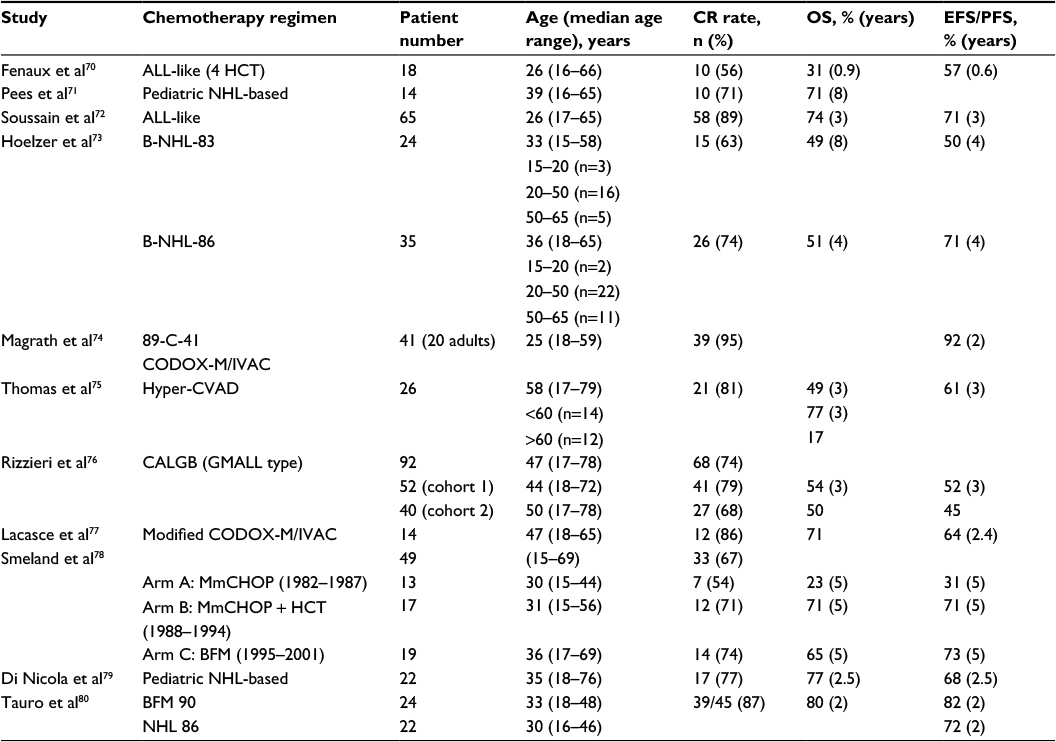 | Table 1 Treatment results in BL prior to rituximab (distribution and outcome of different age subsets indicated when available) Abbreviations: BL, Burkitt lymphoma; CR, complete response; OS, overall survival; EFS, event-free survival; PFS, progression-free survival. |
In addition, Murphy et al introduced another very effective regimen of fractionated high-dose cyclophosphamide and high-dose methotrexate (HD-MTX), plus vincristine, doxorubicin, and cytarabine, with cycles administered in tight rotation. CR rate was 93% with 81% 2-year disease-free survival (DFS).82 A subsequent study from the German Multicenter Study Group for Adult ALL (GMALL), adapting to adults a different multidrug pediatric BFM schedule, demonstrated that increasing MTX dosing did not improve survival while aggravating toxicity in older patients, and established an MTX dosing of 1500 mg/m2 in adolescents (age >15 years) and in general in patients younger than 55 years.83 These and other short-intensive regimens, developed before the rituximab era and adapted to AYAs and adults, reported CR and overall survival (OS) rates of 54%–95% and 31%–80%, respectively. Importantly, all effective regimens include multiple doses of CNS-penetrating agents (HD-MTX, cytarabine) and/or repeated medicated intrathecals to prevent the risk of CNS progression, which is otherwise rather common in BL.
Improving BL therapy with rituximab
The addition to such brief, high-intensity BL regimens of the anti-CD20 antibody rituximab resulted in further prognostic improvement, bringing 3-year survival close to 90% in younger adults (<55–60 years) (Table 2).84–96 To underscore the usefulness of rituximab, a pediatric window study with anti-CD20 rituximab given prior to chemotherapy to 136 patients (22 aged ≥15 years) yielded a response rate of 40% in BL/Burkitt leukemia.97 In adults, rituximab added to the Hyper-CVAD regimen (cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with HD-MTX and cytarabine) improved outcome with no additional toxicity compared to Hyper-CVAD alone: 5-year OS and DFS rates were improved from 60% and 50% to 70% and 70%, respectively.94 In the final analysis of the most recent rituximab-modified GMALL program administered to 363 patients (2014–2016), CR rate was 88%, 5-year OS was 80%, and PFS was 71%.78 Comparable results were reported by others, confirming the value of rituximab and the better outcome of younger patients whenever this type of analysis was available. Lately, an infusional MTX-free regimen, dose-adjusted (DA) EPOCH-R (etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, rituximab), reported by Dunleavy et al, achieved excellent response and survival rates in a small patient cohort, retaining a high therapeutic potential even without dose adjustment.92 This type of treatment has not yet been tested in adolescents and children. The role of additional rituximab in BL was conclusively demonstrated by a phase III trial from France,98 also reporting separately the more favorable outcome of AYAs <40 years compared to older patients (described in the “AYA vs adult results” section).
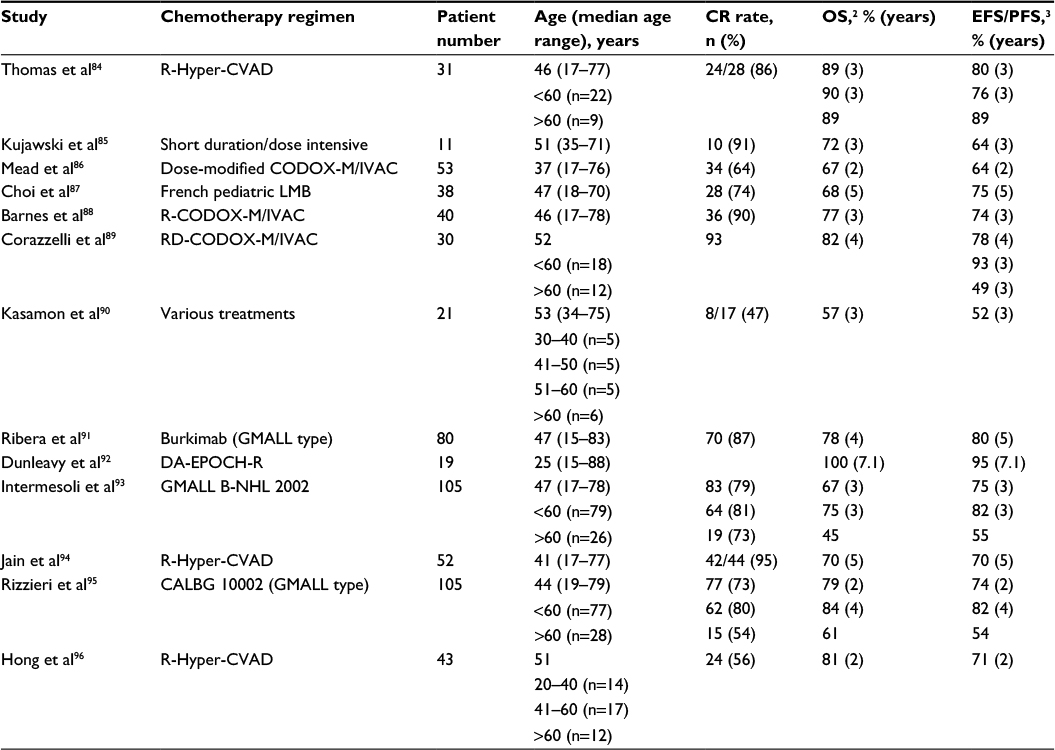 | Table 2 Treatment results in BL with rituximab-based regimens (distribution and outcome of different age subsets indicated when available) Abbreviations: BL, Burkitt lymphoma; CR, complete response; OS, overall survival; EFS, event-free survival; PFS, progression-free survival. |
At present, it is not known whether any of these rituximab-based modern regimens is superior to others, although differences exist (Table 3). This possibility could be suggested by an easier drug scheduling and/or an inferior toxicity profile of a given regimen, provided OS and DFS results are comparable to other top regimens, or alternatively, by a clear therapeutic advantage, which is more difficult to assess in view of the uniform OS/DFS rates around 80%–90% for AYA patients. Of note, a randomized comparison might be difficult to set up not only for the choice of study arms (perhaps having CODOX-M/IVAC as standard arm) but also for the primary objective which, in case of efficacy, would require very high patient numbers, while in case of feasibility and toxicity (SC-EPOCH-RR better?) would imply a different design with a combined end point consisting of multiple grade 3 and 4 toxicities plus a non-inferiority survival assessment.
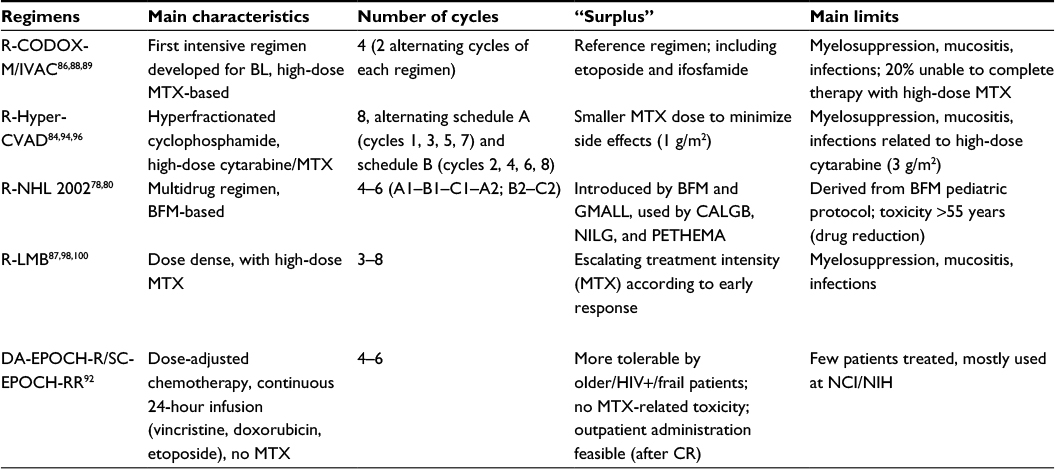 | Table 3 Comparative features of highly effective, modern treatment regimens for BL (all with rituximab) Note: In some regimens, number of cycles may vary depending on disease stage and/or early treatment response. Abbreviations: BL, Burkitt lymphoma; MTX, methotrexate; BFM, Berlin-Frankfurt-Muenster; GMALL, German Multicenter Study Group for Adult ALL; NCI/NIH, National Cancer Institute/National Institutes of Health; CALGB, Cancer and Leukemia Group B; NILG, Northern Italy Leukemia Group; PETHEMA, Programa Español de Tratamientos en Hematologia. |
AYA vs adult results
The more favorable outcome of younger patients in both pre-rituximab and rituximab era was confirmed in more than 1 study and in rather large patient numbers (Table 4).83,98–101 Two no-rituximab study are of historical interest only. With rituximab, the GMALL reported significant outcome difference between AYAs aged 15–25 years, adults between 26 and 55 years, and older patients (OS 90%, 84%, and 62%, respectively).83 The French study randomizing 260 patients to chemotherapy with or without rituximab reported therapeutic results for patients <40 vs >40 years (AYAs 38%). In univariate analysis, regardless of rituximab therapy, EFS and OS of 101 AYAs were statistically better than the figures observed in older patients (P<0.0001).98 Therefore, with modern rituximab-based BL regimens, the expectation is to cure 85%–90% of AYAs in the 15–40 years age range, with a low incidence of failures related to either resistant/progressive disease or treatment complications. Among the latter, tumor lysis syndrome (TLS) associated with acute kidney failure is a well-recognized and most feared potentially fatal complication. Because BL is one of the malignancies at highest risk of TLS, one primary treatment end point is to avoid this possibility and preserve an adequate renal function throughout therapy.
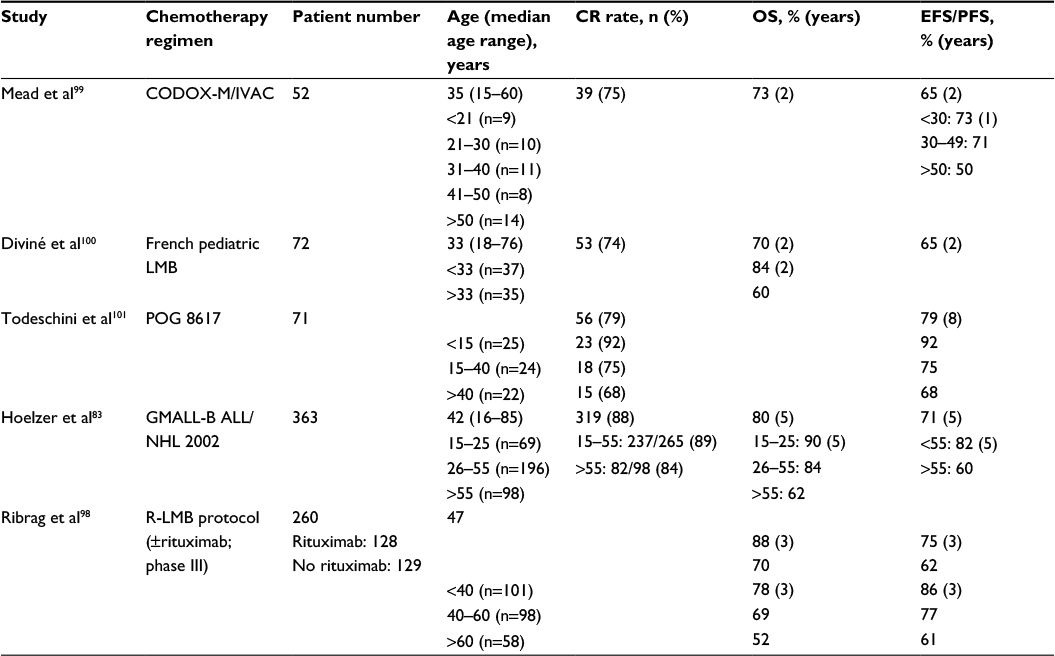 | Table 4 Age-related treatment results in BL with or without rituximab-based regimens Abbreviations: BL, Burkitt lymphoma; CR, complete response; OS, overall survival; EFS, event-free survival; PFS, progression-free survival. |
Tumor lysis syndrome
TLS results from massive cytolysis with release of large amounts of phosphates and chelation of calcium and its precipitation in the kidney tubules. It is characterized by hyperuricemia, hyperkalemia, hyperphosphatemia, and hypocalcemia, which may overwhelm the kidney’s homeostatic mechanisms and capacity of excretion.102,103 TLS can cause acute renal impairment, cardiac rhythm disturbances, CNS toxicity, and eventually death.102 The laboratory TLS is defined by ≥2 of the following metabolic abnormalities occurring simultaneously within 3 days prior to and up to 7 days after treatment initiation: hyperuricemia (>8.0 mg/dl), hyperkalemia (>6.0 mmol/l), hyperphosphatemia (>4.5 mg/dl), and hypocalcemia (corrected Ca <7.0 mg/dl, ionized Ca <1.12 mg/dl). The definition for clinical TLS is as above plus the occurrence of an elevated creatinine, seizures/CNS toxicity, cardiac dysrhythmia, or symptomatic hypocalcemia.103 Risk factors for TLS include preexisting kidney dysfunction, high pretreatment uric acid and LDH, male gender, and splenomegaly.102,103 Appropriate fluid management before and during the administration of chemotherapy is the key to prevent TLS, to maintain an abundant urinary output that will dispose of systemic uric acid and phosphate. Allopurinol and better rasburicase are the cornerstones of TLS prevention, with the latter rapidly inhibiting the uric acid synthesis.22,103 When severe, acute kidney failure develops, hemodialysis is mandatory, preferably with a continuous modality to reduce the risk of “rebound” hyperkalemia or hyperphosphatemia.102
Hematopoietic cell transplantation
Because modern chemotherapy may be curative for the majority of BL patients and up to 90% of AYAs, the interest toward hematopoietic cell transplantation (HCT) has now considerably diminished. Past experience with allogeneic HCT is anecdotal, but some studies evaluated autologous HCT. A Dutch group treated 27 patients in first CR with brief initial high-dose chemotherapy consisting of 2 cycles of cyclophosphamide, doxorubicin, etoposide, mitoxantrone, and prednisone followed by autologous HCT after BEAM conditioning (carmustine, etoposide, cytarabine, melphalan), obtaining a 5-year EFS and OS estimates of 73% and 81%, respectively.104 A retrospective analysis considered 117 patients with BL who underwent autologous HCT between 1984 and 1994, showing a 72% 3-year OS for those in first CR, 37% for those in chemosensitive progression, and only 7% for those with chemoresistant disease.105 HCT is currently of no interest in AYAs responsive to modern treatment regimens and is of unproven value in progressive disease.
Management of BL in HIV+ AYAs
The HIV infection is expected to occur in a fraction of BL patients belonging to the AYA group defined by a maximum age of 40 years. It is however difficult to extrapolate AYA results from the general experience in HIV+ adult patients. Nevertheless, high cure rates were reported in HIV+ patients treated with optimal BL regimens. In a series of 14 HIV+ patients treated with Magrath’s program, 2-year survival was 60%, even though the majority of patients had stage IV disease.6 In other series with a median patient age of 46–52 years, 3-year survival ranged from 82% to 89%.99,106 The major limitation in highly immunocompromised HIV+ patients with clinical AIDS and BL is the higher risk of infections, despite the concurrent administration of highly active antiretroviral therapy (HAART). In this setting, low CD4+ T-cell counts and poor performance status are predictors of inferior survival. In a study from Spain, 118 patients (38 HIV+) were treated with the intensive GMALL program plus rituximab. Four-year DFS and OS were not significantly different between HIV+ and HIV− patients: 77% and 63% vs 80% and 78%.99 A larger study on 81 HIV+ patients treated with the same regimen and including AYAs (about 50% of the cases aged <40 years) reported OS and PFS rates of 72% and 71% at 4 years, and confirmed the greater treatment-related toxicity and the inferior outcome of patients in leukemic presentation, with CD4 T-cell count <200/μl and Eastern Cooperative Group performance score >2.107 Another study evaluated CHOP chemotherapy plus cytarabine and etoposide in 63 HIV+ patients (median age 40 years) with a median CD4 count at diagnosis of 239/μl: CR rate was 70% with only 7 treatment-related deaths though marked cytopenia developed in all patients; 2-year OS was 47%.106
Oriol et al were among the first to show the effectiveness of an intensive chemotherapy regimen in HIV+ individuals with BL/Burkitt leukemia (n=53, median age 35 years, range 15–74 years), with 14 HIV+ patients receiving concurrent HAART.108 Twenty-nine of these patients (55%) completed the 8 scheduled cycles, 28% of HIV+ cases and 64% of HIV− patients. However, there was no difference in CR rate between HIV− (77%) and HIV+ patients (71%), and the 2-year OS probability was 51%, again without significant difference between the 2 subsets (43% vs 55%). Only an advanced age (>60 years) correlated with a lower CR rate and resulted the only adverse prognostic factor.108 Another PETHEMA study, enrolling 18 HIV+ patients, analyzed the relation between HAART and response to treatment: 9 patients (47%) did not receive or discontinued HAART, and 5 were started on HAART before diagnosis. CR was 70% for HAART users (7 of 10) and 71% for HAART responders (5 of 7), with a significant difference in 2-year OS between HAART responders (85%) and nonresponders (27%).109 Despite the apparent benefit of HAART response in HIV-related lymphomas treated with intermediate intensity regimens, there may be concerns about the toxicity of combining antiretroviral treatment with high-dose chemotherapy; however, this study demonstrates, for the first time, that response to HAART prolongs OS in HIV-infected patients treated with a specific BL protocol.109
Recently, the DA-EPOCH-R regimen has been modified for HIV+ patients, aiming to reduce the length of neutropenia and the infectious risk, using a short-course lower dose formulation including a second rituximab administration (SC-EPOCH-RR).92 Five-year OS was 90% in 13 immunodeficient BL patients (11 HIV+). Based on all these studies, HIV+ BL AYAs should be approached like immunocompetent patients with the addition of HAART. A de-intensified, less toxic regimen such as SC-EPOCH-RR deserves further evaluation.
BL in underdeveloped countries
The African BL epidemic affects mainly children with a very low incidence of the disease in AYAs, yet it is highly representative of the problems affecting the underdeveloped world. In a study from Uganda (n=1088), median patient age was 7 years, and apparently, no case was older than 14 years.110 Comparable data were provided by another large study from Northern Tanzania (n=944) and by a collaborative series from equatorial Africa (n=356),111,112 in which only 13 patients (3.7%) were aged ≥15 years. Apart from this uniqueness, delayed referral to treatment center, favoring growth and dissemination of BL,113 low socioeconomic status of patient families, and serious economic constraints of health systems all preclude access to expensive drugs (rituximab, high-dose therapies) and high-level supportive care, excluding these patients from the most effective, modern chemo-immunotherapy regimens. As anticipated by the historical experience, the CHOP regimen is poorly effective in endemic BL.114 However, thanks to an international initiative led by Ian Magrath, the use of a moderately intensive protocol consisting of cyclophosphamide, standard-dose MTX, vincristine, and intrathecal therapy, with a shift to ifosfamide, mesna, etoposide, and cytarabine in refractory cases (INCTR 03-06), this program resulted in an OS rate of 62% at 2 years, definitely an improvement over prior rates of 10%–20% only,112 and a major step forward in the management of endemic BL in underdeveloped areas with hundreds of new cases yearly.
Resistance and relapse
Apart from the occasional refractory case, a few responsive BL patients will relapse soon after treatment completion and generally within the first 6 months of follow-up. Current results in refractory/relapsed BL are extremely poor, and new options are urgently needed. While some patients with chemotherapy-sensitive disease may achieve long-term remissions, the outcome of patients with chemotherapy-resistant disease is dismal.44
The frequency of relapse vs progression of disease varies according to first-line treatment used, and obtaining an objective comparison is difficult. Kim et al reported an incidence of relapse or refractory disease of 6.4% (9 of 140) and 10.1% (33 of 327) in 2 Austrian and Japanese multicenter series, respectively.115 In the Japanese series, 15.2% (19 of 125) of the patients had relapsed or refractory BL, and after heterogenous reinduction chemotherapy, 7 of 19 achieved a second CR (36.8%) while 12 died of rapid disease progression.115 Generally, the salvage regimen incorporates chemotherapeutic agents to which the patient was not or only partially exposed, as the DHAP regimen (dexamethasone, high-dose cytarabine, and cisplatin).105 If the disease demonstrates chemosensitivity, the patients are referred for high-dose chemotherapy and HCT, with a reported 5-year OS of only 21% and 18% for autologous and allogeneic HCT, respectively.116 Patients with chemoresistant disease are given supportive care,105 but could be considered for experimental therapies, which in general, due to the globally poor results of traditional salvage programs, should be considered in all refractory or relapsed cases.
Management challenges in AYAs with BL
Remaining challenges with BL in AYAs are resistance and relapse, induction of death by complications in high-risk patients, and the limited access to an early diagnosis and effective treatments in equatorial Africa and other underdeveloped countries. An early diagnosis with immediate referral is crucial because intensive regimens may be too toxic for patients with advanced disease and organ failure (liver, kidney), or simply not feasible when acute renal failure develops (no HD-MTX). This is of particular concern in HIV+ patients and other frail patients who are more prone to infectious complications and should be preferably treated with less toxic chemotherapy. In this regard, the MTX-free SC-EPOCH-RR regimen could be a valid choice to reduce the duration of neutropenia and the risk of infections.92 The issue of an early treatment failure related to late diagnosis and referral and/or extensive disease with end-stage organ damage is of particular concern in BL therapy as summarized in Table 5.
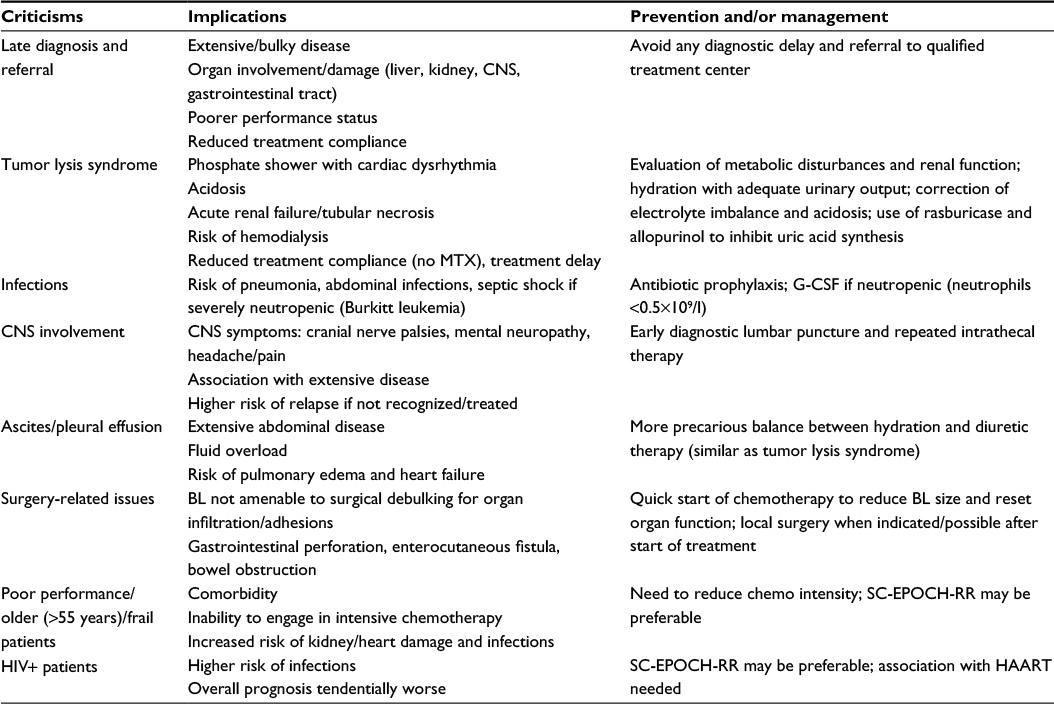 | Table 5 Critical issues and steps to optimize the initial management of BL Abbreviations: BL, Burkitt lymphoma; CNS, central nervous system; MTX, methotrexate; G-CSF, granulocyte colony-stimulating factor; HAART, highly active antiretroviral therapy. |
New drugs and treatment modalities
At present, it is not possible to define which patients will fail therapy due to tumor resistance or progression during or soon after therapy. With an OS rate of 90% in most AYA series, exclusively obtained with highly intensive first-line therapy, resistance and relapse are rare, and no specific retreatment regimen is associated with durable results. Rather, the early identification of cases at high risk of failure could allow a stepwise intensification of therapy with radiotherapy (for localized residual disease on CT/PET reassessment) and/or novel agents such as immunotherapeutics and other (for detection of MRD at critical evaluation timepoints).
Immunotherapy
Immunotherapy is an attractive option, since BL cells express several target antigens and new monoclonals are now available other than rituximab. Ofatumumab and obinutuzumab (GA101), the latter demonstrated more active than rituximab even in rituximab-resistant BL cell lines, are other anti-CD20 antibodies deserving further evaluation in BL therapy.117 Other most promising new agents are blinatumomab, a bispecific antibody targeting CD19 and bridging CD19+ BL cells to CD3+ cytotoxic autologous T cells, and inotuzumab ozogamicin, an anti-CD22 monoclonal conjugated to calicheamicin. Both were demonstrated effective in refractory/relapsed and MRD+ B-precursor ALL,118–120 and could therefore be used in BL to enhance the activity of current first-line regimens and/or improve treatment of advanced or MRD+ disease. Anti-CD19 chimeric antigen receptor T cells could also be exploited therapeutically, although compared to monoclonals they are more cumbersome to obtain, which contrasts with the need for an immediate therapeutic action at first signs of treatment failure in BL.
Experimental agents
Among experimental drugs, evidence is accumulating in favor of histone acetylase inhibitors (valproic acid, tubacin) and mTOR inhibitors (temsirolimus). When used at pharmacologic concentrations, these drugs may interact synergistically and inhibit BL cell growth by induction of autophagy or apoptopsis.121,122 Activity against BL cells was reported for plitidepsin, an antitumor agent of marine origin expressing antiproliferative and antiangiogenic activity, in association with rituximab.123 Of interest are also ascorbic acid in EBV+ BL cells,124 and icaritin, which induces S-phase arrest and cell apoptosis by activation of caspase-8 and -9 and inhibition of c-MYC and BCL2.125
Other highly effective inhibitors of cell proliferation already tested with success in a variety of aggressive B-cell neoplasms may soon lead to further improvement of existing BL chemo-immunotherapy regimens. These agents include the proteasome inhibitor bortezomib,126,127 inhibitors of metabolic pathways related to the B-cell receptor (ibrutinib, idelalisib), and the BCL2 inhibitor venetoclax.128 Blocking the tumor cell escape from the immune response mediated by the PD-1 pathway is another therapeutic opportunity. Several trials with anti-PD-1 and anti-PD-L1 agents in a variety of resistant B-cell malignancies (excluding BL but including DLBCL) were recently completed or are still recruiting.129
Toward MYC inhibition in BL
Because of the functional overexpression and the pathogenetic role of the MYC proto-oncogene in BL, new experimental therapy using direct and indirect MYC inhibitors is highly attractive. Direct inhibitors such as JQ1 and THZ1 target MYC and MAX interactions, since MYC heterodimerizes with MAX to bind a consensus DNA sequence.130 Furthermore, the link between JQ1 and MYC may involve the dependence of MYC on multiple enhancers and “super-enhancers” that are highly dependent on a BET protein BRD443 in combination with the PI3K pathway, mechanistically targeted by rapamycin (mTOR inhibitor) or histone deacetylase (HDAC) inhibitors, which may cause tumor regression. Inhibition of the BET mitigates the effect of MYC overexpression by preventing signal transduction. JQ1 was found to inhibit the bromodomain BRD4, reducing MYC transcription.130–132 Decreased expression of c-MYC and restoration of apoptosis can also be obtained by dual mTOR inhibitors,133 and BET inhibitors can synergize with inhibitors of the upstream replication stress sensor ATR, blocking progression of c-MYC+ lymphoma cells.134 THZ1 was developed as a novel highly selective covalent inhibitor of CDK7, which, by linking to a cysteine residue, specifically downregulates MYC expression.130 Other agents currently being evaluated to modulate MYC indirectly in vivo are Aurora kinases A and B. They are normally upregulated by MYC, and therefore, when blocked, they escape MYC activity leading to cell apoptosis.131 Other BET inhibitors (I-BET 151, GSK525762, CPI-0610) are currently in phase I clinical trials. Another recent and very interesting finding is the loss of expression of the tumor suppressor PTEN, leading to MYC upregulation by constitutive activation of the PI3K/AKT pathway. PI3K inhibition is selectively toxic to PTEN-deficient aggressive lymphoma models, suggesting a role for PI3K inhibitors in these diseases.135 Several PI3K inhibitors directed against different isoforms of PI3K are under development, showing promising activity. Recent studies have demonstrated that BET and HDAC inhibitors have synergistic activity in MYC-induced murine lymphoma suggesting a rationale for testing combination therapy. Other potential targets that may affect MYC overexpression include human mitochondrial peptide deformylase and the mitochondrial sirtuin 4. The optimal approach to the diseases with these new drugs is still unknown, but the development of rationally designed small-molecule inhibitors is a great promise for the future.134
Discovering new therapies
With recently developed technologies, new drugs could undergo rapid preclinical sensitivity screening using ex vivo coculture methods with simultaneous testing in leukemia/lymphoma xenografts.136 Relative to B-lymphoid malignancies, venetoclax was found synergistic with vincristine and dexamethasone in B-precursor ALL expressing TCF3-HLF gene rearrangement,137 and TCF3 mutations are known to occur in BL. Another example concerns birinapant, an SMAC-mimetic inductor of necroptosis found effective against highly MRD-resistant cases of B-lineage ALL.138 In a study regarding another MYC-driven neoplasm (Group 3 medulloblastoma), an in silico analysis method for screening drug sensitivity databases resulted in the identification of new molecular targets (cyclin-dependent kinase) for these c-MYC-transformed cells.139
Conclusion
The availability of several new compounds coupled with these sophisticated techniques will hopefully open a new phase of highly personalized, precision medicine that could warrant the achievement of cure in all AYAs with newly diagnosed BL.
Acknowledgment
Permission for the use of the photo in Figure 1 was provided orally at the time the photo was taken.
Disclosure
The authors report no conflicts of interest in this work.
References
Burkitt D. A sarcoma involving the jaws in African children. Br J Surg. 1958;46(197):218–223. | ||
Burkitt D, O’Conor GT. Malignant lymphoma in African children. I. A clinical syndrome. Cancer. 1961;14:258–269. | ||
Magrath I, Jaffe ES, Bhatia K. Burkitt’s lymphoma. In: Knowles DM, editor. Neoplastic Hematopathology. Philadelphia, PA: Lippincott Williams & Wilkins; 2001:953–986. | ||
Magrath IT. Burkitt’s lymphoma. The small noncleaved cell lymphomas. In: Canellos GP, Lister TA, Sklar JL, editors. The Lymphomas. Philadelphia, PA: WB Saunders; 1998:413–422. | ||
Leoncini L, Raphael M, Stein H, Harris N, Jaffe E, Kluin P. Burkitt lymphoma. In: Swerdlow S, Campo E, Harris N, et al, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2008. | ||
Costa LJ, Xavier AC, Wahlquist AE, Hill EG. Trends in survival of patients with Burkitt lymphoma/leukemia in the USA: an analysis of 3691 cases. Blood. 2013;121(24):4861–4866. | ||
Magrath I. Epidemiology: clues to the pathogenesis of Burkitt lymphoma. Br J Haematol. 2012;156(6):744–756. | ||
Ogwang MD, Bhatia K, Biqqar RJ, Mbulaiteye SM. Incidence and geographic distribution of endemic Burkitt lymphoma in northern Uganda revisited. Int J Cancer. 2008;123(11):2658–2663. | ||
Morton LM, Wang SS, Devesa SS, Hartge P, Weisenburger DD, Linet MS. Lymphoma incidence patterns by WHO subtype in the United States, 1992–2001. Blood. 2006;107(1):265–276. | ||
Sant M, Allemani C, Tereanu C, et al; HAEMACARE Working Group. Incidence of hematologic malignancies in Europe by morphologic subtype: results of the HAEMACARE project. Blood. 2010;116(19):3724–3734. | ||
Mbulaiteye SM, Anderson WF, Ferlay J, et al. Pediatric, elderly, and emerging adult-onset peaks in Burkitt’s lymphoma incidence diagnosed in four continents, excluding Africa. Am J Hematol. 2012;87(6):573–578. | ||
Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press; 2008. | ||
Armitage JO, Weisenburger DD. New approach to classifying non-Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol. 1998;16(8):2780–2795. | ||
Laurini JA, Perry AM, Boilesen E, et al. Classification of non-Hodgkin lymphoma in Central and South America: a review of 1028 cases. Blood. 2012;120(24):4795–4801. | ||
Boerma EG, van Imhoff GW, Appel IM, Veeger NJ, Kluin PM, Kluin-Nelemans JC. Gender and age-related differences in Burkitt lymphoma–epidemiological and clinical data from the Netherlands. Eur J Cancer. 2004;40(18):2781–2787. | ||
Smith A, Howell D, Patmore R, Jack A, Roman E. Incidence of haematological malignancy by sub-type: a report from the Haematological Malignancy Research Network. Br J Cancer. 2011;105(11):1684–1692. | ||
Shiels MS, Pfeiffer RM, Hall HI, et al. Proportions of Kaposi sarcoma, selected non-Hodgkin lymphomas, and cervical cancer in the United States occurring in persons with AIDS, 1980-2007. JAMA. 2011;305(14):1450–1459. | ||
Guech-Ongey M, Simard EP, Anderson WF, et al. AIDS-related Burkitt lymphoma in the United States: what do age and CD4 lymphocyte patterns tell us about etiology and/or biology? Blood. 2010;116(25):5600–5604. | ||
Mbulaiteye SM, Morton LM, Sampson JN, et al. Medical history, lifestyle, family history, and occupational risk factors for sporadic Burkitt lymphoma/leukemia: the interlymph non-Hodgkin Lymphoma Subtypes Project. J Natl Cancer Inst Monogr. 2014;2014(48):106–114. | ||
Burke ME, Albritton K, Marina N. Challenges in the recruitment of adolescents and young adults to cancer clinical trials. Cancer. 2007;110(11):2385–2393. | ||
Castillo JJ, Winer ES, Olszewski AJ. Population-based prognostic factors for survival in patients with Burkitt lymphoma: an analysis from the Surveillance, Epidemiology, and End Results database. Cancer. 2013;119(20):3672–3679. | ||
Bellan C, Lazzi S, Hummel M, et al. Immunoglobulin gene analysis reveals 2 distinct cells of origin for EBV-positive and EBV-negative Burkitt lymphomas. Blood. 2005;106(3):1031–1036. | ||
Hecht JL, Aster JC. Molecular biology of Burkitt’s lymphoma. J Clin Oncol. 2000;18(21):3707–3721. | ||
Klapproth K, Wirth T. Advances in the understanding of MYC-induced lymphomagenesis. Br J Haematol. 2010;149(4):484–497. | ||
Müller JR, Janz S, Goedert JJ, Potter M, Rabkin CS. Persistence of immunoglobulin heavy chain/c-myc recombination-positive lymphocyte clones in the blood of human immunodeficiency virus-infected homosexual men. Proc Natl Acad Sci U S A. 1995;92(14):6577–6581. | ||
Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci U S A. 1982;79(24):7824–7827. | ||
ar-Rushdi A, Nishikura K, Erikson J, Watt R, Rovera G, Croce CM. Differential expression of the translocated and the untranslocated c-myc oncogene in Burkitt lymphoma. Science. 1983;222(4622):390–393. | ||
Pelicci PG, Knowles DM 2nd, Magrath I, Dalla-Favera R. Chromosomal breakpoints and structural alterations of the c-myc locus differ in endemic and sporadic forms of Burkitt lymphoma. Proc Natl Acad Sci U S A. 1986;83(9):2984–2988. | ||
Neri A, Barriga F, Knowles DM, Magrath IT, Dalla-Favera R. Different regions of the immunoglobulin heavy-chain locus are involved in chromosomal translocations in distinct pathogenetic forms of Burkitt lymphoma. Proc Natl Acad Sci U S A. 1988;85(8):2748–2752. | ||
Ramiro AR, Jankovic M, Callen E, et al. Role of genomic instability and p53 in AID-induced c-MYC-Igh translocations. Nature. 2006;440(7080):105–109. | ||
Robbiani DF, Deroubaix S, Feldhahn N, et al. Plasmodium infection promotes genomic instability and AID-dependent B cell lymphoma. Cell. 2015;162(4):727–737. | ||
Mosialos G, Birkenbach M, Yalamanchili R, VanArsdale T, Ware C, Kieff E. The Epstein-Barr virus transforming protein LMP-1 engages signaling proteins for the tumor necrosis factor receptor family. Cell. 1995;80(3):389–399. | ||
Chene A, Donati D, Orem J, et al. Endemic Burkitt’s lymphoma as a polymicrobial disease: new insights on the interaction between Plasmodium falciparum and Epstein-Barr virus. Semin Cancer Biol. 2009;19(6):411–420. | ||
Parkin DM, Sitas F, Chirenje M, Stein L, Abratt R, Wabinga H. Part I: Cancer in Indigenous Africans–burden, distribution, and trends. Lancet Oncol. 2008;9(7):683–692. | ||
Morrow RH Jr. Epidemiological evidence for the role of falciparum malaria in the pathogenesis of Burkitt’s lymphoma. IARC Sci Publ. 1985;(60):177–186. | ||
Aka P, Vila MC, Jariwala A, et al. Endemic Burkitt lymphoma is associated with strength and diversity of Plasmodium falciparum malaria stage-specific antigen antibody response. Blood. 2013;122(5):629–635. | ||
Havelange V, Pepermans X, Ameye G, et al. Genetic differences between paediatric and adult Burkitt lymphomas. Br J Hematol. 2016;173(1):137–144. | ||
Dave SS, Fu K, Wright GW, et al. Molecular diagnosis of Burkitt’s lymphoma. N Engl J Med. 2006;354(23):2431–2442. | ||
Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390. | ||
Hasserjian RP, Ott G, Elenitoba-Johnson KS, Balague-Ponz O, de Jong D, de Leval L. Commentary on the WHO classification of tumors of lymphoid tissues (2008): “gray zone” lymphomas overlapping with Burkitt lymphoma or classical Hodgkin lymphoma. J Hematop. 2009;2(2):89–95. | ||
Gascoyne RD, Magrath IT, Sehn L. Burkitt lymphoma. In: Armitage JO, Mauch PM, Harris NL, Coiffier B, Dalla-Favera R, editors. Non-Hodgkin Lymphomas. 2nd ed. Philadelphia, PA: Wolters Kluwer and Lippincott Williams & Wilkins; 2010:334–357. | ||
Blum KA, Lozanski G, Byrd JC. Adult Burkitt leukemia and lymphoma. Blood. 2004;104(10):3009–3020. | ||
Wilson WH, Bromberg JE, Stetler-Stevenson M, et al. Detection and outcome of occult leptomeningeal disease in diffuse large B-cell lymphoma and Burkitt lymphoma. Haematologica. 2014;99(7):1228–1235. | ||
Jacobson C, LaCasce A. How I treat Burkitt Lymphoma in adults. Blood. 2014;124(19):2913–2920. | ||
Murphy SB. Classification, staging and end results of treatment of childhood non-Hodgkin’s lymphomas: dissimilarities from lymphomas in adults. Semin Oncol. 1980;7(3):332–339. | ||
Waxman IM, Hochberg J, Cairo MS. Non-Hodgkin Lymphoma. In: Kliegman RM, Behrman RE, Jenson HB, Stanton BF, editors. Nelson Textbook of Pediatrics. 19th ed. New York, NY: Elsevier Inc; 2011:1739–1745. | ||
van Dongen JJ, van der Velden VH, Brüggemann M, Orfao A. Minimal residual disease diagnostic in acute lymphoblastic leukemia: need for sensitive, fast, and standardized technologies. Blood. 2015;125(26):3996–4009. | ||
Bassan R, Spinelli O. Minimal residual disease monitoring in adult ALL to determine therapy. Curr Hematol Malig Rep. 2015;10(2):86–95. | ||
Bassan R, Maino E, Cortelazzo S. Lymphoblastic lymphoma: an update review on biology, diagnosis, and treatment. Eur J Haematol. 2016;96(5):447–460. | ||
Coustan-Smith E, Sandlund JT, Perkins SL, et al. Minimal disseminated disease in childhood T-cell lymphoblastic lymphoma: a report from the children’s oncology group. J Clin Oncol. 2009;27(21):3533–3539. | ||
Busch K, Borkhardt A, Wössmann W, Reiter A, Harbott J. Combined polymerase chain reaction methods to detect c-myc/IgH rearrangement in childhood Burkitt’s lymphoma for minimal residual disease analysis. Haematologica. 2004;89(7):818–825. | ||
Lovisa F, Mussolin L, Corral L, et al. IGH and IGK gene rearrangements as PCR targets for pediatric Burkitt’s lymphoma and mature B-ALL MRD analysis. Lab Invest. 2009;89(10):1182–1186. | ||
Mussolin L, Pillon M, d’Amore ES, et al. Minimal disseminated disease in high-risk Burkitt’s lymphoma identifies patients with different prognosis. J Clin Oncol. 2011;29(13):1779–1784. | ||
Pillon M, Mussolin L, Carraro E, et al. Detection of prognostic factors in children and adolescents with Burkitt and diffuse large B-cell lymphoma treated with the AIEOP LNH-97 protocol. Br J Haematol. Epub 2016 Jul 8. | ||
Agsalda M, Kusao I, Troelstrup D, Shiramizu B. Screening for residual disease in pediatric Burkitt lymphoma using consensus primer pools. Adv Hematol. 2009;2009:412163. | ||
Shiramizu B, Goldman S, Kusao I, et al. Minimal disease assessment in the treatment of children and adolescent with intermediate-risk (Stage III/IV) B-cell non-Hodgkin lymphoma: a children’s oncology group report. Br J Haematol. 2011;153(6):758–763. | ||
Cheson BD, Horning SJ, Coiffier B, et al. Report of an international workshop to standardize response criteria for non-Hodgkin’s lymphomas. NCI Sponsored International Working Group. J Clin Oncol. 1999;17(4):1244. | ||
Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25(5):579–586. | ||
Reiter A, Schrappe M, Ludwig WD, et al. Intensive ALL-type therapy without local radiotherapy provides a 90% event-free survival for children with T-cell lymphoblastic lymphoma: a BFM group report. Blood. 2000;95(2):416–421. | ||
Juweid ME, Stroobants S, Hoekstra OS, et al. Use of positron emission tomography for response assessment of lymphoma: consensus of the Imaging Subcommittee of International Harmonization Project in Lymphoma. J Clin Oncol. 2007;25(5):571–578. | ||
Dunleavy K, Mikhaeel G, Sehn LH, Hicks RJ, Wilson WH. The value of positron emission tomography in prognosis and response assessment in non-Hodgkin lymphoma. Leuk Lymphoma. 2010;51 Suppl 1:28–33. | ||
Riad R, Omar W, Sidhom I, et al. False-positive F-18 uptake in PET/CT studies in pediatric patients with abdominal Burkitt’s lymphoma. Nucl Med Commun. 2010;31(3):232–238. | ||
Hutchings M, Mikhaeel NG, Fields PA, Nunan T, Timothy AR. Prognostic value of interim FDG-PET after two or three cycles of chemotherapy in Hodgkin lymphoma. Ann Oncol. 2005;16(7):1160–1168. | ||
Gallamini A, Hutchings M, Rigacci L, et al. Early interim 2-[18F]fluoro-2-deoxy-D-glucose positron emission tomography is prognostically superior to international prognostic score in advanced-stage Hodgkin’s lymphoma: a report from a joint Italian-Danish study. J Clin Oncol. 2007;25(4):3746–3752. | ||
Barrington SF, Qian W, Somer EJ, et al. Concordance between four European centres of PET reporting criteria designed for use in multicentre trials in Hodgkin lymphoma. Eur J Nucl Med Mol Imaging. 2010;37(10):1824–1833. | ||
Meignan M, Gallamini A, Meignan M, Gallamini A, Haioun C. Report on First International Workshop on Interim-PET-Scan in Lymphoma. Leuk Lymphoma. 2009;50(8):1257–1260. | ||
Le Roux PY, Gastinne T, Le Gouill S, et al. Prognostic value of interim FDG PET/CT in Hodgkin’s lymphoma patients treated with interim response-adapted strategy: comparison of International Harmonization Project (IHP), Gallamini and London criteria. Eur J Nucl Med Mol Imaging. 2011;38(6):1064–1071. | ||
Minard-Colin V, Brugières L, Reiter A, et al. Non-Hodgkin lymphoma in children and adolescent: progress through effective collaboration, current knowledge, and challenge ahead. J Clin Oncol. 2015;33(27):2963–2974. | ||
Sandlund JT. Burkitt lymphoma: staging and evaluation. Br J Haematol. 2012;156(6):761–765. | ||
Fenaux P, Lai JL, Miaux O, Zandecki M, Jouet JP, Bauters F. Burkitt cell acute leukaemia (L3 ALL) in adults: a report of 18 cases. Br J Haematol. 1989;71(3):371–376. | ||
Pees HW, Radtke H, Schwamborn J, Graf N. The BMF-protocol for HIV-negative Burkitt’s lymphomas and L3 ALL in adult patients: a high chance for cure. Ann Hematol. 1992;65(5):201–205. | ||
Soussain C, Patte C, Ostronoff M, et al. Small noncleaved cell lymphoma and leukemia in adults. A retrospective study of 65 adults treated with the LMB pediatric protocols. Blood. 1995;85(3):664–674. | ||
Hoelzer D, Ludwig WD, Thiel E, et al. Improved outcome in adult B-cell acute lymphoblastic leukemia. Blood. 1996;87(2):495–508. | ||
Magrath I, Adde M, Shad A, et al. Adults and children with small non-cleaved-cell lymphoma have a similar excellent outcome when treated with the same chemotherapy regimen. J Clin Oncol. 1996;14(3):925–934. | ||
Thomas DA, Cortes J, O’brien S, et al. Hyper-CVAD program in Burkitt’s-type adult acute lymphoblastic leukemia. J Clin Oncol. 1999;17(8):2461–2470. | ||
Rizzieri DA, Johnson JL, Niedzwiecki D, et al. Intensive chemotherapy with and without cranial radiation for Burkitt leukemia and lymphoma: final results of Cancer and Leukemia Group B Study 9251. Cancer. 2004;100(7):1438–1448. | ||
Lacasce A, Howard O, Lib S, et al. Modified Magrath regimens for adults with Burkitt and Burkitt-like lymphomas: preserved efficacy with decreased toxicity. Leuk Lymphoma. 2004;45(4):761–767. | ||
Smeland S, Blystad AK, Kvaloy SO, et al. Treatment of Burkitt’s/Burkitt-like lymphoma in adolescents and adults: a 20-year experience from the Norwegian Radium Hospital with the use of three successive regimens. Ann Oncol. 2004;15(7):1072–1078. | ||
Di Nicola M, Carlo-Stella C, Mariotti J, et al. High response rate and manageable toxicity with an intensive, short term chemotherapy programme for Burkitt’s lymphoma in adults. Br J Haematol. 2004;126(6):815–820. | ||
Tauro S, Cochrane L, Lauritzsen GF, et al. Dose-intensified treatment of Burkitt lymphoma and B-cell lymphoma unclassifiable, (with features intermediate between diffuse large B-cell lymphoma and Burkitt lymphoma) in young adults (<50 years): a comparison of two adapted BFM protocols. Am J Hematol. 2010;85(4):261–263. | ||
Cairo MS, Sposto R, Perkins SL, et al. Burkitt’s and Burkitt-lyke lymphoma in children and adolescents: a review of the Children’s Cancer Group experience. Br J Haematol. 2003;120(4):660–670. | ||
Murphy SB, Bowman WP, Abromowitch M, et al. Results of treatment of advanced-stage Burkitt’s lymphoma and B cell (Sig+) acute lymphoblastic leukemia with high-dose fractionated cyclophosphamide and coordinated high-dose methotrexate and cytarabine. J Clin Oncol. 1986;4(12):1732–1739. | ||
Hoelzer D, Walewski J, Döhner H, et al. Improved outcome of adult Burkitt lymphoma/leukemia with rituximab and chemotherapy: report of a large prospective multicenter trial. Blood. 2014;124(26):3870–3879. | ||
Thomas DA, Faderl S, O’Brien S, et al. Chemoimmunotherapy with hyper-CVAD plus rituximab for the treatment of adult Burkitt and Burkitt-type lymphoma or acute lymphoblastic leukemia. Cancer. 2006;106(7):1569–1580. | ||
Kujawski LA, Longo WL, Williams EC, et al. A 5-drug regimen maximizing the dose of cyclophosphamide is effective therapy for adult Burkitt or Burkitt-like lymphomas. Cancer Invest. 2007;25(2):87–93. | ||
Mead GM, Barrans SL, Qian W, et al. A prospective clinicopathologic study of dose-modified CODOX-M/IVAC in patients with sporadic Burkitt lymphoma defined using cytogenetic and immunophenotypic criteria (MRC/NCRI LY10 trial). Blood. 2008;112(6):2248–2260. | ||
Choi MK, Jun HJ, Lee SY, et al. Treatment outcome of adult patients with Burkitt lymphoma: results using the LMB protocol in Korea. Ann Hematol. 2009;88(11):1099–1106. | ||
Barnes JA, Lacasce AS, Feng Y, et al. Evaluation of the addition of rituximab to CODOX-M/IVAC for Burkitt’s lymphoma: a retrospective analysis. Ann Oncol. 2011;22(8):1859–1864. | ||
Corazzelli G, Frigeri F, Russo F, et al. RDCODOX-M/IVAC with rituximab and intrathecal liposomal cytarabine in adult Burkitt lymphoma and ‘unclassifiable’ highly aggressive B-cell lymphoma. Br J Haematol. 2012;156(2):234–244. | ||
Kasamon YL, Brodsky RA, Borowitz MJ, et al. Brief intensive therapy for older adults with newly diagnosed Burkitt or atypical Burkitt lymphoma/leukemia. Leuk Lymphoma. 2013;54(3):483–490. | ||
Ribera JM, García O, Grande C, et al. Dose-intensive chemotherapy including rituximab in Burkitt’s leukemia or lymphoma regardless of human immunodeficiency virus infection status: final results of phase 2 study (Burkimab). Cancer. 2013;119(9):1660–1668. | ||
Dunleavy K, Pittaluga S, Shovlin M, et al. Low-intensity therapy in adults with Burkitt’s lymphoma. N Engl J Med. 2013;369(20):1915–1925. | ||
Intermesoli T, Rambaldi R, Rossi G, et al. High cure rates in Burkitt lymphoma and leukemia: Northern Italy Leukemia Group study of the German short intensive rituximab-chemotherapy program. Haematologica. 2013;98(11):1718–1725. | ||
Jain P, Kantarjian HM, Cortes JE, et al. Long term follow-up of de novo or minimally treated Burkitt lymphoma/leukemia (BL/B-ALL) after frontline therapy per the hyper-CVAD regimen with or without rituximab: 20-year cumulative experience. Blood. 2013;122:3917. | ||
Rizzieri DA, Johnson JL, Byrd JC, et al. Improved efficacy using rituximab and brief duration, high intensity chemotherapy with filgrastim support for Burkitt or aggressive lymphomas: cancer and Leukemia Group B study 10 002. Br J Haematol. 2014;165(1):102–111. | ||
Hong J, Kim SJ, Ahn JS, et al. Treatment outcomes of rituximab plus hyper-CVAD in Korean patients with sporadic Burkitt or Burkitt-like lymphoma: results of a multicenter analysis. Cancer Res Treat. 2015;47(2):173–181. | ||
Meinhardt A, Burkhardt B, Zimmermann M, et al; Berlin-Frankfurt-Münster Group. Phase II window study on rituximab in newly diagnosed pediatric mature B-cell non-Hodgkin’s lymphoma and Burkitt leukemia. J Clin Oncol. 2010;28(19):3115–3121. | ||
Ribrag V, Koscielny S, Bosq J, et al. Rituximab and dose-dense chemotherapy for adults with Burkitt’s lymphoma: a randomised, controlled, open-label, phase 3 trial. Lancet. 2016;387(10036):2402–2411. | ||
Mead GM, Sydes MR, Walewski J, et al. An international evaluation of CODOX-M and CODOX-M alternating with IVAC in adult Burkitt’s lymphoma: results of United Kingdom Lymphoma Group LY06 study. Ann Oncol. 2002;13(8):1264–1274. | ||
Diviné M, Casassus P, Koscielny S, et al. Burkitt lymphoma in adults: a prospective study of 72 patients treated with an adapted pediatric LMB protocol. Ann Oncol. 2005;16(12):1928–1935. | ||
Todeschini G, Bonifacio M, Tecchio C, et al. Intensive short-term chemotherapy regimen induces high remission rate (over 90%) and event-free survival both in children and adult patients with advanced sporadic Burkitt lymphoma/leukemia. Am J Hematol. 2012;87(1):22–25. | ||
Wilson FP, Berns JS. Tumor lysis syndrome: new challenges and recent advances. Adv Chronic Kidney Dis. 2014;21(1):18–26. | ||
McBride A, Westervelt P. Recognizing and managing the expanded risk of tumor lysis syndrome in hematologic and solid malignancies. J Hematol Oncol. 2012;5:75. | ||
van Imhoff GW, van der Holt B, MacKenzie MA, et al. Short intensive sequential therapy followed autologous stem cell transplantation in adult Burkitt, Burkitt-like and lymphoblastic lymphoma. Leukemia. 2005;19(6):945–952. | ||
Sweetenham JW, Pearce R, Taghipour G, Blaise D, Gisselbrecht C, Goldstone AH. Adult Burkitt’s and Burkitt-like non-Hodgkin’s lymphoma – outcome for patients treated with high-dose therapy and autologous stem-cell transplantation in first remission or at relapse: results from the European Group for Blood and Marrow Transplantation. J Clin Oncol. 1996;14(9):2465–2472. | ||
Hoffmann C, Wolf E, Wyen C, et al. AIDS-associated Burkitt or Burkitt-like lymphoma: short intensive polychemotherapy is feasible and effective. Leuk Lymphoma. 2006;47(9):1872–1880. | ||
Xicoy B, Ribera JM, Müller M, et al; PETHEMA Group and German HIV Lymphoma Cohort. Dose-intensive chemotherapy including rituximab is highly effective but toxic in human immunodeficiency virus-infected patients with Burkitt lymphoma/leukemia: parallel study of 81 patients. Leuk Lymphoma. 2014;55(10):2341–2348. | ||
Oriol A, Ribera JM, Esteve J, et al; PETHEMA Group; Spanish Society of Hematology. Lack of influence of human immunodeficiency virus infection status in the response to therapy and serviva of adult patients with mature B-cell lymphoma or leukemia. Result of the PETHEMA-LAL3/97 study. Haematologica. 2003;88(4):445–453. | ||
Oriol A, Ribera JM, Brunet S, del Potro E, Abella E, Esteve J. Highly active antiretroviral therapy and outcome of AIDS-related Burkitt’s lymphoma or leukemia. Results of the PETHEMA-LAL3/97 study. Haematologica. 2005;90(7):990–992. | ||
Baik S, Mbaziira M, Williams M, et al. A case-control study of Burkitt lymphoma in East Africa: are local health facilities an appropriate source of representative controls? Infect Agents Cancer. 2012;7(1):5. | ||
Aka P, Kawira E, Masalu N, et al. Incidence and trends in Burkitt lymphoma in northern Tanzxania from 2000 to 2009. Pediatr Blood Cancer. 2012;59(7):1234–1238. | ||
Ngoma T, Adde M, Durosinmi M, et al. Treatment of Burkitt lymphoma in equatorial Africa using a simple three-drug combination followed by a salvage regimen for patients with persistent or recurrent disease. Br J Haematol. 2012;158(6):749–762. | ||
Buckle GC, Collins JP, Sumba PO, et al. Factors influencing time to diagnosis and initiation of treatment of endemic Burkitt Lymphoma among children in Uganda and western Kenya: a cross-sectional survey. Infect Agent Cancer. 2013;8(1):36. | ||
Stanley CC, Westmoreland KD, Heimlich BJ, et al. Outcomes for paediatric Burkitt lymphoma treated with anthracycline-based therapy in Malawi. Br J Haematol. 2016;173(5):705–712. | ||
Kim H, Park ES, Lee SH, et al. Clinical outcome of relapsed or refractory Burkitt lymphoma and mature B-cell lymphoblastic leukemia in children and adolescents. Cancer Res Treat. 2014;46(4):358–365. | ||
Peniket AJ, Ruiz de Elvira MC, Taghipour G, et al; European Bone Marrow Transplantation (EBMT) Lymphoma Registry. An EBMT registry matched study of allogeneic stem cell transplants for lymphoma: allogeneic transplantation is associated with a lower relapse rate but a higher procedure-related mortality rate than autologous transplantation. Bone Marrow Transplant. 2003;31(8):667–678. | ||
Awasthi A, Ayello J, Van de Ven C, et al. Obinutuzumab (GA101) compared to rituximab significantly enhances cell death and antibody-dependent cytotoxicity and improves overall survival against CD20(+) rituximab-sensitive/-resistant Burkitt lymphoma (BL) and precursor B-acute lymphoblastic leukaemia (pre-B-ALL): potential targeted therapy in patients with poor risk CD20(+) BL and pre-B-ALL. Br J Haematol. 2015;171(5):763–775. | ||
Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375:740–753. | ||
Topp MS, Gökbuget N, Stein AS, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16(1):57–66. | ||
Gökbuget N, Dombret H, Bonifacio M, et al. Long-term outcomes after blinatumomab treatment: follow-up of a phase 2 study in patients (Pts) with minimal residual disease (MRD) positive B-cell precursor acute lymphoblastic leukemia (ALL). Blood. 2015;126:680. | ||
Dong LH, Cheng S, Zheng Z, et al. Histone deacetylase inhibitor potentiated the ability of MTOR inhibitor to induce autophagic cell death in Burkitt leukemia/lymphoma. J Hematol Oncol. 2013;6:53. | ||
Kawada J, Zou P, Mazitschek R, Bradner JE, Cohen JI. Tubacin kills Epstein-Barr virus (EBV)-Burkitt lymphoma cells by inducing reactive oxygen species and EBV lymphoblastoid cells by inducing apoptosis. J Biol Chem. 2009;284(25):17102–17109. | ||
Barboza NM, Medina DJ, Budak-Alpdogan T, et al. Plitidepsin (Aplidin) is a potent inhibitor of diffuse large cell and Burkitt lymphoma and is synergistic with rituximab. Cancer Biol Ther. 2012;13(2):114–122. | ||
Shatzer AN, Espey MG, Chavez M, Tu H, Levine M, Choen JI. Ascorbic acid kills Epstein-Barr virus (EBV) positive Burkitt lymphoma cells and EBV transformed B-cells in vitro, but not in vivo. Leuk Lymphoma. 2013;54(5):1069–1078. | ||
Li ZJ, Yao C, Liu SF, et al. Cytotoxic effect of icaritin and its mechanisms in inducing apoptosis in human Burkitt lymphoma cell line. Biomed Res Int. 2014;2014:391512. | ||
Shirley CM, Chen J, Shamay M, et al. Bortezomib induction of C/EBPβ mediates Epstein-Barr virus lytic activation in Burkitt lymphoma. Blood. 2011;117(23):6297–6303. | ||
Suk FM, Lin SY, Lin RJ, et al. Bortezomib inhibits Burkitt’s lymphoma cell proliferation by downregulating sumoylated hnRNP K and c-Myc expression. Oncotarget. 2015;6(28):25988–26001. | ||
De Vos S, Flowers C, Wang D, et al. Interim results from a dose-escalation study of the BCL-2 inhibitor venetoclax (ABT-199/GDC-0199) plus bendamustine (B) and rituximab (R) in patients (pts) with relapsed/refractory (R/R) non-Hodgkin’s lymphoma (NHL). J Clin Oncol. 2015;33 Suppl 15:8535. | ||
Bryan LJ, Gordon LI. Blocking tumor escape in hematologic malignancies: the anti-PD-1 strategy. Blood Rev. 2015;29(1):25–32. | ||
Posternak V, Cole MD. Strategically targeting MYC in cancer. F1000Res. 2016;5:F1000. | ||
Anderson MA, Tsui A, Wall M, Huang DC, Roberts AW. Current challenges and novel treatment strategies in double hit lymphomas. Ther Adv Hematol. 2016;7(1):52–64. | ||
Hartl M. The quest of targets executing MYC-dependent cell transformation. Front Oncol. 2016;6:132. | ||
Yun S, Vincelette ND, Knorr KL, et al. 4EBP1/c-MYC/PUMA and NF-κB/EGR1/BIM pathways underlie cytotoxicity of mTOR dual inhibitors in malignant lymphoid cells. Blood. 2016;127(22):2711–2722. | ||
Muralidharan SV, Bhadury J, Nilsson LM, Green LC, McLure KG, Nilsson JA. BET bromodomain inhibitors synergize with ATR inhibitors to induce DNA damage, apoptosis, senescence-associated secretory pathway and ER stress in Myc-induced lymphoma cells. Oncogene. 2016;35(36):4689–4697. | ||
Dunleavy K. Aggressive B cell lymphoma: optimal therapy for MYC-positive double-hit, and triple-hit DLBCL. Curr Treat Options Oncol. 2015;16(12):58. | ||
Frismantas V, Dobay MP, Rinaldi A, et al. Drug response profiling to identify selective pharmacological activity in drug resistant ALL. Blood. 2015;126:2532. | ||
Fischer U, Foster M, Rinaldi A, et al. Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic option. Nat Genet. 2015;47(9):1020–1029. | ||
McComb S, Aguade-Gorgorio J, Harder L, et al. Activation of concurrent apoptosis and necroptosis by SMAC mimetics for the treatment of refractory and relapsed ALL. Sci Transl Med. 2016;8(339):339ra70. | ||
Hanaford AR, Archer TC, Price A, et al. DiSCoVERing innovative therapies for rare tumors: combining genetically accurate disease models with in silico analysis to identify novel therapeutic targets. Clin Cancer Res. 2016;22(15):3903–3914. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.