")
Back to Journals » ImmunoTargets and Therapy » Volume 9
Bruton’s Tyrosine Kinase Inhibitors: A New Therapeutic Target for the Treatment of SLE?
Authors Lorenzo-Vizcaya A, Fasano S, Isenberg DA
Received 29 February 2020
Accepted for publication 19 May 2020
Published 2 June 2020 Volume 2020:9 Pages 105—110
DOI https://doi.org/10.2147/ITT.S240874
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Ana Lorenzo-Vizcaya,1 Serena Fasano,2 David A Isenberg3
1Department of Internal Medicine, Hospital Universitario De Ourense, Ourense, Spain; 2Rheumatology Unit, Department of Clinical and Experimental Medicine, University of Campania L. Vanvitelli, Naples, Italy; 3Centre for Rheumatology, Department of Medicine, University College London, London, UK
Correspondence: David A Isenberg Email [email protected]
Abstract: Systemic lupus erythematosus (SLE) is an autoimmune disease with a complex pathogenesis, which presents a great variability in its presentation and can affect almost all organs and systems. Multiple therapeutic targets have been discovered recently, but there also have been failed attempts to treat SLE using biologic agents. Bruton’s tyrosine kinase (BTK) is a cytoplasmic tyrosine kinase expressed in several types of cells of hematopoietic origin which participate in both innate and adaptive immunity. Ibrutinib, a BTK inhibitor, is approved for the treatment of several B cell malignancies, including some types of lymphoma and leukemia. As BTK is expressed on several immune cell types, the mechanism of action of BTK also suggests the use of BTK inhibitors in the treatment of autoimmune diseases. In this review, we will summarize what is known and what has been published so far about the treatment of mouse models of SLE and the human disease, using BTK inhibitors.
Keywords: lupus, Bruton, tyrosine kinase, therapeutic targeting, ibrutinib, fenebrutinib
Introduction
Bruton’s tyrosine kinase (BTK) is a cytoplasmic tyrosine kinase expressed in several types of cells of hematopoietic origin which participate in both innate and adaptive immunity. Functionally, BTK is an essential intracellular signaling molecule in the development, survival, and activation of B cells. It was first identified as the genetic defect in the primary immunodeficiency X-linked agammaglobulinemia, which is characterized by an almost complete loss of serum immunoglobulins and circulating mature B cells, with a subsequent susceptibility to infections.1 BTK is involved in a variety of B cell functions, including antigen presentation and production of antibodies by B cell antigen receptor (BCR) signaling. In addition, BTK is important in the normal functioning of several immune cells. It is also involved in the activation of innate immune cells (macrophages, neutrophils, and mast cells/basophils) by immune complexes. BTK controls cytokine production, phagocytosis, and production of inflammatory mediators.2 Furthermore, BTK is involved in platelet activation via the glycoprotein VI receptor3 and osteoclast differentiation via the receptor activator of NF-kB (RANK).4
The BTK inhibitor, ibrutinib, is approved for the treatment of several B cell malignancies including some types of lymphoma and leukemia.5 It is widely used in hematological patients. This approach is quite well tolerated in humans; its main reported side effects are mild such upper respiratory infections, fatigue, and diarrhea.6 The mechanism of action of BTK suggests a role for BTK inhibition in the treatment of autoimmune diseases.7 B cells are crucial players in several systemic autoimmune diseases including Systemic Lupus Erythematosus (SLE), Sjogren’s syndrome, and myositis.8–10 These diseases are marked by altered B cell selection leading to the production of autoreactive antibodies, pro-inflammatory cytokines, and the presentation of autoantigens to autoreactive T cells. BTK inhibition in experimental autoimmune diseases in preclinical models has led to a reduction of disease, notably in mouse models of collagen-induced arthritis and anti-glomerular basement membrane glomerulonephritis.11,12 Increased BTK expression and phosphorylation were noted in the peripheral blood B cells of patients with rheumatoid arthritis (RA) compared with healthy controls13 correlating with anti-citrullinated protein antibodies and rheumatoid factors (RF).14 Increased BTK expression and phosphorylation correlated with RF in patients with Sjogren’s syndrome.13
Why Is Blocking BTK a Potentially Good Idea in SLE?
SLE is an autoimmune disease characterized by autoantibodies against nuclear antigens. These autoantibodies promote disease pathogenesis by forming immune complexes that cause inflammation and tissue damage. Several components of BTK signaling pathways are altered in B cells from lupus patients.15,16 It was notable that BTK+ cells in the peripheral blood of SLE patients correlated with disease activity, anti-dsDNA antibodies, proteinuria, and C3 levels.15
The importance of B cells in SLE is highlighted by the efficacy of B cell–targeting therapies. Following successful clinical trials,17,18 Belimumab, a fully human IgG1 monoclonal antibody that selectively targets and inhibits the biological activity of the soluble B lymphocyte stimulator (BLyS), was approved by the Federal Drug Administration (FDA) in the United States for the treatment of lupus in 2011. Belimumab was the first drug approved by the FDA for the treatment of SLE in over 50 years. It does not directly bind to B cells and does not directly reduce B-cell population, but it inhibits B cell survival.
In addition, other agents targeting B cells, e.g., monoclonals against CD20, CD22, and CD19, have also been used for SLE patients.19 The efficacy of anti-CD20 B-cell-depleting therapy (notably rituximab) provides indirect clinical evidence for the likely efficacy of the B cell modulatory effects of BTK inhibition in autoimmune diseases. Rituximab has established efficacy and is approved in RA20 and granulomatosis with polyangiitis/microscopic polyangiitis.21 European League Against Rheumatism (EULAR) and American College of Rheumatology (ACR) guidelines also suggest considering rituximab for refractory or relapsing disease.22,23 Case series and open-label studies have also reported efficacy for rituximab in SLE. Interestingly, it has been shown that B cell depletion is associated with a fall in antibodies to DNA, nucleosomes, and cardiolipin,24,25 and subsequent increase in anti-dsDNA levels may be associated with disease flare.26 In contrast, B cell depletion using rituximab does not alter the levels of antibodies to the extractable nuclear antigens (e.g., Ro, La) and fluctuations in the levels of these antibodies are not clearly associated with disease exacerbation. However, two randomized trials, EXPLORER (Exploratory Phase II/III SLE Evaluation of Rituximab) and LUNAR (Lupus Nephritis Assessment with Rituximab), failed to meet their primary endpoints.27,28 The reasons for these disappointing results are likely to include in part the study design, as substantial amounts of background therapy were allowed, disease heterogeneity, and limitations of the available outcome tools. Nevertheless, a clear restriction of targeting a B cell antigen is the lack of specificity against autoreactive B cells. The CD20 antigen is not expressed on plasmablasts and plasma cells. Consequently, in SLE patients treated with rituximab, persistent long-lived plasma cells survive and continue to produce autoantibodies and the resulting immune complexes have been implicated as triggers of flares.29
There are thus several unmet needs for targeted therapy in SLE. Because of the complexity of SLE pathogenesis, a drug that inhibits more than one pathway would promise a great therapeutic potential. Inhibition of BTK may act via additional mechanisms to anti-CD20 B cell-depleting therapy. As BTK is expressed on several immune cell types, blocking BTK signaling might be expected to impact not only the B cell component but also the innate immunity component activated by pre-existing immune complexes and Toll-like receptors.30 Preliminary data suggest that BTK inhibition may actually promote central tolerance.31
Mouse Model and Clinical Application
In this section, we will review what is known about the treatment of mouse models of SLE and the human disease, using BTK inhibitors.
Antibodies and B Cells
One study conducted in murine model with SLE (NZBxW_F1) had as its primary objective the demonstration of the effects of BTK inhibition on systemic immune activation, as well as its ability to prevent antibody-mediated glomerulonephritis. The mice were treated for 12 weeks, with different doses of BTK inhibitors: 3, 10, or 30 mg/kg.32 It was observed that treatment with 10 or 30 mg/kg significantly reduced the total number of splenic B cells up to almost 10 times in 90% of treated mice,32 and those who received lower doses (3 mg/kg) showed a more modest effect. The levels of anti-dsDNA antibodies were significantly lower in mice that received 10–30 mg/kg. There was no significant reduction in those treated with 3 mg/kg.
The authors concluded that treatment with a BTK inhibitor reduces the formation of plasma cells in the germinal center and limits the spontaneous accumulation of activated B cells and reduced levels of anti-dsDNA and IgG autoantibodies in these mice.
In a human study, the efficacy, safety, and pharmacodynamic profile of fenebrutinib (FEN), a BTK inhibitor, was evaluated in patients with moderate to severe SLE.24 Patients received different doses: 150 mg daily or 200 mg twice a day, for 48 weeks. The safety results were similar between FEN and placebo, although more serious adverse events were observed in the arm of FEN 200 mg. Treatment with both doses of FEN significantly reduced levels of CD19 + B cells and IgG anti-dsDNA autoantibodies compared to placebo-treated mice.33 However, no clinical benefit was identified.
Nephritis
One of the above studies32 also analyzed the effects of the inhibition of BTK on renal damage measuring urinary protein levels every other week to assess the extent of kidney damage and observed that at the end of treatment, 70% of mice treated with placebo developed severe proteinuria while none of those treated with BTK inhibitor had developed it. The development of the disease was prevented by BTK inhibition by inhibiting glomerular antibody and C3 deposition;32 as well as a significant dose-dependent reduction of inflammatory infiltrates and glomerular lesions. Those mice who received 30 mg/kg had no inflammatory infiltrates.
These results demonstrate that BTK inhibition can limit the development of antibody-driven glomerular pathology in this type of SLE prone mice.
Another study in mice using a BTK inhibitor (M7583) had as one of its main objectives the evaluation of the renal involvement in BXSB-Yaa mice, who developed nephritis. When administering treatment with M7583 or standard treatment (mycophenolate mofetil (MMF)) to BXSB-Ya mice; they found that both drugs led to a significant reduction in proteinuria and also showed that the increase in titers of autoantibodies was significantly blocked by inhibition of BTK with M7583.34
In another study of BTK inhibition on renal involvement, MRL/lpr mice were divided into two groups: half were treated with BI-BTK-1 (another BTK inhibitor) at 10 mg/kg, and the other half was given a comparable control.35 The main results were the demonstration of improvement in kidney disease, despite having more serious inflammation and using lower doses of the drug (10 mg/kg) compared to some models previously reported; as well as showing that BI-BTK-1 can reverse the established and severe renal disease of inducible antibody-mediated glomerulonephritis, in one of the mice strains (MRL/lpr).35
Thus, the authors of this study,35 were the first to show the reversal of the disease in a strain of mice with spontaneous lupus (MRL/lpr), demonstrating that BTK inhibition is effective in murine lupus model also in reversing disease and not only preventively.
These results emphasize the therapeutic potential for BTK inhibition in lupus nephritis.
Arthritis
In one of the studies above34 using a BTK inhibitor (M7583), another objective was to determine the role of this drug in preventing the development of arthritis in the DBA/1 mice. In this study, the other drug used was MMF;, and although this is not the drug of choice to treat arthritis, they compared its effectiveness with that of the BTK inhibitor. At the end of the study, they observed that treatment with BTK inhibitor led to a significant reduction in the clinical signs of arthritis34 and also showed greater efficacy than MMF. They also observed that those who received M7583 had a reduction in the levels of anti-dsDNA, anti-histones, and anti-Ro/SSA, but not anti-Sm/RNP. Meanwhile, the MMF did not have a significant impact on any of the autoantibody levels.
Thus, they confirm that blocking the activity of BTK may be a very effective treatment of end-organ disease in BXSB-Yaa and pristane-DBA/1 mice34 in particular reducing kidney disease and arthritis. It gives some hope that this approach might be effective in patients with these problems.
Cutaneous and Neuropsychiatric Manifestations
In another murine study using the MRL/lpr mouse model, the effect of blocking BTK was tested on cutaneous and neuropsychiatric manifestations.
MRL/lpr skin lesions improved significantly with BI-BTK-1 treatment, macroscopically and histologically. At the macroscopic level, the improvement was sustained until the mice were sacrificed, when 42% of the mice treated with BI-BTK-1 had some sign of skin disease, while 92% of the control mice had visible skin involvement (p < 0.0001).36 They observed microscopically that the treated mice had less cell infiltration in the skin, fewer deposit of IgG, and a decrease in the number of macrophages and T cells. In addition, macrophages observed in mice treated with BI-BTK-1 had a less activated phenotype.36 To assess these activated macrophages further in the skin, sections were stained with Mac-2, a macrophage activation marker. Another observation made by the authors was the demonstration of reduced levels of cytokines associated with the pathogenesis of cutaneous lupus, e.g., IL-6, IL-17, and TNF, following therapeutic inhibition of BTK.
MRL/lpr mice also manifest spontaneously a neuropsychiatric phenotype, which includes cognitive abnormalities such as memory deficits and depression-like behavior; in this study36 they also evaluated the effects of the treatment in NPSLE. They found that treatment with a BTK inhibitor improved the manifestations of NPSLE as the treated mice had improved cognitive function showing a significant improvement in the object placement test that indicates preserved spatial memory.
With respect to visual memory, they performed object recognition tests; reporting that the deficits observed were not completely reversed.36 No improvement in depression-like behavior in the mice treated with BTK inhibition was observed. The authors suggested that perhaps longer treatment should be attempted to obtain a better response in these NPSLE manifestations because they did identify a decrease in macrophages in the brain, microglia, are an important source of inflammatory cytokines that are believed that play a fundamental role in NPSLE pathogenesis.
An overview of the murine and human therapeutic attempts is shown in Table 1. The therapeutic target of BTK inhibition may thus be a potentially valuable therapeutic option for NPSLE, in addition to any beneficial effect on skin disease.
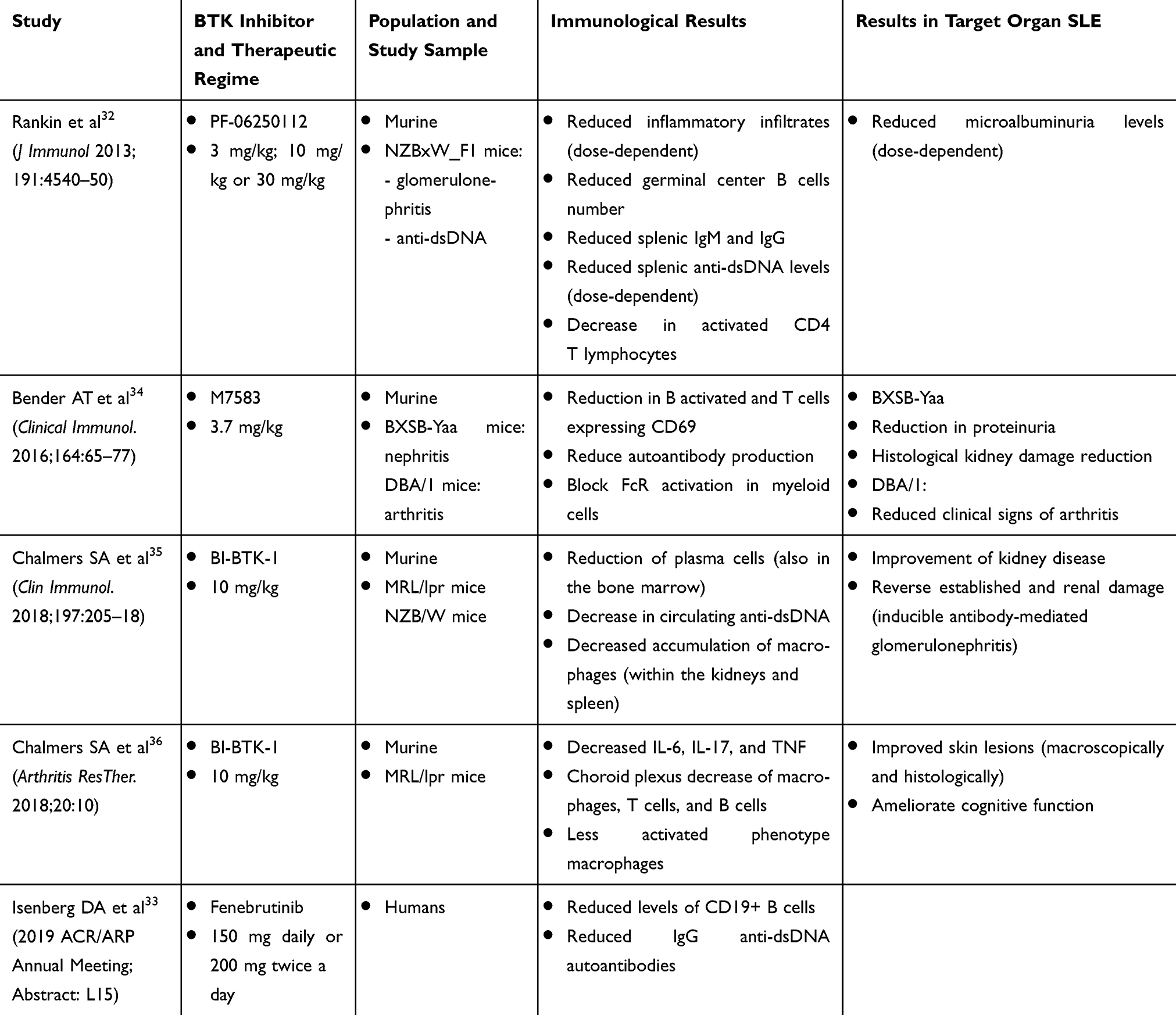 |
Table 1 Summary of the Main Points and Results of the Studies Performed to Know the Role of BTK Inhibitors in SLE, in Murine Models, and in Humans |
Conclusions
BTK inhibition is a promising therapeutic target for patients with SLE due to its capacity to act through several different mechanisms. Clinical trials on patients treated with Ibrutinib showed a reduction in the levels of some autoantibodies (antinucleosome, antihistone, and anti-ssDNA) and improved some clinical manifestations of SLE.
From the data obtained in the studies discussed above, it is evident the effectiveness of BTK inhibition in multiple models of murine lupus, for different manifestations. It will be interesting to discover if similar is observed in current early phase studies of BTK inhibition in human SLE.
Nowadays, drugs that act at this level like Ibrutinib are widely used and well tolerated in humans. Due to this and to the results of the studies published so far, we conclude that BTK may be a successful approach to the treatment of resistant manifestations of SLE, including renal, arthritic, CNS, and skin disorders.
Disclosure
Professor David Isenberg reports personal fees from Roche, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Vetrie D, Vorechovský I, Sideras P, et al. The gene involved in X linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature. 1993;361(6409):226–233. doi:10.1038/361226a0
2. López-Herrera G, Vargas-Hernández A, González-Serrano ME, et al. Bruton’s tyrosine kinase–an integral protein of B cell development that also has an essential role in the innate immune system. J Leukoc Biol. 2014;95(2):243–250. doi:10.1189/jlb.0513307
3. Quek LS, Bolen J, Watson SP. A role for Bruton’s tyrosine kinase (Btk) in platelet activation by collagen. Curr Biol. 1998;8(20):1137–1140. doi:10.1016/S0960-9822(98)70471-3
4. Shinohara M, Koga T, Okamoto K, et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell. 2008;132:794–806. doi:10.1016/j.cell.2007.12.037
5. Wang Y, Zhang LL, Champlin RE, et al. Targeting Bruton’s tyrosine kinase with ibrutinib in B-cell malignancies. Clin Pharmacol Ther. 2015;97(5):455–468. doi:10.1002/cpt.85
6. Carreira PL, Isenberg DA. Recent developments in biologic therapies for the treatment of patients with systemic lupus erythematosus. Rheumatology. 2019;58(3):382–387. doi:10.1093/rheumatology/key064
7. Horwood NJ, Urbaniak AM, Danks L. Tec family kinases in inflammation and disease. Int Rev Immunol. 2012;31(2):87–103. doi:10.3109/08830185.2012.670334
8. Fasano S, Isenberg DA. Present and novel biologic drugs in primary Sjögren’s syndrome. Clin Exp Rheumatol. 2019;37 Supl 118:167–174.
9. Edwards JC, Cambridge G, Leandro MJ. B cell depletion therapy in rheumatic disease. Best Pract Res Clin Rheumatol. 2006;20(5):915–928. doi:10.1016/j.berh.2006.05.010
10. Fasano S, Gordon P, Hajji R, et al. Rituximab in the treatment of inflammatory myopathies: a review. Rheumatology (Oxford). 2017;56(1):26–36. doi:10.1093/rheumatology/kew146
11. Honigberg LA, Smith AM, Sirisawad M, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010;107(29):13075–13080. doi:10.1073/pnas.1004594107
12. Mina-Osorio P, LaStant J, Keirstead N, et al. Suppression of glomerulonephritis in lupus-prone NZB × NZW mice by RN486, a selective inhibitor of Bruton’s tyrosine kinase. Arthritis Rheum. 2013;65(9):2380–2391. doi:10.1002/art.38047
13. Corneth OBJ, Verstappen GMP, Paulissen SMJ, et al. Enhanced Bruton’s tyrosine kinase activity in peripheral blood B lymphocytes from patients with autoimmune disease. Arthritis Rheumatol. 2017;69(6):1313–1324. doi:10.1002/art.40059
14. Wang SP, Iwata S, Nakayamada S, et al. Amplification of IL-21 signalling pathway through Bruton’s tyrosine kinase in human B cell activation. Rheumatology (Oxford). 2015;54(8):1488–1497. doi:10.1093/rheumatology/keu532
15. Kong W, Deng W, Sun Y, et al. Increased expression of Bruton’s tyrosine kinase in peripheral blood is associated with lupus nephritis. Clin Rheumatol. 2018;37:43–49. doi:10.1007/s10067-017-3717-3
16. Wu XN, Ye YX, Niu JW, et al. Defective PTEN regulation contributes to B cell hyperresponsiveness in systemic lupus erythematosus. Sci Transl Med. 2014;6:246–299. doi:10.1126/scitranslmed.3009131
17. Furie R, Petri M, Zamani O, et al. A Phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011;63(12):3918–3930. doi:10.1002/art.30613
18. Navarra SV, Guzmán RM, Gallacher AE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, Phase 3 trial. Lancet. 2016;377(9767):721–731. doi:10.1016/S0140-6736(10)61354-2
19. Mouyis M, Ciutin C, Isenberg DA. Biologic therapies in systemic lupus erythematosus.. Biol Rheumatol Nova Biomed New York. 2016;3–33.
20. Edwards JC, Szczepanski L, Szechinski J, et al. Efficacy of B‐cell‐targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–2581. doi:10.1056/NEJMoa032534
21. Geetha D, Kallenberg C, Stone JH, et al. Current therapy of granulomatosis with polyangiitis and microscopic polyangiitis: the role of rituximab. J Nephrol. 2015;28(1):17–27. doi:10.1007/s40620-014-0135-3
22. Bertsias GK, Tektonidou M, Amoura Z, et al. Joint European League Against Rheumatism and European Renal Association-European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of adult and paediatric lupus nephritis. Ann Rheum Dis. 2012;71(11):1771–1782. doi:10.1136/annrheumdis-2012-201940
23. Hahn BH, McMahon MA, Wilkinson A, et al. American college of rheumatology guidelines for screening, treatment, and management of lupus nephritis. Arthritis Care Res (Hoboken). 2012;64:797–808. doi:10.1002/acr.21664
24. Ng K, Cambridge G, Leandro MJ, et al. B cell depletion therapy in systemic lupus erythematosus: long-term follow-up and predictors of response. Ann Rheum Dis. 2007;66:1259–1262. doi:10.1136/ard.2006.067124
25. Ioannou Y, Lambrianides A, Cambridge G, et al. B cell depletion therapy for patients with systemic lupus erythematosus results in a significant drop in anticardiolipin antibody titres. Ann Rheum Dis. 2008;67:425–426. doi:10.1136/ard.2007.078402
26. Swaak AJ, Groenwold J, Aarden LA, et al. Prognostic value of anti-dsDNA in SLE. Ann Rheum Dis. 1982;41:388–395. doi:10.1136/ard.41.4.388
27. Merrill JT, Neuwelt CM, Wallace DJ, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62(1):222–233. doi:10.1002/art.27233
28. Rovin BH, Furie R, Latinis K, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 2012;64(4):1215–1226. doi:10.1002/art.34359
29. Hiepe F, Dörner T, Hauser AE, et al. Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nat Rev Rheumatol. 2011;7:170–178. doi:10.1038/nrrheum.2011.1
30. Mariño E, Grey ST. B cells as effectors and regulators of autoimmunity. Autoimmunity. 2012;45(5):377–387. doi:10.3109/08916934.2012.665527
31. Sanz I, Lee FE. B cells as therapeutic targets in SLE. Nat Rev Rheumatol. 2010;6(6):326–337. doi:10.1038/nrrheum.2010.68
32. Rankin AL, Seth N, Keegan S, et al. Selective inhibition of BTK prevents murine lupus and antibody-mediated glomerulonephritis. J Immunol. 2013;191(9):4540–4550. doi:10.4049/jimmunol.1301553
33. Isenberg DA, Furie R, Jones N et al. Efficacy, Safety, and Pharmacodynamic Effects of the Bruton’s Tyrosine Kinase Inhibitor,Fenebrutinib (GDC-0853), in Moderate to Severe Systemic Lupus Erythematosus: results of a Phase2 Randomized Controlled Trial. 2019 ACR/ARP Annual Meeting; Abstract number: L15. 2019.
34. Bender AT, Pereira A, Fu K, et al. Btk inhibition treats TLR7/IFN driven murine lupus. Clin Immunol. 2016;164:65–77. doi:10.1016/j.clim.2016.01.012
35. Chalmers SA, Glynn E, Garcia SJ, et al. BTK inhibition ameliorates kidney disease in spontaneous lupus nephritis. Clin Immunol. 2018;197:205–218. doi:10.1016/j.clim.2018.10.008
36. Chalmers SA, Wen J, Doerner J, et al. Highly selective inhibition of Bruton’s tyrosine kinase attenuates skin and brain disease in murine lupus. Arth Res Ther. 2018;20(1):10. doi:10.1186/s13075-017-1500-0
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.