")
Back to Journals » ImmunoTargets and Therapy » Volume 10
Bruton’s Tyrosine Kinase Inhibition for the Treatment of Rheumatoid Arthritis
Authors Arneson LC , Carroll KJ , Ruderman EM
Received 2 June 2021
Accepted for publication 4 August 2021
Published 28 August 2021 Volume 2021:10 Pages 333—342
DOI https://doi.org/10.2147/ITT.S288550
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Laura C Arneson,1,* Kristen J Carroll,1,* Eric M Ruderman2
1Department of Medicine, Northwestern University Feinberg School of Medicine, Chicago, IL, USA; 2Department of Medicine, Division of Rheumatology, Northwestern University Feinberg School of Medicine, Chicago, IL, USA
*These authors contributed equally to this work
Correspondence: Eric M Ruderman
Department of Medicine, Division of Rheumatology, Northwestern University Feinberg School of Medicine, 675 N. St. Clair, Suite 14-100, Chicago, IL, 60611, USA
Tel +1 312-503-3188
Fax +1 312-695-0114
Email [email protected]
Abstract: Bruton’s tyrosine kinase (BTK) inhibitors are an emerging class of drugs that inhibit B cell receptor activation, FC-γ receptor signaling, and osteoclast proliferation. Following on approval for treatment of hematologic malignancies, BTK inhibitors are now under investigation to treat a number of different autoimmune diseases, including rheumatoid arthritis (RA). While the results of BTK inhibitors in RA animal models have been promising, the ensuing human clinical trial outcomes have been rather equivocal. This review will outline the mechanisms of BTK inhibition and its potential impact on immune mediated disease, the types of BTK inhibitors being studied for RA, the findings from both preclinical and clinical trials of BTK inhibitors in RA, and directions for future research.
Keywords: evobrutinib, spebrutinib, acalabrutinib, fenebrutinib, rheumatoid arthritis, Bruton’s tyrosine kinase
Introduction
Kinase inhibition has become a promising therapeutic target in many diseases, including rheumatoid arthritis. Kinase inhibitors, which disrupt the intracellular signaling leading to cell activation, have been shown to be effective in treating several diseases for which this signaling cascade drives pathogenesis. Bruton’s tyrosine kinase (BTK), a member of the Tec family of nonreceptor tyrosine kinases, is a cytoplasmic protein found in cells of myeloid lineage, including neutrophils, macrophages, monocytes, and mast cells, and in B cells, but not in T cells, plasma cells, or natural killer cells.1–4 BTK plays an important role in B cell regulation, making it a strong candidate for targeting inhibition of B cells in a number of inflammatory diseases, including rheumatoid arthritis (RA). In this manuscript, we will review the mechanisms of BTK inhibition, the results seen with BTK inhibitors for RA in both preclinical and clinical studies to date, and the potential future role of BTK inhibitors in the management of RA and other systemic rheumatic diseases.
Methodology
A review of the literature on BTK inhibitors for the treatment of RA was conducted through April 1st 2021. PubMed was searched using the terms “‘BTK inhibitor’ AND ‘rheumatoid arthritis,’” yielding 31 papers, and “‘Bruton’s tyrosine kinase inhibitor’ AND ‘rheumatoid arthritis,’” yielding 9 additional papers. All 40 papers were reviewed. Clinicaltrials.gov was also searched using the same terms, yielding five studies, for which the publicly available results were reviewed. A synthesis of the reviewed literature and trials is presented herein.
Mechanisms of BTK Inhibition
Mechanistically, BTK plays a role in three independent pathways. These pathways include 1) B cell activation via the B cell receptor (BCR) signaling pathway, 2) immune complex-driven monocyte and macrophage activation via the Fc-γ receptor signaling pathway, ultimately driving the production of inflammatory cytokines (namely interleukin (IL)-1β, IL-6, and tumor necrosis factor α), and 3) osteogenesis via osteoclast proliferation.5–7 BTK’s distinct role in B cell proliferation has been well studied. Upon antigen binding to the BCR, BTK becomes activated downstream of Src family kinases, leading to phospholipase-Cγ (PLC-γ) signaling that activates the NFκ-B and MAP kinase pathways, thereby driving transcriptional alterations that promote B cell activation, proliferation, and expression of CD40, CD69 and CD86 (Figure 1).5,6,8,9 There is also evidence that BTK signaling may act in synergy with Toll-like receptor pathways in driving autoimmune disease.10 Additionally, BTK contributes to B cell differentiation and chemotaxis.11 Accordingly, inhibition of BTK has been found to hinder B cell proliferation and decrease the formation of inflammatory cytokines.2,7 IL-21 plays a key role in B cell differentiation and class-switch DNA recombination, and BTK appears to mediate IL-21 signaling by facilitating STAT1 phosphorylation and translocation to the nucleus.12
 |
Figure 1 Simplified schematic of the proposed mechanism of BTK inhibition in RA. (Figure modified and used with permission from American Laboratory Products Company. BTK Inhibitors as Cancer Drug Treatments. Available from: https://www.alpco.com/btk-inhibitors-as-cancer-drug-treatments).44 |
Potential Benefits of BTK Inhibition in RA
RA is a systemic autoimmune disease that involves dysregulated T and B lymphocyte proliferation. Specifically, B cell dysregulation via BCR signaling, as previously detailed, drives the production of autoantibodies and inflammatory cytokines, contributing to the development and progression of RA (Figure 1).11,13 Myeloid cells can invade the synovium in RA, thus providing another target, given BTK’s expression in these cells.13 Additionally, BTK mediates bone resorption by activating the receptor activator of nuclear factor kappa B (RANK), which in turn stimulates osteoclast proliferation and differentiation, a key contributor to RA pathophysiology.13,14 Higher levels of phosphorylated BTK have been demonstrated in peripheral B cells from RA patients, compared to healthy control patients. Among rheumatoid factor (RF)-positive RA patients, phosphorylated BTK levels correlate with RF titers, though not with other measures of disease activity (ESR, CRP, MMP-3, or Simple Disease Activity Index).12
While multiple disease-modifying anti-rheumatic drugs (DMARDs) and biologics are available for the treatment of RA, many patients still do not achieve disease remission with available agents. At least one currently available biologic, rituximab, can achieve some degree of disease control by targeting B cell-dependent pathways. This suggests that additional agents targeting B cell pathways, including BTK inhibitors, might play a role as independent therapies or adjuncts to current treatment paradigms.
BTK Inhibitors in Development
Molecular Characteristics
At present, there are several orally administered BTK inhibitors in development, including both reversible and irreversible inhibitors, many of which have been studied in RA (Tables 1 and 2).15,16 The irreversible inhibitors covalently bind with cysteine-481 in the active site of the kinase protein.17 Ibrutinib (PCI-32765) is a first-generation irreversible BTK inhibitor that is FDA approved for treatment of certain forms of leukemia and lymphoma. Its irreversible binding to cysteine leads to off-target effects on kinases with similarly positioned cysteine residues, including EGFR, ITK, and TEC,15 while its promiscuous hinge-binding moiety can interact with kinases, such as Lck, Lyn, Fyn, Src, and Abl.18 Prominent and more selective second-generation irreversible inhibitors of BTK include tirabrutinib (ONO/GS-4059), a dual BTK and Tec inhibitor currently in Phase I clinical trials for RA, as well as spebrutinib (CC-292, AVL-292), acalabrutinib (ACP-196), and evobrutinib (M-2951), all of which are actively undergoing Phase II RA trials.15,19,20 Evobrutinib is notable for its high potency in inhibiting both BCR and Fc receptor signaling, strongly favoring BTK as a target over similar kinases in the Tec family.21–23 Poseltinib (previously LY3337641/HM71224) is a selective irreversible BTK inhibitor that completed a phase I trial in RA, leading to a phase II trial that was aborted due to low likelihood of achieving adequate clinical response in interim analysis.24 Recent drug development has yielded a group of irreversible BTK inhibitors with tricyclic structures, which have shown efficacy in preclinical RA models. These include SOMCL-17-016, formed by modifying the side chain of ibrutinib,25 and compounds with pyranochromenone scaffolds that occupy the adenine rather than cysteine-binding site of BTK.26
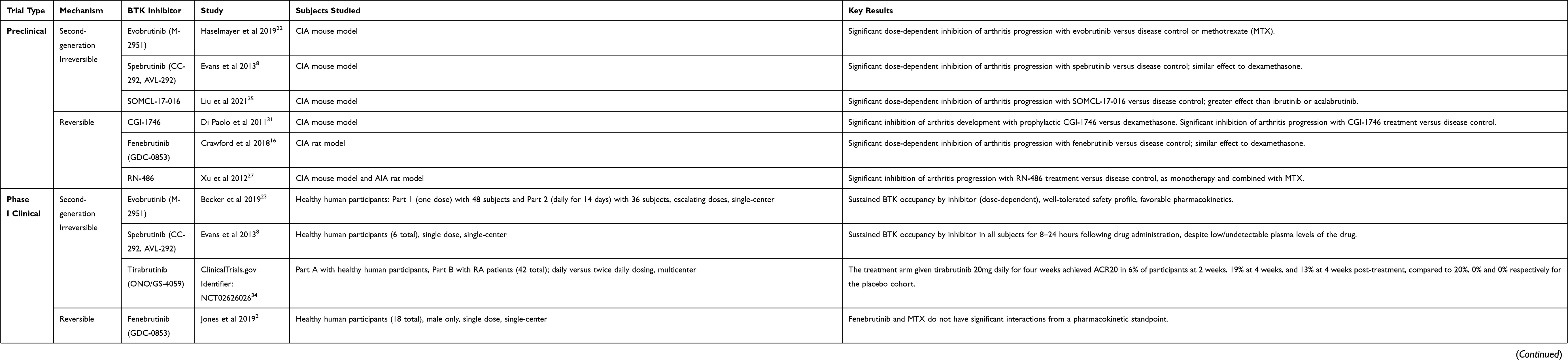 |
Table 1 BTK Inhibitors Studied for Rheumatoid Arthritis |
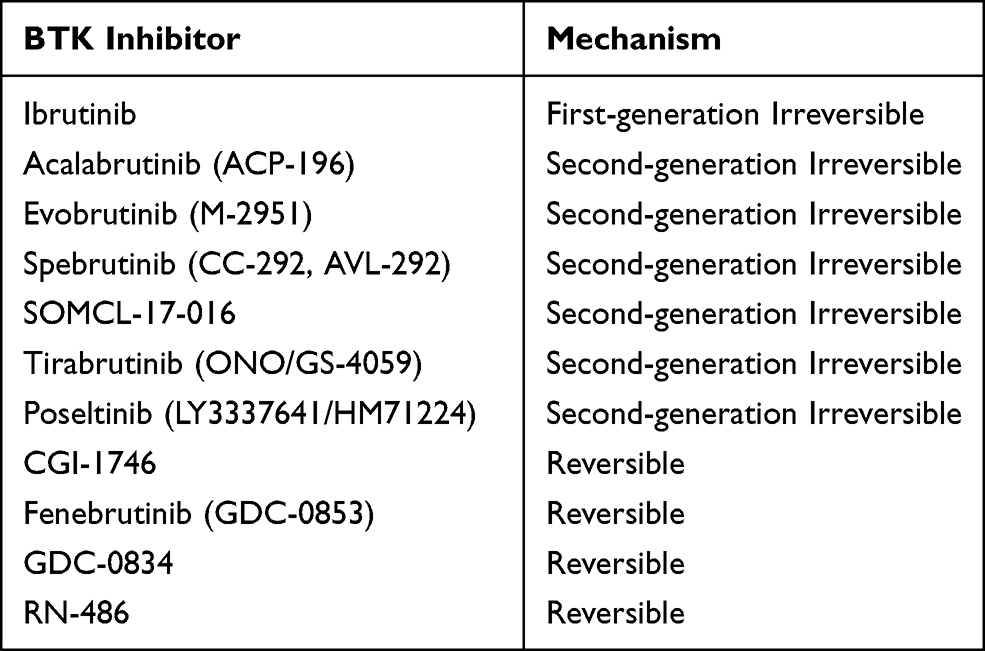 |
Table 2 Mechanisms of BTK Inhibitors Studied for Rheumatoid Arthritis |
Reversible inhibitors, while potentially requiring more frequent and sustained dosing, raise less concern for long-lasting off-target effects on other kinases. Fenebrutinib (GDC-0853) is a reversible inhibitor that binds to BTK with a precise orientation, ensuring high potency and selectivity.16 Other reversible inhibitors, including CGI-1746 and RN-486, are currently being studied in preclinical models.15,27 RN-486 has demonstrated in vitro selectivity for B-cell inhibition without affecting T-cell, endothelial, fibroblast, myeloid, or epithelial cell function.27 GDC-0834 is a highly potent and selective reversible BTK inhibitor that initially showed promise but was clinically suspended upon demonstrating metabolic instability in a phase I trial.15,28
Preclinical Data
Several of the aforementioned reversible BTK inhibitors, including fenebrutinib and GDC-0834, have shown favorable pharmacokinetics, pharmacodynamics, and safety outcomes in preclinical testing (Table 1).6,16 GDC-0834 demonstrated exceptional oral bioavailability in preclinical studies in various species, including rodents, dogs, and monkeys, without clinically significant metabolism of the drug.29 Spebrutinib continued to occupy BTK in vivo despite undetectable plasma levels of the drug with a prolonged recovery, reaching only 50% of baseline BTK protein levels within 1–2 days post administration of a single dose.8 BTK itself has a considerably long half-life, documented to extend past 12 hours in human B lymphocytes, thus permitting prolonged biologic activity even with reversible inhibitors of the protein.8,30
Recent preclinical data also highlight the potential role of BTK in RA therapy. The collagen-induced arthritis (CIA) rodent model is a well-established animal model for RA in humans.6 Clinical polyarthritis in CIA rodents presents as cartilage breakdown, resorption of bone, and pronounced synovitis and joint inflammation, identified by increases in paw diameter.6,22 Interestingly, mice with deletions or point mutations of BTK exhibit an X-linked immunodeficiency (Xid), resulting in a reduced B lymphocyte population with overall lower immunoglobulin levels. Mice with Xid appear to harbor resistance to CIA.17 Numerous studies using the CIA model demonstrate the potential multifaceted role of BTK inhibitors in RA, notably inhibition of B cell activation, formation of myeloid immune complexes and inflammatory cytokines, and osteoclastogenesis as previously described.11,13,25,31 Evobrutinib and spebrutinib have both demonstrated significant dose-dependent reduction in the development of arthritis in CIA mouse models.8,22 While MTX alone reduced arthritis by only 13%, evobrutinib accounted for a 69% to 92% reduction in disease activity with 1 mg/kg or 3 mg/kg dosing, respectively.22 The reversible BTK inhibitor GDC-0834 exhibited a marked decrease in arthritis severity in a CIA rat model following drug administration, but was comparable to a methotrexate (MTX)-treated control group. This study also demonstrated that 73% of BTK inhibition equated to “half-maximal activity,” or 50% of the achievable disease reduction, suggesting that a significant proportion of BTK needs to be inhibited to achieve maximal therapeutic benefit in the rodent model.6 Of note, trials of GDC-0834 have since been discontinued due to rapid metabolism when tested in humans.28 The irreversible tricyclic BTK inhibitor SOMCL-17-016 demonstrated dose-dependent decreases in progression of arthritis severity and bone damage in CIA mice, with greater effect than equivalent doses of ibrutinib or acalabrutinib.25 Finally, CGI-1746 prophylaxis produced 96% inhibition of clinical arthritis in CIA mice, compared to 56% in the positive control animals administered steroids.31
Clinical Data
BTK inhibitors currently being studied in the clinical setting for RA include spebrutinib, acalabrutinib, fenebrutinib, evobrutinib, and tirabrutinib. Evans et al performed a preliminary phase I double-blinded randomized control trial (RCT) of spebrutinib in 2013, including 6 subjects at a single center. This trial demonstrated sustained BTK occupancy by inhibitor in all subjects for 8–24 hours following drug administration, despite low, even undetectable plasma levels of the drug, thereby largely mirroring data seen in the preclinical setting.8 Unfortunately, spebrutinib did not produce a significant clinical response in a phase IIa randomized, double-blinded, placebo-controlled study of 47 RA patients at 12 United States academic centers. While there was a numerical trend toward clinical improvement in active RA symptoms with spebrutinib treatment, the effect was not statistically significant.11 Acalabrutinib was studied in a phase IIa trial with 31 RA patients on background MTX, assessed with a composite index of active rheumatoid arthritis, known as disease activity score 28-C-reactive protein (DAS28-CRP). After four weeks of treatment, DAS28-CRP scores were comparable between the acalabrutinib and placebo arms, suggesting a lack of meaningful clinical response.32
Fenebrutinib was studied in the phase II ANDES trial, an international multicenter double-blinded RCT involving two separate cohorts of patients. All patients continued MTX therapy in addition to their randomized drug. The primary outcome was the percentage of patients fulfilling the American College of Rheumatology criteria for 50% clinical improvement (ACR50) in rheumatoid arthritis symptoms at 12 weeks of treatment. Cohort I compared fenebrutinib, adalimumab, and placebo in 480 patients with previous suboptimal response to MTX. Lower dose fenebrutinib (50 mg daily) performed similarly to placebo, but higher doses of fenebrutinib (150–200 mg twice daily) demonstrated statistically significant improvement in ACR50 compared to placebo. The highest dose of fenebrutinib (200 mg twice daily) was noninferior to adalimumab. Cohort II consisted of 98 patients taking MTX, with previous TNF inhibitor inadequate response, who were randomized to fenebrutinib or placebo. In this cohort, there was a trend toward improved ACR50 with the highest dose of fenebrutinib (200 twice daily) compared to placebo, but this difference was not statistically significant (p = 0.072).33
In the spebrutinib and fenebrutinib phase II trials above, favorable changes in biomarkers were noted beyond clinical outcomes. In the ANDES trial, both fenebrutinib and adalimumab treatment resulted in statistically significant reductions in RF and Myeloid/FcγR biomarkers (CCL4, IL-6) compared to placebo, but only the fenebrutinib group showed a statistically significant decrease in total IgG and IgM.33 In the phase II spebrutinib trial, despite the lack of significant clinical response, patients treated with spebrutinib (compared to placebo) showed statistically significant reductions in serum CTX-I (a measure of osteoclastic activity) and chemokines CXCL13 and MIP-1β (implicated in B-cell trafficking).11
The clinical roles of tirabrutinib and evobrutinib in RA are also under investigation, pending the publication of finalized results from phase I and phase II clinical trials, respectively. Tirabrutinib entered a randomized phase I clinical trial in 2016. Part A of the trial explored the pharmacokinetics, pharmacodynamics, and safety profile of tirabrutinib in healthy subjects, given at doses of either 20 mg daily or 10 mg twice daily. Of note, 38% of patients (n=16) in the treatment arm administered tirabrutinib 20 mg daily for four total weeks experienced a treatment emergent adverse event (TEAE), compared to 40% of placebo participants (n=5). Part B of the trial assessed the tolerability, safety profile, and resultant inflammatory marker response with tirabrutinib administration in participants with RA. The treatment arm given tirabrutinib 20 mg daily for four weeks achieved an ACR 20% improvement response (ACR20) in 6% of participants at 2 weeks, 19% at 4 weeks, and 13% at 4 weeks post-treatment, compared to 20%, 0% and 0%, respectively, for the placebo cohort. Only one participant in treatment arm (6%), however, achieved an ACR 70% improvement response (ACR70).34 Further synthesis of prospective results and future higher-powered studies, notably phase II trials, are thereby needed to determine the clinical efficacy of tirabrutinib.
Evobrutinib was studied in a placebo-controlled, randomized, double-blinded phase IIa clinical trial (2016–2017) that analyzed the safety profile and effectiveness of the drug in participants with RA who were already on a stable dose of MTX. Enrollment included 65 participants who were randomized into two groups and administered either evobrutinib 50 mg twice daily or placebo for a total of 12 weeks. The primary outcome of ACR20 was achieved by 52% (n=33) of the evobrutinib group and 42% (n=31) of the placebo group. The difference in responder proportion (0.10), however, did not reach statistical significance (80% CI −0.07 to 0.25). In terms of safety profile, 67% (n=22) of the evobrutinib group experienced TEAEs, compared to 50% (n=16) of the placebo group. One serious adverse event (SAE) occurred in the evobrutinib group (vestibular disorder/vertigo), compared to none in the placebo group.35 Evobrutinib subsequently entered a phase IIb clinical trial (2017–2019), which assessed its efficacy and safety specifically in RA patients non-responsive to MTX. The study enrolled 390 participants who were uniformly randomized into four groups: one control group and three treatment arms that were administered evobrutinib at 25 mg daily, 75 mg daily, or 50 mg twice daily. The primary outcome was again ACR20 response within the 12-week study period. None of the three treatment groups demonstrated statistically significant improvements in ACR20 when compared to placebo (p =0.17, p=0.83, and p=0.13, respectively, for escalating doses of evobrutinib), nor in secondary outcomes ACR50 or ACR70. An additional secondary outcome analyzed in this study was a change in Disease Activity Score (DAS). Interestingly, discordant with the composite DAS28-CRP results of the previously mentioned acalabrutinib phase IIa trial, each of the three treatment arms demonstrated greater achievement of low DAS and remission DAS after 12 weeks of evobrutinib as compared to the placebo group. This finding suggests the potential for evobrutinib as RA therapy for MTX non-responders in clinical practice, despite the lack of statistical significance in the primary outcome. The three treatment groups and placebo group demonstrated comparable safety profiles in terms of TEAEs and SAEs. No participants in any group experienced clinically significant electrocardiogram, laboratory, or vital sign changes from baseline, and all-cause mortality remained at 0% for all groups, highlighting the overall high tolerability of evobrutinib without overt short-term toxicity concerns.36
The remaining BTK inhibitors in clinical trials to date have corroborated the favorable side effect profile of evobrutinib. For example, only one serious adverse event (stomatitis) was documented in the spebrutinib multicenter phase IIa trial.11 A Phase I trial of fenebrutinib in 111 healthy volunteers showed no serious or dose-limiting adverse events, so a maximum tolerated dose (MTD) was not delineated.37 The phase II trial of fenebrutinib noted three serious infections in the cohort I fenebrutinib groups, compared to one in the placebo group, and rare, reversible, asymptomatic grade 3 transaminase elevations in the fenebrutinib and adalimumab groups.33 The most commonly observed side effects in both phase I and phase II trials with fenebrutinib were nausea and headaches.11,33,37
Limitations and Future Directions
Overall, the promising preclinical data on BTK inhibitors in RA have not been fully replicated in the clinical trials to date. Several factors could account for this discrepancy. These include limitations of the clinical studies, as well as the CIA rodent models themselves, which both pose significant barriers to bridging the gap between underlying biologic mechanisms and clinical implementation. The phase I and II clinical studies to date have had relatively small sample sizes, limiting their power to identify positive effects, and short durations, which may underestimate potential delayed treatment effects.11,33 In addition, the rodent CIA model does not account for certain metabolic differences between rodents and humans, such as significant amide hydrolysis detected when GDC-0834 transitioned from animal to human trials, which prompted abandonment of that drug and re-design of alternate compounds.28,29,38
Additionally, covalent binding by irreversible inhibitors to the cysteine-481 residue in the ATP binding site of BTK poses a safety concern, as many other human kinases have a similar cysteine residue at their active binding site, increasing the risk of off-target effects.16 Murine models simply cannot replicate the complexity of these enzyme interactions. Thus, the specific mechanism of a drug’s BTK inhibition likely plays a role in the challenges seen in the transition from pre-clinical to clinical studies. For example, the poseltinib (irreversible) phase II trial was terminated given lack of efficacy, while the fenebrutinib (reversible) phase II trial showed some benefit.33
The existing preclinical and clinical data on BTK inhibitors in RA reveal several nuances that could be pursued to optimize therapeutic benefit. The timing of BTK inhibitor therapy within a patient’s RA disease course may be a key factor influencing clinical efficacy. In a study of peripheral B cells from RA patients, phosphorylated BTK levels correlated with RF titers among rheumatoid factor (RF)-positive patients, but did not correlate with measures of disease activity including ESR, CRP, MMP-3, or Simple Disease Activity Index.12 As BTK is known to play a role in B cell differentiation and DNA class-switching pathways, this finding suggests that BTK inhibition may be most relevant in the early stages of disease when autoantibodies are first developing, rather than in the later stages of arthritis.12 Supporting this hypothesis, successful preclinical studies of BTK inhibitors have administered treatment either prophylactically or within a few days of disease induction.8,14,31 Therefore, the limited efficacy of BTK inhibitors seen in clinical trials may relate to the timing of therapy and the duration of the RA diagnosis. Moreover, the correlation between phosphorylated BTK and RF titers suggests that BTK inhibitors may be specifically beneficial in RF-positive patients, such as those in the positive fenebrutinib phase II trial, which enrolled only seropositive patients.33 Other biomarkers appeared to correlate with enhanced response in the phase II trial of spebrutinib, including high total plasma CD19+ B lymphocytes and low serum CTX-I levels at baseline.11 Finally, the optimal dosing of BTK inhibitors in humans could be further clarified. For example, the spebrutinib phase II trial, which displayed a non-significant trend toward efficacy, administered doses lower than the amounts shown to be safe in a phase I study of spebrutinib for chronic lymphocytic leukemia.11 Meanwhile, the positive fenebrutinib phase II trial demonstrated greater efficacy at higher doses, and benefits manifested after 8–12 weeks of treatment, in contrast to the 4-week treatment period in the spebrutinib trial.33 Clinical trials of BTK inhibitors in RA may still have the potential to mirror the efficacy shown in preclinical trials, if treatment is started earlier in the disease course, at the optimal dose and duration, and with patients of the right phenotype.
Further studies should also focus on potential combination therapies that function synergistically. Fenebrutinib’s phase II efficacy in combination with MTX (ANDES cohort I) highlights one potential synergy.33 Another possible synergy might be the combination of BTK inhibitors with TNF inhibitors. In one study, RA patients with a “lymphoid” phenotype (synovial histology with B cell and plasmablast predominance) responded less well to TNF inhibitor therapy than those with a “myeloid” phenotype.13 Combining a BTK inhibitor with TNF inhibitor therapy in such patients could address B cell pathogenetic pathways in pathogenesis that may be of importance. Fenebrutinib has shown a trend toward clinical benefit among patients with previous sub-optimal TNF inhibitor response (ANDES cohort II).33
BTK inhibitors are also being studied for other autoimmune diseases, most notably systemic lupus erythematosus (SLE). In mouse models of SLE, the BTK inhibitors PF-06250112, M7583, BI-BTK-1, RN-486, ibrutinib, poseltinib, and evobrutinib have demonstrated prevention or amelioration of lupus nephritis, M7583 has shown prevention of arthritis, and BI-BTK-1 and poseltinib have shown reductions in skin inflammation.22,39–42 These clinical benefits were associated with reduced IgG anti-dsDNA antibody levels.22,40–42 A phase II randomized, placebo-controlled trial of fenebrutinib in 260 patients with SLE demonstrated significant reductions in CD19+ B cells and anti-dsDNA autoantibodies, though no significant clinical response.43
Conclusion
Considering its essential role in B cell function and other immune pathways, Bruton’s tyrosine kinase represents an attractive target in the treatment of RA. Pre-clinical data, particularly in murine models, appear to confirm this potential and have prompted a number of clinical RA trials focused on BTK inhibitors. Unfortunately, despite a reassuring safety profile, the efficacy of these BTK inhibitors in clinical trials was not as strong as anticipated. Whether this lack of clinical response stems from unique issues inherent to the individual molecules, or suggests a gap in our current understanding of the implicated signaling pathways in RA, remains unclear. Additional trials may help determine whether BTK inhibition can be helpful in a subset of patients with RA, or perhaps effective in combination with other therapies. There is also continued interest in exploring the efficacy of BTK inhibition in other rheumatic diseases. For the moment, however, the promise of this particular pathway remains unrealized.
Disclosure
Dr Eric M Ruderman reports on personal fees from Genentech, outside of the submitted work. The authors report no other conflicts of interest in this work.
References
1. Ruderman EM, Pope RM. More than just B-cell inhibition. Arthritis Res Ther. 2011;13(4):125. doi:10.1186/ar3439
2. Jones NS, Winter H, Katsumoto TR, et al. Absence of pharmacokinetic interactions between the Bruton’s tyrosine kinase inhibitor fenebrutinib and methotrexate. J Pharmacol Exp Ther. 2019;371(1):202–207. doi:10.1124/jpet.119.257089
3. Satterthwaite AB, Witte ON. The role of Bruton’s tyrosine kinase in B-cell development and function: a genetic perspective. Immunol Rev. 2000;175:120–127. doi:10.1111/j.1600-065X.2000.imr017504.x
4. Brunner C, Müller B, Wirth T. Bruton’s tyrosine kinase is involved in innate and adaptive immunity. Histol Histopathol. 2005;20(3):945–955. doi:10.14670/hh-20.945
5. Park JK, Byun JY, Park JA, et al. HM71224, a novel Bruton’s tyrosine kinase inhibitor, suppresses B cell and monocyte activation and ameliorates arthritis in a mouse model: a potential drug for rheumatoid arthritis. Arthritis Res Ther. 2016;18:91. doi:10.1186/s13075-016-0988-z
6. Liu L, Di Paolo J, Barbosa J, Rong H, Reif K, Wong H. Antiarthritis effect of a novel Bruton’s tyrosine kinase (BTK) inhibitor in rat collagen-induced arthritis and mechanism-based pharmacokinetic/pharmacodynamic modeling: relationships between inhibition of BTK phosphorylation and efficacy. J Pharmacol Exp Ther. 2011;338(1):154–163. doi:10.1124/jpet.111.181545
7. Kelly V, Genovese M. Novel small molecule therapeutics in rheumatoid arthritis. Rheumatology. 2013;52(7):1155–1162. doi:10.1093/rheumatology/kes367
8. Evans EK, Tester R, Aslanian S, et al. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J Pharmacol Exp Ther. 2013;346(2):219–228. doi:10.1124/jpet.113.203489
9. Niiro H, Clark EA. Regulation of B-cell fate by antigen-receptor signals. Nat Rev Immunol. 2002;2(12):945–956. doi:10.1038/nri955
10. Rip J, de Bruijn MJW, Appelman MK, Pal Singh S, Hendriks RW, Corneth OBJ. Toll-like receptor signaling drives btk-mediated autoimmune disease. Front Immunol. 2019;10:95. doi:10.3389/fimmu.2019.00095
11. Schafer PH, Kivitz AJ, Ma J, et al. Spebrutinib (CC-292) affects markers of B cell activation, chemotaxis, and osteoclasts in patients with rheumatoid arthritis: Results from a Mechanistic Study. Rheumatol Ther. 2020;7(1):101–119. doi:10.1007/s40744-019-00182-7
12. Wang SP, Iwata S, Nakayamada S, et al. Amplification of IL-21 signalling pathway through Bruton’s tyrosine kinase in human B cell activation. Rheumatology. 2015;54(8):1488–1497. doi:10.1093/rheumatology/keu532
13. Gillooly KM, Pulicicchio C, Pattoli MA, et al. Bruton’s tyrosine kinase inhibitor BMS-986142 in experimental models of rheumatoid arthritis enhances efficacy of agents representing clinical standard-of-care. PLoS One. 2017;12(7):e0181782. doi:10.1371/journal.pone.0181782
14. Shinohara M, Chang BY, Buggy JJ, et al. The orally available Btk inhibitor ibrutinib (PCI-32765) protects against osteoclast-mediated bone loss. Bone. 2014;60:8–15. doi:10.1016/j.bone.2013.11.025
15. Wu J, Liu C, Tsui ST, Liu D. Second-generation inhibitors of Bruton tyrosine kinase. J Hematol Oncol. 2016;9(1):80. doi:10.1186/s13045-016-0313-y
16. Crawford JJ, Johnson AR, Misner DL, et al. Discovery of GDC-0853: a potent, selective, and noncovalent Bruton’s tyrosine kinase inhibitor in early clinical development. J Med Chem. 2018;61(6):2227–2245. doi:10.1021/acs.jmedchem.7b01712
17. Boga SB, Alhassan AB, Liu J, et al. Discovery of 3-morpholino-imidazole[1,5-a]pyrazine BTK inhibitors for rheumatoid arthritis. Bioorg Med Chem Lett. 2017;27(16):3939–3943. doi:10.1016/j.bmcl.2017.03.040
18. Caldwell R, Liu-Bujalski L, Qiu H, et al. Discovery of a novel series of pyridine and pyrimidine carboxamides as potent and selective covalent inhibitors of Btk. Bioorg Med Chem Lett. 2018;28(21):3419–3424. doi:10.1016/j.bmcl.2018.09.033
19. Abdelhameed AS, Attwa MW, Al-Shaklia NS, Kadi AA. A highly sensitive LC-MS/MS method to determine novel Bruton’s tyrosine kinase inhibitor spebrutinib: application to metabolic stability evaluation. R Soc Open Sci. 2019;6(6):190434. doi:10.1098/rsos.190434
20. Ariza Y, Murata M, Ueda Y, Yoshizawa T. Bruton’s tyrosine kinase (Btk) inhibitor tirabrutinib suppresses osteoclastic bone resorption. Bone Rep. 2019;10:100201. doi:10.1016/j.bonr.2019.100201
21. Caldwell RD, Qiu H, Askew BC, et al. Discovery of evobrutinib: an oral, potent, and highly selective, covalent Bruton’s Tyrosine Kinase (BTK) inhibitor for the treatment of immunological diseases. J Med Chem. 2019;62(17):7643–7655. doi:10.1021/acs.jmedchem.9b00794
22. Haselmayer P, Camps M, Liu-Bujalski L, et al. Efficacy and pharmacodynamic modeling of the BTK inhibitor evobrutinib in autoimmune disease models. J Immunol. 2019;202(10):2888–2906. doi:10.4049/jimmunol.1800583
23. Becker A, Martin EC, Mitchell DY, et al. Safety, tolerability, pharmacokinetics, target occupancy, and concentration-QT analysis of the novel BTK inhibitor evobrutinib in healthy volunteers. Clin Transl Sci. 2020;13(2):325–336. doi:10.1111/cts.12713
24. Genovese M, Spindler A, Sagawa A, et al. Safety and efficacy of poseltinib, Bruton’s tyrosine kinase inhibitor, in patients with rheumatoid arthritis: a randomized, double-blind, placebo-controlled, 2-part Phase II Study. J Rheumatol. 2021;48(7):969–976. doi:10.3899/jrheum.200893
25. Liu YT, Ding HH, Lin ZM, et al. A novel tricyclic BTK inhibitor suppresses B cell responses and osteoclastic bone erosion in rheumatoid arthritis. Acta Pharmacol Sin. 2021. doi:10.1038/s41401-020-00578-0
26. Cho H, Lee E, Kwon HA, et al. Discovery of tricyclic pyranochromenone as novel Bruton’s tyrosine kinase inhibitors with in vivo antirheumatic activity. Int J Mol Sci. 2020;21(21):7919. doi:10.3390/ijms21217919
27. Xu D, Kim Y, Postelnek J, et al. RN486, a selective Bruton’s tyrosine kinase inhibitor, abrogates immune hypersensitivity responses and arthritis in rodents. J Pharmacol Exp Ther. 2012;341(1):90–103. doi:10.1124/jpet.111.187740
28. Young WB, Barbosa J, Blomgren P, et al. Potent and selective Bruton’s tyrosine kinase inhibitors: discovery of GDC-0834. Bioorg Med Chem Lett. 2015;25(6):1333–1337. doi:10.1016/j.bmcl.2015.01.032
29. Sodhi JK, Wong S, Kirkpatrick DS, et al. A novel reaction mediated by human aldehyde oxidase: amide hydrolysis of GDC-0834. Drug Metab Dispos. 2015;43(6):908–915. doi:10.1124/dmd.114.061804
30. Saffran DC, Parolini O, Fitch-Hilgenberg ME, et al. Brief report: a point mutation in the SH2 domain of Bruton’s tyrosine kinase in atypical X-linked agammaglobulinemia. N Engl J Med. 1994;330(21):1488–1491. doi:10.1056/NEJM199405263302104
31. Di Paolo JA, Huang T, Balazs M, et al. Specific Btk inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat Chem Biol. 2011;7(1):41–50. doi:10.1038/nchembio.481
32. ACP-196 versus placebo in subjects with rheumatoid arthritis on background methotrexate. Available from: https://ClinicalTrials.gov/show/NCT02387762.
33. Cohen S, Tuckwell K, Katsumoto TR, et al. Fenebrutinib versus placebo or adalimumab in rheumatoid arthritis: a randomized, double-blind, Phase II trial. Arthritis Rheumatol. 2020;72(9):1435–1446. doi:10.1002/art.41275
34. Study to evaluate safety and pharmacokinetics of GS-4059 (tirabrutinib) in healthy volunteers and participants with Rheumatoid Arthritis (RA). Available from: https://ClinicalTrials.gov/show/NCT02626026.
35. Safety and Efficacy Study of M2951 in participants with rheumatoid arthritis. Available from: https://ClinicalTrials.gov/show/NCT02784106.
36. Phase IIb Study of evobrutinib in subjects with rheumatoid arthritis. Available from: https://ClinicalTrials.gov/show/NCT03233230.
37. Herman AE, Chinn LW, Kotwal SG, et al. Safety, pharmacokinetics, and pharmacodynamics in healthy volunteers treated with GDC-0853, a selective reversible Bruton’s tyrosine kinase inhibitor. Clin Pharmacol Ther. 2018;103(6):1020–1028. doi:10.1002/cpt.1056
38. Liu L, Halladay JS, Shin Y, et al. Significant species difference in amide hydrolysis of GDC-0834, a novel potent and selective Bruton’s tyrosine kinase inhibitor. Drug Metab Dispos. 2011;39(10):1840–1849. doi:10.1124/dmd.111.040840
39. Lorenzo-Vizcaya A, Fasano S, Isenberg DA. Bruton’s tyrosine kinase inhibitors: a new therapeutic target for the treatment of SLE? Immunotargets Ther. 2020;9:105–110. doi:10.2147/itt.S240874
40. Mina-Osorio P, LaStant J, Keirstead N, et al. Suppression of glomerulonephritis in lupus-prone NZB x NZW mice by RN486, a selective inhibitor of Bruton’s tyrosine kinase. Arthritis Rheum. 2013;65(9):2380–2391. doi:10.1002/art.38047
41. Hutcheson J, Vanarsa K, Bashmakov A, et al. Modulating proximal cell signaling by targeting Btk ameliorates humoral autoimmunity and end-organ disease in murine lupus. Arthritis Res Ther. 2012;14(6):R243. doi:10.1186/ar4086
42. Kim YY, Park KT, Jang SY, et al. HM71224, a selective Bruton’s tyrosine kinase inhibitor, attenuates the development of murine lupus. Arthritis Res Ther. 2017;19(1):211. doi:10.1186/s13075-017-1402-1
43. Isenberg D, Furie R, Jones N, et al. Efficacy, safety, and pharmacodynamic effects of the Bruton’s tyrosine kinase inhibitor, fenebrutinib (GDC-0853), in moderate to severe systemic lupus erythematosus: results of a phase 2 randomized controlled trial [abstract]. Arthritis Rheum. 2019;71(10).
44. American Laboratory Products Company. BTK Inhibitors as Cancer Drug Treatments. Available from: https://www.alpco.com/btk-inhibitors-as-cancer-drug-treatments. Accessed August 13, 2021.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.