")
Back to Archived Journals » Core Evidence » Volume 9
Brodalumab: an evidence-based review of its potential in the treatment of moderate-to-severe psoriasis
Authors Coimbra S, Figueiredo A, Santos-Silva A
Received 21 May 2014
Accepted for publication 13 June 2014
Published 21 July 2014 Volume 2014:9 Pages 89—97
DOI https://doi.org/10.2147/CE.S33940
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Susana Coimbra,1,2 Américo Figueiredo,3 Alice Santos-Silva2,4
1CESPU (Advanced Polytechnic and University Cooperative), Institute of Research and Advanced Training in Health Sciences and Technologies, Gandra-PRD, 2IBMC, Institute for Molecular and Cell Biology, University of Porto, Porto, 3Service of Dermatology, Hospital and University Centre of Coimbra (CHUC), University of Coimbra, Coimbra, 4Biochemistry Laboratory, Biological Sciences Department, Faculty of Pharmacy (FFUP), University of Porto, Porto, Portugal
Abstract: Advances in knowledge regarding the pathogenesis of psoriasis have allowed the development of a new class of agents known as biologic drugs. Data confirm that T helper (Th)17 and interleukin (IL)-17 signaling has a crucial role in the pathogenesis of the disease. High levels of IL-17 and Th17-related cytokines have been reported in psoriasis, leading to the suggestion of agents targeting IL-17 as a potential therapeutic strategy in psoriasis. Brodalumab is a human monoclonal antibody that targets IL-17 receptor A, blocking the effects of IL-17A, IL-17F, and IL-17E. Data from Phase I and Phase II clinical trials indicate that brodalumab has a favorable safety and tolerability profile, with strong clinical activity, suggesting that it is a potential tool for use in the treatment of moderate-to-severe psoriasis.
Keywords: interleukin-17, interleukin-17 receptor, monoclonal antibody, T helper 17 pathway
Introduction
Psoriasis is a chronic, recurrent, inflammatory cutaneous disease that affects 1%–3% of the general population. It is characterized by an abnormal cycle of epidermal development, with epidermal hyperproliferation, altered maturation of skin cells, vascular changes, and marked inflammation. Histologically, there is marked acanthosis, accompanied by parakeratosis and a mixed dermal infiltrate, including CD4+ T-cells, dendritic cells, macrophages, and mast cells. In the epidermis, neutrophilic exudates and CD8+ T-cells are often seen. Dermal papillary blood vessels are dilated and tortuous.1
Psoriasis is associated with significant impairment of health-related quality of life, especially in the more severe and nonresponder forms.2 The severity of psoriasis is graded as mild, moderate, or severe, mainly in accordance with the area of skin surface affected, the degree of scaling, and the type of infiltration.3 Disease severity usually reflects the extent of skin involvement and is often associated with poor quality of life.
The pathogenesis of psoriasis was initially described as an abnormal proliferation of epidermal keratinocytes, resulting from epidermal keratinocyte hyperplasia, parakeratosis, leukocyte infiltration, and neoangiogenesis. Later, T-cells were identified as crucial to the initiation and maintenance of psoriasis.4 Nowadays, it is accepted that genetic, environmental, and immunologic factors are involved in the disease onset and course. Moreover, different cells are important key players at different stages, and the onset of psoriasis is similar to an immune reaction.5
The T helper (Th)1 pathway is believed to be important in the pathogenesis of psoriasis, as suggested by the increase in cytokines of this axis, such as interferon (IFN)-γ, tumor necrosis factor (TNF)-α, interleukin (IL)-2, and IL-12, observed in plaques and in the serum of patients with psoriasis.6–8 Levels of these cytokines have been correlated with severity of psoriasis,6,9 and the success of some therapies was associated with deviation of Th1 to a Th2 response.10
Currently, psoriasis is known to be a complex disease in which the cytokine network is disturbed, and the IL-23/Th17 axis, as well as the Th1 pathway, appear to be crucial for its pathogenic mechanisms.11,12 T-cell cytokines induce proliferation and activation of keratinocytes, also producing several proinflammatory cytokines and chemokines that sustain and amplify the inflammatory response by recruiting, retaining, and activating immune cells in the skin. The relevance of the IL-23/Th17 axis (Figure 1) was highlighted by the successful treatment of moderate-to-severe psoriasis with drugs targeting the p40 subunit of IL-12 and IL-23.13
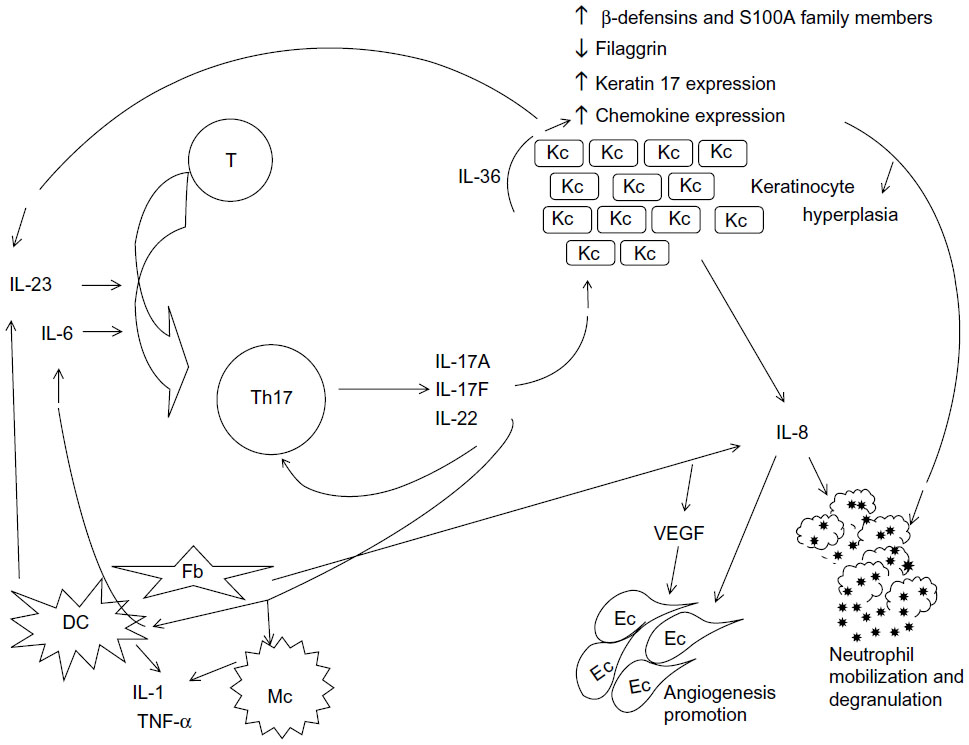 | Figure 1 T helper 17 signaling pathway. |
IL-17 produced by Th17 cells has a crucial role in the inflammatory process, by stimulating production of several proinflammatory cytokines and chemokines by keratinocytes, dendritic cells, and other immune cells. Moreover, both IL-17 and Th17 cells have been found to have a role in the pathology of several other autoimmune disorders.14,15 Therefore, the Th17 pathway is considered to be an attractive potential target for the treatment of several pathologic conditions, including psoriasis.
Th17 pathway
Th17-type T-cells produce IL-17 and are a distinct Th cell population that plays a crucial role in CD4+ T-cell-mediated adaptive immunity. Human CD4+ T-cells may differentiate into a Th1 or Th17 phenotype. The transcription factors retinoid-related orphan receptor (ROR)c and T-bet contribute to both phenotypes, and both phenotypes may produce IL-17A and IFN-γ.16 IL-12 or IL-23 mediates the differentiation of these cells into Th1 or Th17, respectively.16
Mediators of the Th17 immune system include IL-1, IL-6, IL-21, IL-23, and transforming growth factor (TGF)-β, which stimulate the differentiation of naïve CD4+ T-cells into activated memory Th17 cells.17,18 IL-1β is involved in the activation of naïve T-cells from peripheral blood, and seems to be the strongest inducer of IL-17 production. IL-6 and IL-21 further enhance IL-17 production.19 In opposition, development of Th17 cells from naïve precursors is inhibited by IFN-γ and IL-4. IL-23, another Th17 system mediator, drives proliferation of Th17 cells.20 In humans, the lineage-specific transcription factor RORc leads to the developmental program of Th17 cells.
In psoriasis, at least in the severe form of the disease, CD4+ regulatory T-cells also showed a capacity to differentiate into Th17 cells.21 The differentiation of CD4+ T-cells into regulatory T-cells is stimulated by TGF-β in the absence of IL-6. However, regulatory T-cells may be induced to differentiate into IL-17-producing Th17 cells in the presence of an inflammatory stimulus.
Apart from Th17 cells, IL-17-secreting CD8+ T-cells (Tc) are also potent producers of IL-17.22 IL-6 and TGF-β, which promote expression of the Th17 lineage-specific transcription factor ROR, are also required for induction of Tc17 cells.23 Moreover, IL-23 supports the development of Tc17 cells, which is inhibited by IFN-γ.24 Tγδ cells appear to be another source of IL-17, particularly the early source of IL-17 in response to infection. Natural killer T-cells are also another population of innate T lymphocytes known to produce IL-17. Tγδ and natural killer T-cells induce secretion of IL-17 through stimulation of IL-23.25,26 However, the activities and functions of these cells as a source of IL-17 are not entirely understood.
The IL-17 family comprises IL-17A through IL-17F. IL-17A is a proinflammatory cytokine, and IL-17F seems to act in parallel with IL-17A, but is less potent.27 Apparently, in certain circumstances, IL-17A may also be expressed by neutrophils, mast cells, and macrophages.28 Mast cells and neutrophils discharge IL-17 during formation of specialized structures, ie, extracellular traps, by the classical degranulation pathways.29 In opposition, IL-17E (also known as IL-25) is an anti-inflammatory cytokine, inducing a deviation to a Th2 response.30 IL-17B, IL-17C, and IL-17D are poorly characterized. IL-23 stimulates the production of IL-17A and other Th17 cytokines associated with the Th17 pathway, namely IL-17F.31
The IL-17 receptor family includes IL-17 receptors A–E (IL-17RA, IL-17RB, IL-17RC, IL-17RD, and IL-17RE). The IL-17 cytokines seem to bind to their receptors with different affinities, inducing production of proinflammatory cytokines and chemokines. The proinflammatory cytokines IL-17A and IL-17F, as well as the 17A/F heterodimer ligands, stimulate a receptor complex consisting of IL-17RA and IL-17RC subunits.27,32 The IL-17RA/IL-17RC complex induces signaling through a pathway that depends on the similar expression to FGF genes (SEF)/IL-17R containing the adaptor protein ACT1 (nuclear factor κB activator 1 [NF-κB1]).32
Th17 and Tc17 cells produce IL-17A and IL-22, but subsets of Th17 and Tc17 cells also secret IFN-γ and TNF-α.33,34 Recently, another T-cell population was described, ie, Th22 cells that produce IL-22 and TNF-α.35 Th17 cells also produce IL-21, which has a crucial role in the Th17 pathway, amplifying Th17 differentiation.36
Associations between psoriasis and gene regions involved in the Th17 pathway have been established. In genes encoding for the IL-12B, IL-23A, and IL-23 receptor, single nucleotide polymorphisms have been identified in psoriasis.37,38 The genes encoding for TNF-α-induced protein 3 (TNFIP3), TNFIP3-interacting protein 1, and TNF receptor-associated factor 3 interacting protein 2 (TRAF3IP2) are associated with risk of psoriasis.37,38 The gene TRAF3IP2 codes for the adaptor protein ACT1 (NF-κB1), a regulator of the NF-κB pathway that is involved in IL-17 signaling. Moreover, diseases causing variants of TRAF3IP2 are associated with altered IL-17R adaptor protein interactions.39
IL-17 signaling in psoriasis
IL-17, namely IL-17A, has an important role in host defense, inducing IL-6 production to enhance acute-phase responses and differentiation of additional Th17 cells, thereby intensifying the response against pathogens.40 However, regardless of its protective effects, in some autoimmune and immunoinflammatory diseases IL-17 can be deregulated, contributing to the pathogenesis and/or maintenance of these disorders. Indeed, deregulation of IL-17A favors chronic inflammation and tumor development.41
IL-17 activates keratinocytes to produce interleukins and chemokines, such as IL-8, which provides a strong chemotactic signal for neutrophil recruitment.42 It was reported that administration of IL-17F to mouse skin increases the expression of IL-8,43 which is known to be elevated in psoriasis.12 IL-17 also upregulates keratinocyte expression of other chemokines (eg, C-X-C motif ligand [CXCL]1, CXCL3, CXCL5, CXCL5, and CXCL6), which have been associated with recruitment of neutrophils.44
IL-17A exerts its effects in multiple cell types, namely in macrophages, dendritic cells, neutrophils, fibroblasts, endothelial cells, epithelial cells, keratinocytes, and lymphocytes, leading to production of several cytokines and chemokines.45
Using a human monolayer model, Th17 cytokines (eg, IL-17A, IL-22, TNF-α) stimulated the upregulation of chemokine (C-C motif) ligand (CCL)20.46 In psoriasis, IL-17A induces keratinocytes to express CCL20, recruiting Th17 cells and dendritic cells to the skin,44 which may contribute to maintain both cells in psoriatic lesions. A study of psoriatic dermal dendritic cells cultured with allogenic CD4+ T-cells showed that these cells induced a higher number of CD4+ T-cells to produce IL-17 than normal dendritic cells.47
Moreover, in keratinocytes, IL-17A upregulates antimicrobial peptides such as β-defensins and S100A family members, providing a stimulus for the innate immune system,44,46 downregulates filaggrin and other factors involved in cell adhesion, contributing to skin barrier disruption,48 and increases expression of keratin 17, contributing to epidermal hyperproliferation.49 IL-17A also stimulates keratinocytes to express IL-36 that, by acting synergistically with IL-17A, promotes expression of the antimicrobial peptides CXCL8, IL-6, and TNF-α.50
IL-17A stimulates fibroblasts and dendritic cells to produce IL-6, which favors the commitment of more T-cells to the Th17 phenotype (Figure 1). Dendritic cells and macrophages are stimulated to produce IL-1 and TNF-α by IL-17A.36
In summary, IL-17 and Th17-related cytokines, such as IL-23 and IL-22, contribute to the pathologic alterations found in psoriasis (Figure 1). IL-17 is a critical component in the establishment and perpetuation of inflammation, inducing production of proinflammatory cytokines such as IL-6, IL-8, and prostaglandin E2,51 and also stimulates secretion of proinflammatory cytokines by other cells, namely endothelial cells and macrophages.36 IL-22 induces epidermal hyperplasia and hypogranulosis; it also induces proinflammatory responses, such as the production of cytokines, chemokines, and acute-phase proteins from many cell types, and regulates the differentiation and migration of keratinocytes. Production of IL-22 is directly induced by IL-23, and IL-22 can mediate IL-23-induced acanthosis and dermal inflammation.52,53
IL-17 also seems to promote angiogenesis. IL-17 indirectly enhances the proliferation of endothelial cells via induction of vascular endothelial growth factor and IL-8 by fibroblasts.54
These cytokines can also induce production of chemokines and subsequently increase the recruitment of endothelial progenitor cells to support angiogenesis.
IL-17 interacts with several cytokines. In psoriatic skin, IL-17A and TNF-α act synergistically or additively on keratinocytes to upregulate several genes, many of which are expressed significantly in psoriatic skin, such as S100A7, β-defensin, IL-23, CCL20, and CXCL1.55 IL-17A also acts together with IFN-γ to increase production of IL-6 and CXCL8,56 and acts in synergy with other proinflammatory cytokines, such as IL-1 and IL-6.
Circulating Th17 cells are increased in psoriasis, as well as Th22 and Th1 cells, although to a lesser extent.57 As referred, increased levels of IL-17 were found in skin lesions and in the blood of patients with psoriasis, and were correlated with disease severity;6,58–60 moreover, the levels decreased after successful treatment,12,58 confirming the role of IL-17 in the pathogenesis of psoriasis.
Expression of IL-17E, IL-17B, and IL-17D does not seem to increase in psoriatic skin lesions, while expression of IL-17A, IL-17C, and IL-17F increases.61 Apparently, IL-17E, IL-17B, and IL-17D do not have a significant role in the development of psoriasis. However, IL-17A in particular has been demonstrated to have a key role in the chronic inflammatory process of several immune-mediated diseases, including psoriasis. Therefore, blocking IL-17A appears to be a sustainable approach for the treatment of such pathologies.
Blocking IL-17 signaling
The activity of IL-17 and the effect of its inhibition have been evaluated in animal models of psoriasis. Induction of inflammation with TPA (12-O-tetradecanoylphorbol-13-acetate) seems to lead to a Th17-like response in transgenic keratin 14/vascular endothelial growth factor mice, a potential animal model for psoriasis, and an increase in the ear tissue of TNF-α, IL-1β, IL-6, IL-12/23p40, IL-12p70, IL-22, and IL-17 levels was reported.62 Imiquimod-induced psoriasis-like skin inflammation was almost blocked in mice deficient in IL-17RA and IL-23.63 In K5.hTGF-β1 transgenic mice, presenting a skin phenotype and cytokine alterations analogous to those found in psoriasis, administration of anti-IL-17A blocked progression of disease.64 K5.Stat3C mice treated with TPA developed psoriasis-like skin lesions, and treatment with anti-IL-17A reduced epidermal hyperplasia.65 In mice, administration of IL-23 induced epidermal hyperplasia mediated by IL-17 and IL-22. In mice deficient in these cytokines or in IL-17RA, epidermal hyperplasia is minor, while antibodies against these cytokines completely inhibit IL-23-induced hyperplasia.66,67
Inhibition of IL-23, acting upstream from IL-17, has been also studied, both in animals and in clinical studies. Mice deficient in IL-23 showed defects in terminal differentiation and function of Th17, namely a decrease in production of IL-17.68 In clinical studies, agents that target IL-23 have been shown to inhibit IL-17, eg, ustekinumab, which is used to treat moderate and severe forms of psoriasis.13
Successful conventional therapies, such as psoralen plus ultraviolet (UV) light A, narrow-band UVB (NB-UVB), and NB-UVB combined with calcipotriol, lead to a decrease in circulating levels of IL-17 and Th17-related cytokines, but acitretin had no such effect.12,59,69 Treatment with cyclosporin normalized the IL-17 and IL-22 mRNA levels that were increased in psoriatic skin.33 NB-UVB inhibited the IL-23/Th17 axis, with a decrease observed in CD11c+ dendritic cells and their products, namely IL-20, inducible nitric oxide synthase, IL-12/23p40, and IL-23p19, as well as suppression of IL-17 and IL-22 mRNA.70
Concerning treatment with biologics, infliximab decreased the high Th17 and Th1 cell levels observed before treatment.57 Moreover, early improvement was significantly correlated with a reduction in IL-17 and its signaling pathway when anti-TNF agents were used as a treatment for psoriasis.59,71
According to a systematic review by Tausend et al, biologic agents specifically targeting IL-12, IL-17, and IL-23 are efficacious and safe for the treatment of moderate-to-severe psoriasis in adults, although long-term data are still needed.72
Agents that target IL-17 are under clinical investigation, namely brodalumab (AMG-827), secukinumab (AIN457), and ixekizumab (LY2439821). Secukinumab and ixekizumab are human monoclonal antibodies against IL-17A,73,74 whereas brodalumab is a fully human monoclonal antibody that targets IL-17RA.75
Brodalumab
As referred, brodalumab (previously designated as AMG-827) targets IL-17RA, preventing its activation by IL-17A, IL-17A/F heterodimer, IL-17F, and IL-17E (IL-25).75 In a placebo-controlled Phase I study (Table 1), 25 patients with moderate-to-severe psoriasis were randomized to receive a single subcutaneous (SC) dose of brodalumab 140 mg (n=4), a single SC dose of 350 mg (n=8), a single intravenous (IV) dose of 700 mg (n=8), or placebo (n=5), and were followed for 85 days. Two weeks after receiving a single dose of 350 mg SC or 700 mg IV, the patients showed a dose-dependent improvement in PASI (Psoriasis Area and Severity Index) score and on the static Physician Global Assessment. The eight subjects who received brodalumab 700 mg IV achieved a 50% improvement in PASI (PASI 50) by day 29; at day 43, 88% achieved PASI 75 and 38% reached PASI 90. During the study, 75% and 62.5% of patients receiving 350 mg SC achieved PASI 50 and PASI 75, respectively. PASI 50 was achieved by two of the four patients who received brodalumab 140 mg SC. None of the placebo subjects achieved PASI 50.76
 | Table 1 Randomized, placebo-controlled clinical trials of brodalumab in patients with psoriasis |
The SC 350 mg and IV 700 mg cohorts also showed significant reductions in epidermal thickening, keratin 16 levels, and Ki67-expressing cells. Skin lesions in these cohorts showed an improvement in mRNA levels of several IL-17-modulated keratinocyte-derived factors (eg, cathelicidin, keratin 16, CCL18, and CCL20) and cytokines known to be directly regulated by IL-17R (IL-22, IL-23A, IL-12B). IL-17A, IL-17C, and IL-17F mRNA levels were reduced to nonlesional levels during 6 weeks of treatment. The safety profiles for the subjects who received brodalumab and placebo were similar.76 In this study, a single IV dose of brodalumab up to 700 mg produced substantial improvements in clinical and histopathologic variables, with an acceptable safety profile.
In a randomized, double-blind, placebo-controlled, dose-ranging Phase II study (Table 1), involving 198 adult patients with moderate-to-severe psoriasis, placebo or brodalumab 70, 140, or 210 mg was administered SC on day 1 and at weeks 1, 2, 4, 6, 8, and 10, or at a dose of 280 mg SC on day 1 and at weeks 4 and 8. At week 12, brodalumab reduced the PASI scores by 45.0% (70 mg), 85.9% (140 mg), 86.3% (210 mg), and 76.0% (280 mg), while improvement in PASI on placebo was 16.0% (P<0.001 for all comparisons with placebo). At week 12, PASI 75 and PASI 90 was observed in 77% and 72% of patients receiving brodalumab 140 mg, respectively; 82% and 75% of those receiving 210 mg had PASI 75 and PASI 90, respectively; and the placebo group showed no improvement (P<0.001 for all comparisons).75
Improved values for static Physician Global Assessment, Dermatology Quality of Life Index, and the Medical Outcomes Study 36-Item Short-Form Health Survey (SF-36) were also reported for the 140 mg and 210 mg groups when compared with placebo.75 Moreover, at week 12, subjects receiving brodalumab showed significant improvements in mean Psoriasis Symptom Inventory total score when compared with placebo (8.5 [70 mg], 15.8 [140 mg], 16.2 [210 mg], 12.7 [280 mg], and 4.8 [placebo]), and all the eight item scores improved significantly in patients on brodalumab.77 These data confirm that brodalumab is associated with improvement of the disease from the patient point of view, with an increase in quality of life also reported.
Endres et al studied the pharmacokinetics of brodalumab and assessed the effects of covariates at the doses used in the Phase I and Phase II clinical studies. For doses between 140 mg and 210 mg, they reported that the area under the curve was more than two-fold higher in subjects weighing less than 75 kg as compared with reference subjects; age and diagnosis had a smaller influence on exposure, and were not clinically significant. Their data suggest that body weight is an important covariate, explaining some of the pharmacokinetic variability observed in human clinical trials with brodalumab.78
Recent data show that blockade of IL-17 signaling by brodalumab in psoriatic skin leads to rapid transcriptional changes, initially in keratinocyte-expressed genes, followed by normalization of leukocyte abnormalities, and also established the essential role of IL-17R in keratinocytes by driving the pathogenesis of psoriasis.79 IL-17A has an important role in host defense, so its inhibition raises concern about a potential risk of serious infection and other immune-mediated diseases.
Studies in animal models showed that IL-17A contributes to host defense and to the expression of antimicrobial peptides;46,80,81 however, absence of IL-17A has not been associated with infection or autoimmune manifestations,80 suggesting that inhibition of IL-17 has a low potential for adverse effects on the immune system. In the clinical trials with secukinumab, ixekizumab, and brodalumab, infection rates were only slightly higher than those seen with placebo or than the rates expected for a comparable patient population. Indeed, in the Phase II study reported by Papp et al, two cases of grade 3 neutropenia were found in the 210 mg brodalumab group; the adverse events reported more frequently in the brodalumab groups were nasopharyngitis (8%), upper respiratory tract infection (8%), and injection site erythema (6%).75
The costs of treating psoriasis with biologics are high, but their contribution to resolution of lesions and improved quality of life, especially in patients who are currently undertreated, might reduce the need for inpatient care and improve the outcome for patients. Indeed, according to Schmitt and Ford, the indirect costs of productivity lost exceed the direct costs, which may justify the use of more expensive medications.82
It is important to note that psoriasis is often associated with several comorbidities, in particular cardiovascular disease. Several risk factors for cardiovascular events have been reported in psoriasis, and some treatments for the disease appear to enhance the cardiovascular risk profile. IL-17 has been reported to be associated with atherosclerosis by favoring chronic vascular inflammation,83,84 so might contribute to the increased risk of cardiovascular disease found in patients with psoriasis. Brodalumab, and eventually the other anti-IL-17 agents, decrease vascular inflammation by blocking IL-17 signaling, and may have a positive effect on the risk of cardiovascular events by reducing the development of atherosclerotic lesions.
Nonetheless, the efficacy and safety profile of brodalumab needs to be evaluated in larger clinical trials of longer duration. Currently, Phase III clinical trials for brodalumab are ongoing.
Conclusion
Advances in our understanding of the pathology of psoriasis have allowed the development of new therapeutic strategies and the prospect of new agents, especially for severe cases and nonresponders with psoriasis. For instance, anti-TNF agents are important therapeutic options for psoriasis, but for patients in whom response is lost or does not exist, other treatment options are required. These options must have a good safety profile, with few effects in the immune system.
Considering that IL-17 signaling has a crucial role in the pathogenesis of psoriasis, its inhibition has emerged as a critical target. Phase III clinical trials for brodalumab, secukinumab and ixekizumab, agents that target IL-17 signaling, are ongoing. While secukinumab and ixekizumab target IL-17A, brodalumab targets IL-17RA, raising the question as to whether there are differences in antagonizing IL-17A or the IL-17 receptor.
So far, brodalumab has demonstrated a favorable safety and tolerability profile, robust clinical activity, significant improvements in PASI and other scores for psoriasis severity, and a great impact on patient quality of life. Data for its long-term efficacy and safety will be provided by the Phase III clinical trial that is ongoing. Considering the published data, brodalumab appears to be a potential tool for use in the treatment of moderate-to-severe psoriasis.
Disclosure
The authors report no conflicts of interest in this work.
References
Murphy M, Kerr P, Grant-Kels JM. The histopathologic spectrum of psoriasis. Clin Dermatol. 2007;25(6):524–528. | |
Coimbra S, Oliveira H, Reis F, et al. Health-related quality of life in Portuguese psoriatic patients: relation with Psoriasis Area and Severity Index and different types of classical psoriatic treatment. J Dermatol. 2011;38(8):816–819. | |
Naldi L, Gambini D. The clinical spectrum of psoriasis. Clin Dermatol. 2007;25(6):510–518. | |
Gottlieb SL, Gilleaudeau P, Johnson R, et al. Response of psoriasis to a lymphocyte-selective toxin (DAB389IL-2) suggests a primary immune, but not keratinocyte, pathogenic basis. Nat Med. 1995;1(5):442–447. | |
Sabat R, Philipp S, Hoflich C, et al. Immunopathogenesis of psoriasis. Exp Dermatol. 2007;16(10):779–798. | |
Arican O, Aral M, Sasmaz S, Ciragil P. Serum levels of TNF-alpha, IFN-gamma, IL-6, IL-8, IL-12, IL-17, and IL-18 in patients with active psoriasis and correlation with disease severity. Mediators Inflamm. 2005;2005(5):273–279. | |
Szegedi A, Aleksza M, Gonda A, et al. Elevated rate of T helper1 (T(H)1) lymphocytes and serum IFN-gamma levels in psoriatic patients. Immunol Lett. 2003;86(3):277–280. | |
Lowes MA, Lew W, Krueger JG. Current concepts in the immunopathogenesis of psoriasis. Dermatol Clin. 2004;22(4):349–369. | |
Jacob SE, Nassiri M, Kerdel FA, Vincek V. Simultaneous measurement of multiple Th1 and Th2 serum cytokines in psoriasis and correlation with disease severity. Mediators Inflamm. 2003;12(5):309–313. | |
Ghoreschi K, Thomas P, Breit S, et al. Interleukin-4 therapy of psoriasis induces Th2 responses and improves human autoimmune disease. Nat Med. 2003;9(1):40–46. | |
Di Cesare A, Di Meglio P, Nestle FO. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol. 2009;129(6):1339–1350. | |
Coimbra S, Oliveira H, Reis F, et al. Interleukin (IL)-22, IL-17, IL-23, IL-8, vascular endothelial growth factor and tumour necrosis factor-alpha levels in patients with psoriasis before, during and after psoralen-ultraviolet A and narrowband ultraviolet B therapy. Br J Dermatol. 2010;163(6):1282–1290. | |
Kimball AB, Gordon KB, Fakharzadeh S, et al. Long-term efficacy of ustekinumab in patients with moderate-to-severe psoriasis: results from the PHOENIX 1 trial through up to 3 years. Br J Dermatol. 2012;166(4):861–872. | |
Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201(2):233–240. | |
Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171(11):6173–6177. | |
Annunziato F, Romagnani S. Heterogeneity of human effector CD4+ T-cells. Arthritis Res Ther. 2009;11(6):257. | |
Annunziato F, Cosmi L, Santarlasci V, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204(8):1849–1861. | |
Volpe E, Servant N, Zollinger R, et al. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9(6):650–657. | |
Wilson NJ, Boniface K, Chan JR, et al. Development, cytokine profile and function of human interleukin 17-producing helper T-cells. Nat Immunol. 2007;8(9):950–957. | |
Vanden Eijnden S, Goriely S, De Wit D, Willems F, Goldman M. IL-23 up-regulates IL-10 and induces IL-17 synthesis by polyclonally activated naive T-cells in human. Eur J Immunol. 2005;35(2):469–475. | |
Bovenschen HJ, van de Kerkhof PC, van Erp PE, Woestenenk R, Joosten I, Koenen HJ. Foxp3+ regulatory T-cells of psoriasis patients easily differentiate into IL-17A-producing cells and are found in lesional skin. J Invest Dermatol. 2011;131(9):1853–1860. | |
Shin HC, Benbernou N, Fekkar H, Esnault S, Guenounou M. Regulation of IL-17, IFN-gamma and IL-10 in human CD8(+) T-cells by cyclic AMP-dependent signal transduction pathway. Cytokine. 1998;10(11):841–850. | |
Liu SJ, Tsai JP, Shen CR, et al. Induction of a distinct CD8 Tnc17 subset by transforming growth factor-beta and interleukin-6. J Leukoc Biol. 2007;82(2):354–360. | |
He D, Wu L, Kim HK, Li H, Elmets CA, Xu H. CD8+ IL-17-producing T-cells are important in effector functions for the elicitation of contact hypersensitivity responses. J Immunol. 2006;177(10):6852–6858. | |
Rachitskaya AV, Hansen AM, Horai R, et al. Cutting edge: NKT-cells constitutively express IL-23 receptor and RORgammat and rapidly produce IL-17 upon receptor ligation in an IL-6-independent fashion. J Immunol. 2008;180(8):5167–5171. | |
Roark CL, French JD, Taylor MA, Bendele AM, Born WK, O’Brien RL. Exacerbation of collagen-induced arthritis by oligoclonal, IL-17-producing gamma delta T-cells. J Immunol. 2007;179(8):5576–5783. | |
Chang SH, Dong C. A novel heterodimeric cytokine consisting of IL-17 and IL-17F regulates inflammatory responses. Cell Res. 2007;17(5):435–440. | |
Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10(7):479–489. | |
Lin AM, Rubin CJ, Khandpur R, et al. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol. 2011;187(1):490–500. | |
Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21(4):467–476. | |
Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. | |
Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9(8):556–567. | |
Lowes MA, Kikuchi T, Fuentes-Duculan J, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T-cells. J Invest Dermatol. 2008;128(5):1207–1211. | |
Ortega C, Fernandez AS, Carrillo JM, et al. IL-17-producing CD8+ T lymphocytes from psoriasis skin plaques are cytotoxic effector cells that secrete Th17-related cytokines. J Leukoc Biol. 2009;86(2):435–443. | |
Eyerich S, Eyerich K, Pennino D, et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest. 2009;119(12):3573–3585. | |
Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T-cells. N Engl J Med. 2009;361(9):888–898. | |
Cargill M, Schrodi SJ, Chang M, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80(2):273–290. | |
Nair RP, Duffin KC, Helms C, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet. 2009;41(2):199–204. | |
Bohm B, Burkhardt H, Uebe S, et al. Identification of low-frequency TRAF3IP2 coding variants in psoriatic arthritis patients and functional characterization. Arthritis Res Ther. 2012;14(2):R84. | |
Onishi RM, Gaffen SL. Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology. 2010;129(3):311–321. | |
Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity. 2011;34(2):149–162. | |
Pietrzak AT, Zalewska A, Chodorowska G, et al. Cytokines and anticytokines in psoriasis. Clin Chim Acta. 2008;394(1−2):7–21. | |
Watanabe H, Kawaguchi M, Fujishima S, et al. Functional characterization of IL-17F as a selective neutrophil attractant in psoriasis. J Invest Dermatol. 2009;129(3):650–656. | |
Nograles KE, Zaba LC, Guttman-Yassky E, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol. 2008;159(5):1092–1102. | |
Iwakura Y, Nakae S, Saijo S, Ishigame H. The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol Rev. 2008;226:57–79. | |
Harper EG, Guo C, Rizzo H, et al. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Invest Dermatol. 2009;129(9):2175–2183. | |
Zaba LC, Fuentes-Duculan J, Eungdamrong NJ, et al. Psoriasis is characterized by accumulation of immunostimulatory and Th1/Th17 cell-polarizing myeloid dendritic cells. J Invest Dermatol. 2009;129(1):79–88. | |
Gutowska-Owsiak D, Schaupp AL, Salimi M, et al. IL-17 downregulates filaggrin and affects keratinocyte expression of genes associated with cellular adhesion. Exp Dermatol. 2012;21(2):104–110. | |
Shi X, Jin L, Dang E, et al. IL-17A upregulates keratin 17 expression in keratinocytes through STAT1- and STAT3-dependent mechanisms. J Invest Dermatol. 2011;131(12):2401–2408. | |
Carrier Y, Ma HL, Ramon HE, et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J Invest Dermatol. 2011;131(12):2428–2437. | |
Fossiez F, Djossou O, Chomarat P, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996;183(6):2593–2603. | |
Zheng Y, Danilenko DM, Valdez P, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445(7128):648–651. | |
Fitch E, Harper E, Skorcheva I, Kurtz SE, Blauvelt A. Pathophysiology of psoriasis: recent advances on IL-23 and Th17 cytokines. Curr Rheumatol Rep. 2007;9(6):461–467. | |
Numasaki M, Lotze MT, Sasaki H. Interleukin-17 augments tumor necrosis factor-alpha-induced elaboration of proangiogenic factors from fibroblasts. Immunol Lett. 2004;93(1):39–43. | |
Chiricozzi A, Guttman-Yassky E, Suarez-Farinas M, et al. Integrative responses to IL-17 and TNF-alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol. 2011;131(3):677–687. | |
Teunissen MB, Koomen CW, de Waal Malefyt R, Wierenga EA, Bos JD. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. 1998;111(4):645–649. | |
Kagami S, Rizzo HL, Lee JJ, Koguchi Y, Blauvelt A. Circulating Th17, Th22, and Th1 cells are increased in psoriasis. J Invest Dermatol. 2010;130(5):1373–1383. | |
Takahashi H, Tsuji H, Hashimoto Y, Ishida-Yamamoto A, Iizuka H. Serum cytokines and growth factor levels in Japanese patients with psoriasis. Clin Exp Dermatol. 2010;35(6):645–649. | |
Caproni M, Antiga E, Melani L, Volpi W, Del Bianco E, Fabbri P. Serum levels of IL-17 and IL-22 are reduced by etanercept, but not by acitretin, in patients with psoriasis: a randomized-controlled trial. J Clin Immunol. 2009;29(2):210–214. | |
Vahavihu K, Ala-Houhala M, Peric M, et al. Narrowband ultraviolet B treatment improves vitamin D balance and alters antimicrobial peptide expression in skin lesions of psoriasis and atopic dermatitis. Br J Dermatol. 2010;163(2):321–328. | |
Johansen C, Usher PA, Kjellerup RB, Lundsgaard D, Iversen L, Kragballe K. Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br J Dermatol. 2009;160(2):319–324. | |
Hvid H, Teige I, Kvist PH, Svensson L, Kemp K. TPA induction leads to a Th17-like response in transgenic K14/VEGF mice: a novel in vivo screening model of psoriasis. Int Immunol. 2008;20(8):1097–1106. | |
van der Fits L, Mourits S, Voerman JS, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol. 2009;182(9):5836–5845. | |
Singh TP, Schon MP, Wallbrecht K, et al. 8-methoxypsoralen plus ultraviolet A therapy acts via inhibition of the IL-23/Th17 axis and induction of Foxp3+ regulatory T-cells involving CTLA4 signaling in a psoriasis-like skin disorder. J Immunol. 2010;184(12):7257–7267. | |
Nakajima K, Kanda T, Takaishi M, et al. Distinct roles of IL-23 and IL-17 in the development of psoriasis-like lesions in a mouse model. J Immunol. 2011;186(7):4481–4489. | |
Rizzo HL, Kagami S, Phillips KG, Kurtz SE, Jacques SL, Blauvelt A. IL-23-mediated psoriasis-like epidermal hyperplasia is dependent on IL-17A. J Immunol. 2011;186(3):1495–1502. | |
Cai Y, Shen X, Ding C, et al. Pivotal role of dermal IL-17-producing gammadelta T-cells in skin inflammation. Immunity. 2011;35(4):596–610. | |
McGeachy MJ, Chen Y, Tato CM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10(3):314–324. | |
Takahashi H, Tsuji H, Ishida-Yamamoto A, Iizuka H. Comparison of clinical effects of psoriasis treatment regimens among calcipotriol alone, narrowband ultraviolet B phototherapy alone, combination of calcipotriol and narrowband ultraviolet B phototherapy once a week, and combination of calcipotriol and narrowband ultraviolet B phototherapy more than twice a week. J Dermatol. 2013;40(6):424–427. | |
Johnson-Huang LM, Suarez-Farinas M, Sullivan-Whalen M, Gilleaudeau P, Krueger JG, Lowes MA. Effective narrow-band UVB radiation therapy suppresses the IL-23/IL-17 axis in normalized psoriasis plaques. J Invest Dermatol. 2010;130(11):2654–2663. | |
Zaba LC, Suarez-Farinas M, Fuentes-Duculan J, et al. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J Allergy Clin Immunol. 2009;124(5):1022–1010. e1–e395. | |
Tausend W, Downing C, Tyring S. Systematic review of interleukin-12, interleukin-17, and interleukin-23 pathway inhibitors for the treatment of moderate-to-severe chronic plaque psoriasis: ustekinumab, briakinumab, tildrakizumab, guselkumab, secukinumab, ixekizumab, and brodalumab. J Cutan Med Surg. 2014;18(3):156–169. | |
Hueber W, Patel DD, Dryja T, et al. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med. 2010;2(52):52ra72. | |
Genovese MC, Van den Bosch F, Roberson SA, et al. LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: a Phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum. 2010;62(4):929–939. | |
Papp KA, Leonardi C, Menter A, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med. 2012;366(13):1181–1189. | |
Papp KA, Reid C, Foley P, et al. Anti-IL-17 receptor antibody AMG 827 leads to rapid clinical response in subjects with moderate to severe psoriasis: results from a Phase I, randomized, placebo-controlled trial. J Invest Dermatol. 2012;132(10):2466–2469. | |
Gordon KB, Kimball AB, Chau D, et al. Impact of brodalumab treatment on psoriasis symptoms and health-related quality of life: use of a novel patient-reported outcome measure, the Psoriasis Symptom Inventory. Br J Dermatol. 2014;170(3):705–715. | |
Endres CJ, Salinger DH, Kock K, et al. Population pharmacokinetics of brodalumab in healthy adults and adults with psoriasis from single and multiple dose studies. J Clin Pharmacol. Epub 2014 May 20. | |
Russell CB, Rand H, Bigler J, et al. Gene expression profiles normalized in psoriatic skin by treatment with brodalumab, a human anti-IL-17 receptor monoclonal antibody. J Immunol. 2014;192(8):3828–3836. | |
Puel A, Cypowyj S, Bustamante J, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011;332(6025):65–68. | |
Cho JS, Pietras EM, Garcia NC, et al. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J Clin Invest. 2010;120(5):1762–1773. | |
Schmitt JM, Ford DE. Work limitations and productivity loss are associated with health-related quality of life but not with clinical severity in patients with psoriasis. Dermatology. 2006;213(2):102–110. | |
Chen S, Crother TR, Arditi M. Emerging role of IL-17 in atherosclerosis. J Innate Immun. 2010;2(4):325–333. | |
Erbel C, Dengler TJ, Wangler S, et al. Expression of IL-17A in human atherosclerotic lesions is associated with increased inflammation and plaque vulnerability. Basic Res Cardiol. 2011;106(1):125–134. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.