")
Back to Archived Journals » Antibody Technology Journal » Volume 6
Broadly neutralizing antibodies for therapy of viral infections
Authors Srinivasan S, Ghosh M, Maity S, Varadarajan R
Received 10 July 2015
Accepted for publication 29 October 2015
Published 8 January 2016 Volume 2016:6 Pages 1—15
DOI https://doi.org/10.2147/ANTI.S92190
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shixia Wang
Sankaranarayanan Srinivasan,1 Maloy Ghosh,1 Sunit Maity,1 Raghavan Varadarajan2
1Theramyt Novobiologics Private Limited, 2Molecular Biophysics Unit, Indian Institute of Science, Bangalore, India
Abstract: Neutralizing antibodies (NAbs) are an essential part of the human immune response that involves an intricate relationship between the innate and adaptive immune system to prevent infection. The appearance of NAbs is a hallmark of viral infection in patients with HIV, dengue, hepatitis C virus, Ebola, and influenza. These viruses are characterized by high genetic diversity of viral epitopes arising due to a high replication rate and an error-prone replication machinery. In general, almost all infected individuals develop strain-specific NAbs in a fairly short period of time. In contrast, broadly neutralizing antibodies (bNAbs) show subdominant responses and are found only in a subset of patients, typically after a lengthy period of infection. Epitopes targeted by specific NAbs and bNAbs evolved thereafter provide useful information for vaccine design. In this review, we discuss the isolation and utility of bNAbs against HIV-1, dengue, hepatitis C virus, influenza, and Ebola. Passive antibody therapy and the economics of NAb therapy are also discussed.
Keywords: neutralizing antibodies, HIV, dengue, hepatitis C, influenza
Introduction
Some antiviral antibodies perform their function by binding to antigens on the viral surface and signaling to white blood cells (WBCs) for viral destruction. Subsequently, these antigens are processed and destroyed. Neutralizing antibodies (NAbs) prevent viral entry by binding to regions on the virus involved in the entry process. Of these, in the case of viral infections, broadly neutralizing antibodies (bNAbs) can recognize a wide variety of viral glycoproteins (GPs) on the surface of enveloped viruses or the protein shell of nonenveloped viruses. Many viruses, including HIV-1, dengue, influenza, hepatitis C virus (HCV), and Ebola, evade the antibody response through mutations in their surface GPs. This has proved to be a major obstacle in the development of therapies and vaccines for such viruses. However, the recent isolation of bNAbs, from infected individuals that can bind to conserved regions of these viruses and neutralize them, is an encouraging development. The major relevance of isolating these bNAbs is that their epitopes can provide valuable information for vaccine development. Furthermore, in some cases, passive immunization with such antibodies can both prevent and help clear existing infection.
bNAbs inhibit infection through: 1) blocking binding to cell surface receptors;1,2 and 2) inhibition of the viral fusion machinery for enveloped viruses or penetration for nonenveloped viruses. In vitro studies with bNAbs are carried out using susceptible cells and infectious viruses. In some cases, it is still unclear if binding to any site on a functional viral spike is sufficient to prevent infection or if neutralization requires binding to specific critical sites on the viral surface.2,3 In some special cases of neutralization, antibodies target host molecules that are displayed either on the surface of viral particles, such as human leukocyte antigen and ICAM-1 molecules for HIV-1,4,5 or viral receptors on host cells. Only a fraction of memory B-cells contribute to the pool of bNAbs as a majority of antibodies are made toward denatured or internal proteins.6 A desirable feature for an effective vaccine candidate would be the ability to elicit a high proportion of bNAbs.
Viral serotypes are determined by the immunogenic and variable regions of viral GPs.7 Diversification of these regions enables the virus to adapt to the host and evade the immune system. The size of the receptor binding site is determined by the ligands that it binds, which include sugar moieties, proteins, and glycolipids. However, the antibody binding site is mostly constant around 700 Å2.1 Consequently for a small receptor binding site, mutations of surrounding residues that form part of the neutralizing epitope can lead to immune evasion. In the case of viruses that undergo fusion, tight packing of the viral GPs leads to their poor accessibility for neutralization activity.
In this review, we discuss some recent work on the isolation and characterization of bNAbs including several preclinical and clinical candidates for treatment of HIV-1, dengue, HCV, Ebola, and influenza. Additionally, the business case of bNAb therapy and passive antibody treatment are addressed.
NAbs for therapy (AIDS, dengue, and HCV)
Binding antibodies bind to antigens without necessarily preventing entry. However, through binding, antigens are marked through processes mediated via antibody effector functions, for processing by WBCs. This is in contrast to bNAbs that bind at epitopes, which is important in the entry process. While in principle, bNAbs can exert important biological effects without the need for further interaction with WBCs, in practice both neutralizing and effector functions of bNAbs contribute to their biological efficacy in vivo. Both these antibody functionalities aid the immune system to control and prevent viral infection. The human immune system is capable of developing potent NAbs to restrict viral infection, but the transition from strain-specific to bNAbs is not well understood. The process is likely to be constrained by factors such as appropriate germ line availability, autoreactivity, epitope exposure, immune dominance, etc. In addition, duration of infection also affects development of bNAbs as longer infection time produces long-term antigenic stimulation and allows for multiple rounds of antibody maturation. One of the biggest barriers to elicitation of bNAbs is that many viruses show extreme variability of their surface antigens with rapid emergence of resistant mutants. As a result, it is very important that bNAbs that are either in preclinical or clinical development demonstrate a broad level of effectiveness in potently neutralizing genetically diverse viral isolates. Such effectiveness of bNAbs is greatly needed, especially with respect to viruses such as HIV-1, dengue, and HCV that show wide genetic diversity among strains isolated from various patients.
bNAbs against HIV-1
HIV-1 is part of the Lentivirus genus and belongs to the retroviridae family with RNA as its genetic material. HIV-1 entry starts with binding of the envelope GP (Env) gp120 to the CD4 receptor on macrophages and CD4+ T-cells. Subsequently, a co-receptor-binding site is formed in gp120 through extensive conformational changes to enable binding to a co-receptor that includes either C–C chemokine receptor type 5 or C–X–C chemokine receptor type 4. This is followed by substantial structural rearrangements in Env gp41 that finally cause virus–cell fusion and injection of the viral RNA into target cells.8 Of the currently available treatments for HIV infection, a highly active antiretroviral therapy (HAART) or “cocktail therapy” includes two nucleoside/nucleotide reverse-transcriptase inhibitors (NRTIs/NtRTIs) plus a non-NRTI or a protease inhibitor (PI) or another NRTI. Although this method is highly efficient in reducing the number of HIV-1 particles in the bloodstream to undetectable levels and providing clinical benefits, it can also lead to adverse effects and drug-resistant mutants. In addition, drugs currently need to be taken lifelong as they do not completely eliminate the virus from the system.
HIV-1 exhibits extensive genetic diversity for the Env gene between clades, within clades, and also in the same infected individual. Different HIV-1 strains show variable responses to antibody-mediated neutralization, leading them to be classified into three tiers, with Tier1 being the most sensitive and Tier3 the most resistant.9 The first-generation bNAbs for HIV include 2G12, targeting a complex gp120-glycan,10 2F5, Z13, m66, 4E10, specific for the membrane-proximal external region (MPER) of gp41,11 and b12 targeting the CD4 binding site (CD4bs). These antibodies had restricted neutralization breadth and potency.12 This was partly due to limited accessibility as well as variation of the corresponding epitopes on the viral surface.
In recent years, a combination of different approaches involving: 1) novel screening and selection; 2) efficient methods to isolate human monoclonal antibodies (mAbs); and 3) identifying chronically infected individuals possessing potent, and cross-clade reactive serum antibodies have led to the development of the second generation of human monoclonal bNAbs for HIV-113 with improved breadth and potency. Epitope regions of a few bNAbs mapped on the crystal structure of the ectodomain of the HIV-1 Env trimer (Protein Data Bank ID: 4TVP14) are shown in Figure 1. PG9 and PG16 (Figure 1A) bNAbs neutralize 75%–80% of isolates by recognizing and binding a quaternary proteoglycan epitope on gp120. These were developed by isolating antibodies from clade-A-infected donors.15 A pseudovirus-based neutralization assay to select mAbs from in vitro activated memory B- cells was used here. Employing a similar approach, four sets of clonally related antibodies PGT121–123, PGT125–131, PGT135–137, and PGT141–145 have also been developed. These antibodies show higher potency than PG9 and a comparable breadth.16 All of these antibodies show a slightly different mode of glycan recognition for gp120 sugars. PGT121 binds to complex-type N-glycans near the V3 loop of gp120 (Figure 1A), whereas PG9 binds to a proteoglycan epitope through an extended CDR3 region of the heavy chain. This is characterized by a hammerhead-like conformation penetrating N156 and N160 glycans to reach strand C of the V1/V2 region (Figure 1A). PGT128 interacts with high mannose N-glycans near the V3 loop17,18 (Figure 1A). In another study, four bNAbs CH01–CH04 with similar specificity to PG9 and PG16 were isolated (Figure 1A). However, they demonstrated more limited breadth of neutralization.19
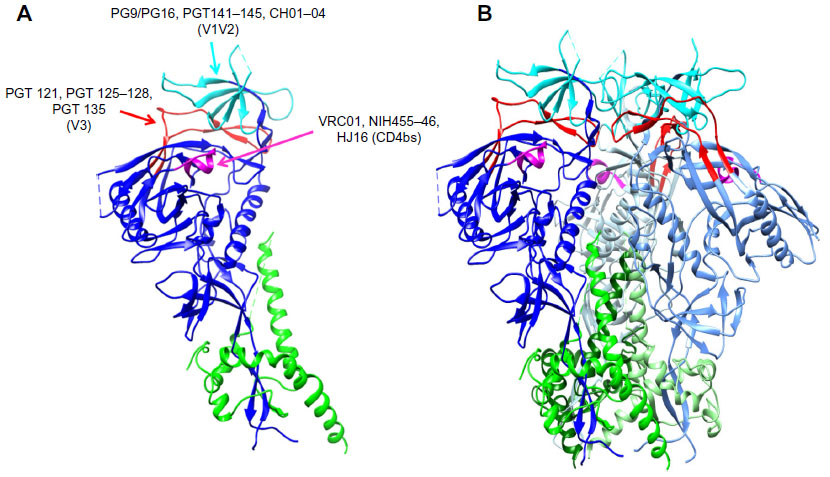 | Figure 1 Binding sites for bNAbs on HIV-1 envelope. |
Some unusual genetic features of bNAbs are the presence of high levels of somatic hypermutation and selective germ line gene usage. Another critical feature is the presence of long CDRH3s for antibodies that penetrate the glycan shield, as well as polyreactivity for some MPER antibodies. Multiple rounds of affinity maturation are required for hypermutation of bNAbs to acquire breadth, which leads to neutralization of autologous, followed by heterologous viruses. Recently, a promising bNAb 10E8, which neutralizes almost 98% of viruses with minimal cross-reactivity, has been identified. This binds to conserved residues in the membrane proximal region of gp41.20 Using an immortalization method, bNAb HJ16 was isolated. This binds to the CD4bs epitope on HIV-1 and is especially effective against Tier2 viruses21 (Figure 1A). The mature VRC01 CD4bs-directed antibody neutralizes more than 90% of clinical HIV isolates. Similar to VRC01, three other potent antibodies that target the CD4bs, 3BNC117, 3BNC60, and NIH45-46, have been identified.22,23 bNAbs such as 3BC176, 3BC315, PGT145, and PGT151 that specifically target intact Env trimers have been isolated. PGT141–145 bind the V1/V2 loop (Figure 1A). Some of the above bNAbs bind to a neutralizing epitope in the proximity of the V3 loop and the CD4 interaction site, while others such as PGT151 bind an epitope that is close to the gp120:gp41 interface. These bNAbs neutralize 60%–70% of HIV-1 isolates.24 Figure 1A and B depict the binding sites of various NAbs. Overall it has been suggested that using a cocktail of potent and bNAbs (specifically NIH45-46, PG16, PGT128, 10-1074, and 3BC176) would provide an alternative to or complement small-molecule antiretroviral therapy by acting in a synergistic manner.24 However, at present this does not appear to be an economically feasible alternative to antiretrovirals.
Recently, the results of a first dose escalation Phase I clinical trial of 3BNC117, a potent human CD4bs antibody, in uninfected and HIV-1 infected individuals were reported.25 3BNC117 infusion was well tolerated and demonstrated favorable pharmacokinetics. In this trial, uninfected and HIV-infected individuals were intravenously given a single dose of the antibody and monitored for 56 days. At the highest dosage level tested in the study, 30 mg/kg of body weight, all eight infected individuals treated showed up to a 300-fold decrease in the amount of virus measured in their blood, with most reaching their lowest viral load in 7–21 days after treatment.25 The viral load did not completely rebound to pretreatment levels even after 56 days of observation in a few patients. This human study also revealed that HIV-1 developed a high level of resistance to 3BNC117 in certain individuals. Analysis of HIV-1 isolated from these patients indicates critical mutations in the envelope sequences known to alter CD4 binding. The study concluded that monotherapy with 3BNC117 would not provide complete viremic control. Therefore, it was suggested to use antibody–drug and/or antibody–antibody combination treatment to achieve better clinical outcomes.24 Table 1 categorizes bNAbs against HIV.
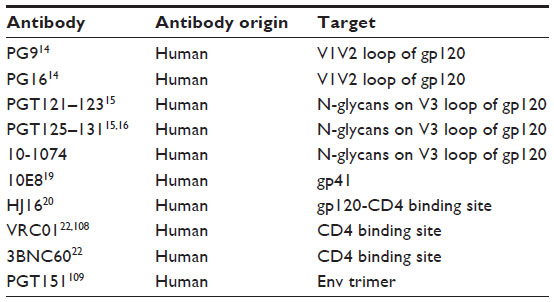 | Table 1 bNAbs against HIV and their corresponding epitopes |
bNAbs against dengue virus
Dengue virus (DENV) belongs to the Flavivirus genus and comprises of four serotypes 1–4. It is the cause of dengue disease, infecting 50–100 million people worldwide.26 It has three major proteins: capsid (C), premembrane (prM), and envelope (E) protein, with the E and prM proteins being the main targets of the humoral immune response.27 The prM protein is involved in assembly of the viral RNA with E protein and association with membranes derived from the endoplasmic reticulum. The E protein undergoes conformational changes and promotes fusion with the endosomes after viral entry. Another protein, NS1, that is expressed after replication is also a target of the immune response. The E GP with its distinctive domains is not only the primary determinant of the four serotypes of DENV, but also the sole target of NAbs.28 It mediates the fusion of the viral and cellular membranes during viral entry. Domain I at the N-terminus possesses glycosylation sites, and Domain II has a conserved fusion loop undergoing structural rearrangements at a low pH. Overlapping epitopes are present in the hinge region of Domains I and II.28 The β barrel-type Domain III is also known to participate in receptor binding and plays an important role in defining the DENV serotype-specific epitopes. It is also a major target for NAbs.29 There are currently no dengue-specific antiviral drugs. Domain II residues 98–110, which are exposed at low pH, form a fusion loop that binds a NAb 2A10G6. 2A10G6 acts by inhibiting fusion with the virus30 (Figure 2A). Another mouse antibody 4G2 that also inhibits viral fusion is known to have neutralizing effects on patients of all the four serotypes.31 Antibody 4.8A that was isolated from a single human donor showed potent neutralizing activity against both DENV-1 and -3.32
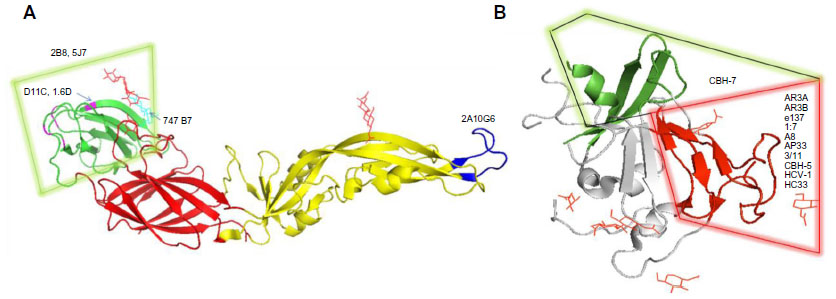 | Figure 2 NAb epitopes on dengue and HCV. |
Similar to 4.8A, antibodies D11C and 1.6D also show broad neutralizing activity against DENV-1 and -3 serotypes32 (Figure 2A). They act by inhibiting viral fusion. Domain III residues 307, 330, 332, 383, and 384 have been shown to be important for recognizing NAbs. Ab 2B8 recognizes Domain III of DENV E protein and inhibits viral binding30 (Figure 2A).
Four new NAbs 747(4) A11, 747 B7, 752-2 C8, and 753(3) C10 targeting the E GP have recently been isolated from patients.33,34 The first two antibodies A11 and B7 (Figure 2A) belong to the E-dimer-dependent epitope 2 group (Ede2) and were isolated from a single patient infected with the DENV-2 serotype. Antibodies of the subgroup Ede2 require glycosylation of the E protein at position 153 for efficient binding. These are somatic variants of immunoglobulin-G (IgG) clones, derived from the IGHV3-74 and IGLV2-23 germ lines. Ab C8 belongs to the Ede1 group with efficient binding regardless of glycosylation at N153. The IgG clones are derived from VH and VL genes IGHV3-64 and IGKV3-11. The serotype of the infecting virus was not determined. Ab C10 also belongs to the Ede1 group with clones derived from the IGHV1-3 and IGLV2-14 germ lines. They were isolated from a patient with secondary infection of the DENV-1 serotype.
A very potent human antibody 5J7 (Figure 2A) has been isolated in a recent study and can neutralize DENV-3 serotypes at nanomolar concentrations. The cryoelectron microscopy structure of 5J7 with the E protein indicates that a single Fab molecule binds across three envelope proteins and thus engages three functionally important domains, each from a different envelope protein. These domains are critical for receptor binding and fusion to the endosomal membrane.34 Very recently, a structure-based design has been used to engineer a bNAb against all four subtypes of dengue.35 Table 2 categorizes bNAbs against DENV.
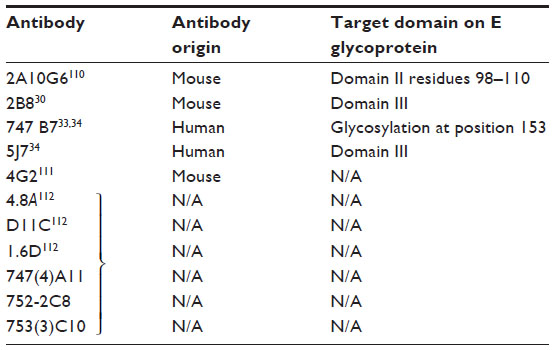 | Table 2 bNAbs against DENV and their corresponding epitopes |
bNAbs against HCV
HCV belongs to the Flaviviridae family and is a positive-strand RNA virus. One of the major causes of chronic liver disease, affecting an estimated 170 million people all over the world, is HCV.36 Murex HCV serotyping classifies them into six serotypes37 of which genotype 1 HCV is the most prevalent one. Current treatment options include HCV/NS3 PIs and interferon-based therapy. Both these therapies have disadvantages with regard to the many side effects associated with interferons, and with the PIs being limited to patients infected with genotype 1.38,39 Small molecules including nucleoside and nucleotide NS5B polymerase inhibitors such as Sovaldi® (Gilead, Foster City, CA, USA) have provided relief across all genotypes. Given the success of these small-molecules inhibitors, bNAbs that can target the HCV GP E2 would not be the first therapeutic option. Nevertheless they might be useful for drug-resistant mutants and more importantly as tools to guide vaccine development. An HCV vaccine is important to prevent primary infection as well as recurrence of infection, especially in resource-poor settings.40 Studies in chimpanzees suggested immunization with E1/E2 Env and has the ability to protect the animal from autologous HCV infection, albeit partially.41 Recent studies in humans reported that most patients failed to develop NAbs and progressed to chronic infection, whereas in certain patients the virus was cleared spontaneously after 62 weeks of infection.42 The only E1-specific bNAbs currently available are IGH505 and IGH526, which neutralize HCV infection by targeting a 15 amino acid region near the C-terminus of E1.38 These are human in origin, and neutralization was characterized by in vitro assays.
The HCV GP E2 plays a major role in infection by forming noncovalent homodimers on the surface and mediating viral endocytosis through interaction with various cellular receptors. The E2 ectodomain has three variable regions that are under constant selection pressure and tolerate great genetic diversity. In particular, the C-terminal region of hypervariable region 1 is a target for genotype-restricted NAbs.43,44 The E2 protein is mainly globular and the surface features include a neutralizing face, containing cross-reactive and strain-restricted (variable) epitopes, a highly glycosylated face, and regions believed to be occluded on the intact virus particle.43
A majority of bNAbs are directed against conformational epitopes for the CD81 binding sites on HCV/E2. Particularly, the domain on E2, encompassing residues 529, 530, and 535, is considered as the core CD81 binding region that is targeted by many bNAbs. bNAbs e137, AR3A, CBH-5, 1:7, and A8 are some well-characterized potent candidates for mAb-based therapy for HCV infection that target this core CD81 binding region44,45 (Figure 2B). All these antibodies possess cross-neutralizing activity. Other bNAbs are targeted toward conserved linear epitopes of E2 encompassing amino acids 412–423, which bind the murine NAb AP33 and 3/11. Human mAbs HCV146 and HC33 bind to linear epitopes of the E2 GP. Antibodies targeting this region (412–423) of E2 are capable of potent and broad neutralization activities. However, their differential neutralizing targeting breadth and potency can be explained on the basis of their distinct but overlapping epitopes.47
A recently isolated antibody HC33.1 (Figure 2B) from an HCV-infected blood donor can neutralize the virus bearing the E2 Asn 417Ser and Asn417Thr adaptive mutations with the glycosylation shift to Asn 415. This antibody is able to penetrate the glycan shield of the HCV E2 protein by disrupting the β-hairpin formed by residues 412–423.48 This is possible due to the unusually long CDR3 region of the HC33.1 heavy chain.49 A recent report has demonstrated the use of an adenoviral vector to abrogate HCV infection in hepatocytes by expressing the bNAbs AR3A, AR3B, and AR4A.47 AR4A is believed to interact with the E2 membrane proximal region that is involved in membrane fusion. Table 3 categorizes bNAbs against HCV.
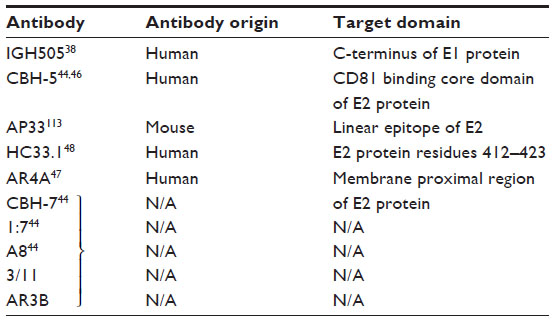 | Table 3 bNAbs against HCV and their corresponding epitopes |
NAbs for Ebola virus
Ebola is a filamentous pleiomorphic virus of the Filoviridae family, with a negative-sense RNA genome that causes severe hemorrhagic fever with high lethality. There are five different classes of Ebola viruses (EBOVs), each named after the location of the outbreak where it was first discovered: EBOV (formerly known as Zaire EBOV), Sudan virus, Reston virus, Taï Forest virus, and Bundibugyo virus.50,51 These viral sequences differ by 40%–50% in their overall primary amino acid sequence and are antigenically distinct. Among all these species, Zaire EBOV is the most common and the most lethal. Survival of EBOV infection appears to depend on the ability of the host to mount an early and strong immune response. It has been observed that even those people that survive EBOV infection often have low-to-insignificant titers of NAbs.50,52
The trimeric, membrane-attached GP1,2 is the only virally encoded protein on the surface and is crucial for viral adhesion, entry, and internalization into host cells. In infected cells, GP1,2 is cleaved by furin yielding two subunits, GP1 and GP2, that remain disulfide linked. GP1 is responsible for recognition and engagement of new target cells, whereas GP2 drives fusion of the viral and endosomal membrane of the target cell. Antibodies against EBOV GP1,2 have been tested for protection against EBOV challenge in different models with mixed outcomes, although a recent study has been quite promising.53
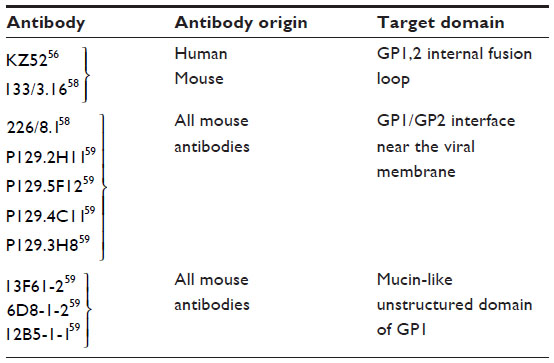
A promising NAb for EBOV was identified from the bone marrow of a human survivor of the Zaire outbreak (Zaire EBOV species). This mAb, termed KZ52, neutralizes Zaire EBOV in vitro54 and offers protection from lethal EBOV challenge in a rodent model.55 KZ52 is specific for the complex of GP1 and GP2 together56 (Figure 3). A murine antibody, termed 133/3.16, neutralizes and protects mice (5/5) from a lethal challenge of EBOV even 2 days postexposure.57 This antibody recognizes a conformation-dependent epitope that could overlap with that of the KZ52 bNAb.58
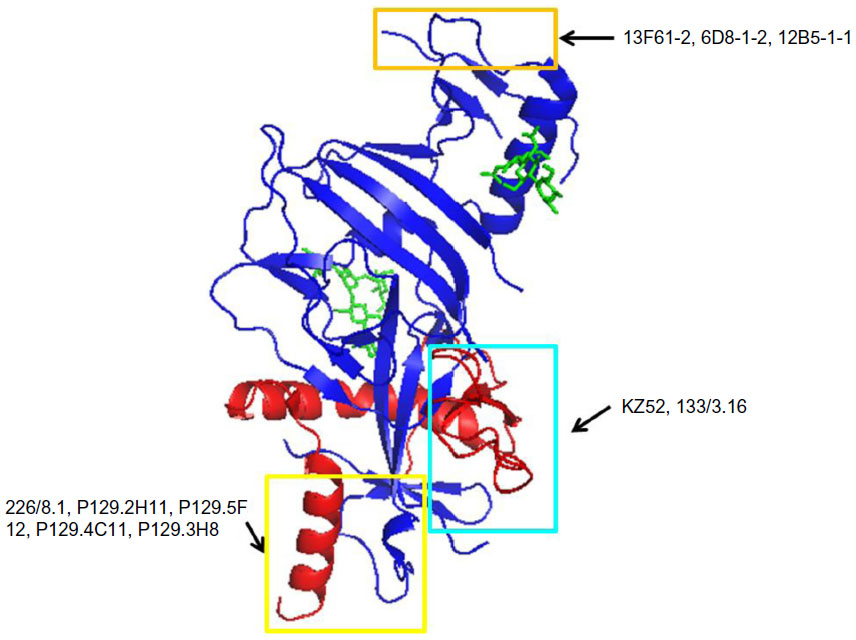 | Figure 3 Binding sites for bNAbs on EBOV GP. |
Antibody 226/8.1, directed toward GP1, was shown to confer protection in mice with a single passive immunization.57 Two other mAbs, 13C6-1-2 and 6D3-1-1, recognize conformation- dependent epitopes shared between soluble GP and GP.59 Another set of antibodies P129.2H11, P129.5F12, P129.4C11, and P129.3H8 are directed against GP1 and bind to the epitopes that overlap with that of mAb 226/8.159 (Figure 3).
The mucin-like domain of GP is predicted to be mostly unstructured and highly N- and O-glycosylated. A set of antibodies have been identified that react with glycans in this domain. They are divided into three groups. Group 1 includes the antibody 13F6-1-2, and is directed against GP1 residues 401–417. Group 2 includes the antibody 6D8-1-2, and is directed against GP1 residues 389–405. Group 3, which includes the antibody 12B5-1-1, is directed against GP1 residues 477–49359 (Figure 3).
In spite of all the available bNAbs, currently there are no licensed treatments or vaccines against EBOV, though there have encouraging results from a recent human vaccine trial60 as well as nonhuman primate (NHP) immunizations.61 In recent years, it has been shown that the administration of polyclonal antibodies or combinations of mAbs help prevent the fatal disease when administered to EBOV-infected individuals. Treatment with these antibody-based therapies results in complete survival when administered at 24 hours postinfection.62 These treatments also provide partial protection when treatment starts as late as 5 days postinfection. More recently, a combination of the best two cocktails (ZMAb and MB-003) called ZMapp fully protects NHPs when the treatment is initiated at 5 days postinfection.63 ZMAb combines three mouse-derived mAbs: 1H3, 2G4, and 4G7. All three antibodies neutralize EBOV variant Mayinga (also known as the Zaire EBOV) by binding to the Ebola GP protein.64 The MB-003 cocktail consists of human–mouse chimeric antibodies 13C6, 13F6, and 6D8. ZMapp offers the best experimental therapeutic option currently available for treating EBOV-infected individuals. Table 4 categorizes bNAbs against EBOV.
 | Table 4 bNAbs against EBOV and their corresponding epitopes |
NAbs in influenza
Influenza viruses belong to the Orthomyxoviridae family of enveloped viruses and have RNA as its genetic material. Influenza occurs globally with an annual occurrence rate estimated at 5%–10% in adults and 20%–30% in children.65 Illnesses can result in hospitalization and death, mainly among high-risk groups (the very young, elderly, or chronically ill). Worldwide, these annual epidemics are estimated to result in approximately 3–5 million cases of severe illness, and approximately 250,000–500,000 deaths.66 Seasonal influenza is caused by influenza A and B viruses, both belonging to the Orthomyxoviridae family.67 During the 20th century, three major influenza pandemics occurred with a total mortality of 50–100 million people.68 Influenza types A and B are enveloped RNA viruses that can lead to respiratory or gastrointestinal tract infections in mammalian or avian species. Influenza A has caused pandemics, though both types are responsible for epidemics. Influenza A viruses circulate in a number of animals, including birds, humans, horses, pigs, and sea mammals, while influenza B is restricted to humans and seals.67
For the last several years, the World Health Organization has recommended a trivalent vaccine composition biannually that targets the three most representative virus types in circulation (two subtypes of influenza A viruses and one B virus). Recently, a quadrivalent vaccine composition has been recommended with a second influenza B virus in addition to the viruses in the conventional trivalent vaccines. It is expected that quadrivalent influenza vaccines will provide better and wider protection against influenza B virus infections than the trivalent one. A major challenge is to design and develop antibodies that can neutralize several different strains.
Recently, several new influenza mAbs have been discovered that are capable of neutralizing a wide range of influenza viruses.69–74 These bNAbs neutralize many different strains and subtypes of influenza virus. Some of them, such as 5J8 and 1F1, bind the head domain of the hemagglutinin (HA) protein (Figure 4). Using a single-cell culture method for screening large numbers of human plasma cells, Corti et al69 isolated a neutralizing mAb F16v3 (Figure 4) that recognized the HA GP of 16 different subtypes and neutralized both group 1 and group 2 influenza A viruses. The binding epitope of F16v3 is localized to the stem domain of HA protein (Figure 4). Similarly, the epitope of another bNAb CR8020 is also located on the stem domain of HA (Figure 4). To develop a broad-spectrum influenza vaccine, the identification and characterization of conserved neutralization epitopes is important. In this regard, by boosting the humoral response against conserved regions of the HA GP, the mouse mAb 9H10 (Figure 4) was generated, which had broad and potent in vitro neutralizing activity against both H3 and H10 group 2 influenza A subtypes.75 The broad binding profiles observed, positively correlated with broad neutralization, demonstrating that MAb 9H10 is indeed a pan-H3 and an H10 NAb. A new class of rare influenza-NAbs that target a conserved site in the HA stem has been described by Pappas et al.76 Most of these antibodies use the heavy-chain variable region VH1-69 gene, and structural data demonstrate that they bind to the hemagglutinin stem through conserved heavy-chain complementarity determining region residues.76
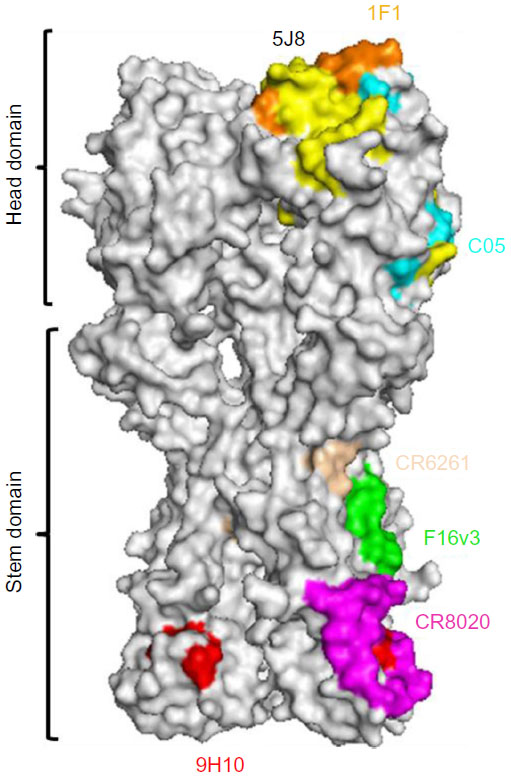 | Figure 4 Epitopes for bNAbs on influenza virus HA protein. |
Benjamin et al77 described the discovery of a unique anti-HA mAb, D1-8, that was derived from human B-cells and exhibits potent, broad neutralizing activity across antigenically diverse influenza H3 subtype viruses. D1-8 targets a novel epitope on the globular head region of the influenza virus HA protein.77
More recently, Skountzou et al78 have demonstrated in mice that IgM antibodies are functionally similar to IgG, as they neutralized the influenza virus in the presence of complement.
Passive antibody therapy
Passive antibody therapy was first introduced in early 1900 to treat against infections when clear understanding of causative agents were not discovered.79 The treatment was known at that time as “serum therapy”, where infected patients were transfused with serum from patients recovered from similar ailments.
In the case of Ebola infection, convalescent therapy was first used for a young woman in the Democratic Republic of Congo (then Zaire) in 1976. She had less clinical bleeding than other Ebola patients, but died within days. Later, during the 1995 Ebola epidemic outbreak in Congo, whole blood collected from recovered patients was administered in eight patients, seven of them recovered. This therapy option was discontinued with the discovery of antibiotic-based treatments because of various reasons such as cost, ease of application, improved safety, consistency in clinical outcome, and avoidance of “serum sickness” – a phenomena arising out of unwarranted antigen–antibody reactions.80 Only in certain very specific cases such as tetanus, botulism, snakebite, and for certain viral diseases where no drugs are available, passive antibody treatment is still a relevant treatment option.
With great technological inventions in developing and manufacturing unique mAb therapeutics, it was hypothesized such treatments can be used to treat debilitating viral infectious diseases like HIV. Interestingly, the majority of the new mAb-based drugs are developed to target various types of cancer and rheumatoid arthritis. Only in certain scenarios, immune gamma globulin preparations have been recommended in clinical settings for prevention as well as in therapy for hepatitis B virus, cytomegalovirus, rabies virus, tetanus toxin, and botulinum toxin with limited success rate.81–84 Being a natural product, antibody treatment in viral diseases poses great advantages like reduced side effects and high specificity toward viral antigens. A drawback of high specificity of antibody drugs is that hypermutation of the viral antigens can result in loss of antibody binding and viral escape. One solution is to use a cocktail of mAbs against the same target antigen, a perfect example of this is the ZMapp product85,86 used against Ebola infection – as already discussed in “NAbs for Ebola virus” section. The recent Ebola epidemic in West Africa has resulted in renewed interest in passive antibody therapy as a potential option, although there is need for more data and especially large-scale clinical trials.86
Another critical limitation of passive antibody therapy is the time of administration. The therapy works best as a prophylactic measure, ie, administered at very early stages of infection. For example, treatment of pneumococcal pneumonia was effective only when the antibody was given within the first 3 days of symptoms. The mechanism is not well understood to date, but is probably because of the fact that antibodies do not cope with high microorganism burden at an advanced stage of disease.87
In the case of HIV, several animal studies using the macaque model showed that NAbs could potently prevent infection with chimeric simian–human immunodeficiency virus. Infusion of 2F5 (origin species: human), 2G12 (origin species: human), and HIVIG (origin species: human), alone or in combination, protected macaques against a highly pathogenic intravenous or vaginal88,89 virus challenge.
Among the bNAb category for HIV-1, recombinant B12 antibody (origin species: human) was one of the first candidates identified through phage display screening.90 Structural analysis of B12 with two other weak NAbs B13 and F105 suggested that very precise targeting of gp120 is responsible for its broadly neutralizing property.91 B12 was subsequently tested in mice and NHPs with positive outcomes, suggesting successful antibody activity against free virus and virus infected cells.92,93 Another antibody 4E10 exhibited broader inter clade activity than others by neutralizing nearly 90 isolates tested, in comparison B12 was effective against 50% of viruses of all clades.12 A Phase I study conducted in 2002 used human mAbs 2F5 and 2G12 in asymptomatic HIV patients. Although the initial results were promising, among seven patients only one virus was neutralized by both antibodies, others were sensitive only to a single antibody.94 While it might be useful to employ combinatorial treatment with HAART and antibody drugs to develop an effective treatment regime, at current market price, this is a cost-prohibitive approach, especially considering the economic status of the majority of HIV patients. Furthermore, given the presence of significant viral reservoirs that are inaccessible to antibodies, it is unclear how useful antibodies will be in the treatment of HIV-1. In an exciting recent development, an adeno-associated virus (AAV) vector expressing an engineered CD4 derivative95 was shown to protect macaques from multiple challenges with a chimeric simian–human immunodeficiency virus. Animals inoculated with the vector stably expressed the CD4 derivative for over 40 weeks. However, the long-term efficacy of such an approach remains to be tested and potential immunogenicity of the vector is a possible concern.
In the recent case of the Ebola epidemic, many deaths could have been prevented if an effective antibody stockpile was available. Administration of these preparations to contacts might have provided them with immediate immunity, possibly resulting in early containment of the epidemic. We have learned a valuable lesson from this tragic epidemic. Government and nongovernment agencies have now initiated development of antibody-based therapies for Ebola.85,96 Clinical trials of sera are being designed, and compassionate use of these therapies has been employed. It is likely that clinically useful preparations may be identified and will be available shortly.62,79
NAb therapy business case – what are we missing?
Historically, antibody therapy was adopted as early as 190079 in the form of serum therapy, predating the current enthusiasm for biotherapeutic products. Between then and now, enormous technological advancement has helped to identify targets and develop mAb therapy in cancer, rheumatoid arthritis, and various other diseases. We now have production facilities for manufacturing mAbs at a scale of hundreds of kilos, globally. In comparisons to tens of marketed mAb products and almost a hundred products under various stages of development in oncology and rheumatology segment, we have just two licensed mAb products in the infectious disease segment, one for anthrax infection – raxibacumab, and one for respiratory syncytial virus infection – palivizumab. Why are we facing such a disparity even after the monumental successes for mAb therapy in cancer and arthritis? The answer is not simple. It is a combination of factors such as cost, research information, patient strata, market dynamics, and the interests of the global pharmaceutical industry. We must consider this multifaceted problem holistically.
Cost
mAb-mediated therapies are costly propositions. The product is manufactured through specialized animal or mammalian cell culture facilities with enormously expensive infrastructure. The manufacturing processes for biologicals are highly complex, compared to generic small molecules. We are yet to achieve simple chemistry-based processes that attain very high yield at a very low unit price. For example, typical annual cost of infliximab (Remicade®, Centocor, Inc., Malvern, PA, USA) treatment, a rheumatoid arthritis drug, is US$14,00097 for patients in the US. This cost is reduced significantly with the introduction of the follow on biologic Remsima™ (Celltrion, Incheon City, Republic of Korea), to an estimated $8,500 for patients in Europe.97 These cost estimates are based on usual recommended doses for each product. Even if we assume such products are financed through governmental and nongovernmental resources for specific indications, it will still remain >100 times costlier than the current antiviral therapy. In addition, recommended dosages for many NAb products are comparatively high. For Ebola treatment, a report98 suggests costs up to $100,000 to treat a single patient, and it takes approximately 78 tobacco plants and up to 10 days to harvest just one dose of the Ebola-curing antibodies.98 Currently, efforts are ongoing with the support from Bill and Melinda Gates foundation and National Institutes of Health (NIH) to establish efficient manufacturing processes to scale-up production facilities. In a recently published Phase I clinical study of HIV patients with bNAbs 3BNC117, a human IgG, a single dose with 30 mg/kg of body weight resulted in significant reduction in viral load and viremia under control for 28 days.25 This translates to a single infusion of 1.8 g of antibody per patient, considering average body weight of 60 kg. Currently, no information is available on how many such doses will be required to prevent the disease rebound for multiple years. The high-dosage requirement multiplies the cost burden significantly. For comparison, to achieve significant disease-free survival, the antibody drug requirement per patient per month for common oncology and rheumatology products like Herceptin (4 mg/kg +2 mg/kg), Avastin (5 mg/kg), and Rituxan (375 mg/m2) (Hoffman-La Roche Ltd., Basel, Switzerland) is 1.5, 0.6, and 2.8 g, respectively.
In the case of palivizumab, average wholesale prices of 50 mg vial and 100 mg vial are $780 and $1,416, respectively. Cost–benefit analyses gave results ranging from significant cost saving to overall cost increase in certain cases.99,100 Therefore, limited use of such drugs exclusively in high-risk patients is probably justifiable.
In recent years, the average cost of first-line antiretroviral treatment (ART) for HIV patients in low- and medium-income countries was estimated at $115 per patient per year, the second-line ART treatment cost estimate $330 per patient per year, and for third-line ART is estimated at $1,500 per patient per year. The price drop from $10,000 per patient per year happened through availability of generic drugs.
Research information
In the last decade, a large body of scientific evidence has become available for NAbs-mediated therapy. We now understand that inherent viral heterogeneity, hypermutation of target antigens, and complex host factor interactions determine the success of such treatment. It is only possible to achieve remission with antibody cocktails, ie, mixture of multiple mAbs. Such antibody cocktails would likely encounter stiff regulatory hurdles. In essence, it is much more complex to develop monoclonal NAb products for viral infections, both acute and chronic, than for disease indications such as cancer and arthritis where single mAbs can successfully control disease progression.
Precise diagnostic platforms for specific microbial infectious diseases are an important prerequisite for adopting antibody-based therapy because of high specificity of the drug substance and sequence diversity in the target antigen. Currently, diagnostic approaches are still dependent on microbial culture techniques that are qualitative and time consuming. Fortunately, with our improved knowledge on genomic information, new rapid diagnostic protocols are becoming available, which may help in adopting antibody-based therapy to infectious diseases.
mAbs product development in oncology and other indications were tremendously hastened due to large multicentric clinical trials with thousands of patients covering large geographies to address patient population variability.101,102 In cases of NAbs, such studies remain to be carried out. Smaller restricted clinical study groups will lead to limited success in developing effective medications.
Patient strata
Infectious disease incidences are high mostly in underdeveloped or developing countries, barring a few disease epidemics like the flu. Economic situations, standard of living, and affordability indices of patients in these geographies are a barrier to adoption of such complex and costly antibody-based treatment options. Even if government and nongovernment resources are available, it is simply beyond the financial reach of most people in such countries. Recent developments permit mAbs or other biologics to be generated in situ using AAV vectors.95,103 This is a promising and cost-effective approach, but long-term efficacy and safety of such treatments remains to be demonstrated.
Market dynamics
In today’s world, large pharmaceutical companies control much of the global market. To become successful in the global pharmaceutical market, it is necessary to focus on chronic diseases of the developed world. Infectious diseases by their very nature are less prevalent where public health is improved and higher health standards are maintained. Additionally, in the majority of countries in Europe and North America, the health insurance safety net would allow patients to access high-quality treatment options irrespective of the cost of the product. As the incidence level is low, insurance companies may be willing to bear the cost burden. In addition, the exquisite specificity of antibody therapy leads to applicability in a specific set of patients, which in turn means smaller market size, increased cost, and restricted profitability. All these factors contribute to a relatively low, market-driven need for affordable antibody product development against infectious diseases such as HIV, influenza, HCV, dengue, and Ebola. In spite of the current market scenario, bNAb drugs could likely bring significant relief to critical high-risk patients and contain disease conditions, and even prevent loss of life in certain situation.
Role of global pharmaceutical companies
Pharmaceutical companies will always focus on products with high-revenue potential to excel in their business and maintain a competitive edge. Therefore, disease indications and corresponding therapeutic products are targeted for higher market penetration. Many of the infectious diseases where NAbs might bring significant therapeutic advantages do not fit these business criteria in a global perspective. This uncomfortable situation is changing slowly with refined information on global epidemics affecting larger human populations.
In today’s connected world, the risks of infectious diseases crossing geographical boundaries are high. NAbs in combination with small molecules could manage and contain disease situations from spreading and affecting larger populations. There is, therefore, a need for appropriate partnerships to arrive at effective solutions against infectious diseases.
Conclusion
The human immune system is multifactorial where no single attribute determines the desired final outcome of developing bNAbs and clearance of viral infection. In the last few years, there is exciting progress in understanding the process of how our immune system is sensitized to develop bNAbs in viral infections. Host factors, for example the specific germ line precursor alleles of known bNAbs104 in combination with viral factors, allow the immune system to produce strain-specific and bNAbs against specific viral epitopes using multiple germ line sequences. Therefore, host factors must be studied using specific individuals with limited or no breadth of bNAb responses to examine whether an effective bNAb response is possible through vaccination in such individuals.
Isolation of bNAbs and determination of their epitopes enables the design of immunogens that can elicit such antibodies.105–107 In addition, bNAbs can potentially be used for therapy, preferably for acute conditions where viral escape is unlikely. However, even in such cases, cost remains a concern. There has been exciting progress recently in the use of AAV vectors expressing entry inhibitors to prevent viral infection. The possibility of such an approach postinfection as a therapeutic option in humans remains to be convincingly demonstrated.
Acknowledgment
We thank Shruti Khare for assistance with the preparation of Figure 1 and for helpful discussions.
Disclosure
The authors of this publication SS, MG, and SM are employees of Theramyt Novobiologics Pvt Ltd. RV is a professor at the Molecular Biophysics Unit, Indian Institute of Science, Bangalore. The authors report no other conflicts of interest in this work.
References
Burton DR, Desrosiers RC, Doms RW, et al. HIV vaccine design and the neutralizing antibody problem. Nat Immunol. 2004;5(3):233–236. | |
Parren PW, Burton DR. The antiviral activity of antibodies in vitro and in vivo. Adv Immunol. 2001;77:195–262. | |
Klasse PJ, Sattentau QJ. Occupancy and mechanism in antibody-mediated neutralization of animal viruses. J Gen Virol. 2002;83(Pt 9):2091–2108. | |
Rizzuto CD, Sodroski JG. Contribution of virion ICAM-1 to human immunodeficiency virus infectivity and sensitivity to neutralization. J Virol. 1997;71(6):4847–4851. | |
Arthur LO, Bess JW Jr, Sowder RC 2nd, et al. Cellular proteins bound to immunodeficiency viruses: implications for pathogenesis and vaccines. Science. 1992;258(5090):1935–1938. | |
Pinna D, Corti D, Jarrossay D, Sallusto F, Lanzavecchia A. Clonal dissection of the human memory B-cell repertoire following infection and vaccination. Eur J Immunol. 2009;39(5):1260–1270. | |
Corti D, Lanzavecchia A. Broadly neutralizing antiviral antibodies. Annu Rev Immunol. 2013;31:705–742. | |
Gong RCW, Dimitrov DS. Candidate antibody-based therapeutics against HIV-1. BioDrugs. 2012;26(3):143–162. | |
Seaman MS, Janes H, Hawkins N, Grandpre LE, Devoy C. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J Virol. 2010;84(3):1439–1452. | |
Kunert R, Rüker F, Katinger H. Molecular characterization of five neutralizing anti-HIV type 1antibodies: identification of nonconventional D segments in the human monoclonal antibodies 2G12 and 2F5. AIDS Res Hum Retroviruses. 1998;14(13):1115–1128. | |
Zwick MB, Jensen R, Church S, et al. Anti-human immunodeficiency virus type 1 (HIV-1) antibodies 2F5 and 4E10 require surprisingly few crucial residues in the membrane-proximal external region of glycoprotein gp41 to neutralize HIV-1. J Virol. 2005;79(2):1252–1261. | |
Binley JM, Wrin T, Korber B, et al. Comprehensive cross-clade neutralization analysis of a panel of anti-human immunodeficiency virus type 1 monoclonal antibodies. J Virol. 2004;78(23):13232–13252. | |
Walker LM, Phogat SK, Chan-Hui PY, et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009;326(5950):285–289. | |
Pancera M, Zhou T, Druz A, et al. Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature. 2014;514(7523):455–461. | |
Walker LM, Huber M, Doores KJ, et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature. 2011;477(7365):466–470. | |
Pejchal R, Doores KJ, Walker LM, et al. A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science. 2011;334(6059):1097–1103. | |
Mouquet H, Scharf L, Euler Z, et al. Complex-type N-glycan recognition by potent broadly neutralizing HIV antibodies. Proc Natl Acad Sci U S A. 2012;109(47):E3268–E3277. | |
Bonsignori M, Hwang KK, Chen X, et al. Analysis of a clonal lineage of HIV-1 envelope V2/V3 conformational epitope-specific broadly neutralizing antibodies and their inferred unmutated common ancestors. J Virol. 2011;85(19):9998–10009. | |
Huang J, Ofek G, Laub L, et al. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature. 2012;491(7424):406–412. | |
Corti D, Langedijk JP, Hinz A, et al. Analysis of memory B cell responses and isolation of novel monoclonal antibodies with neutralizing breadth from HIV-1-infected individuals. PLoS One. 2010;5(1):e8805. | |
Li Y, Migueles SA, Welcher B, et al. Broad HIV-1 neutralization mediated by CD4-binding site antibodies. Nat Med. 2007;13(9):1032–1034. | |
Scheid JF, Mouquet H, Ueberheide B, et al. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science. 2011;333(6049):1633–1637. | |
Falkowska E, Ramos A, Feng Y, et al. PGV04, an HIV-1 gp120 CD4 binding site antibody, is broad and potent in neutralization but does not induce conformational changes characteristic of CD4. J Virol. 2012;86(8):4394–4403. | |
Klein F, Halper-Stromberg A, Horwitz JA, et al. HIV therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature. 2012;492(7427):118–122. | |
Caskey M, Florian K, Lorenzi JC, et al. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature. 2015;522(7557):487–491. doi: 10.1038/nature14411. | |
Roehrig JT, Bolin RA, Kelly RG. Monoclonal antibody mapping of the envelope glycoprotein of the dengue 2 virus, Jamaica. Virology. 1998;246(2):317–328. | |
Cedillo-Barron L, Garcia-Cordero J, Bustos-Arriaga J, Leon-Juarez M, Gutierrez-Castaneda B. Antibody response to dengue virus. Microbes Infect. 2014;16(9):711–720. | |
Modis Y, Ogata S, Clements D, Harrison SC. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci U S A. 2003;100(12):6986–6991. | |
Modis Y, Ogata S, Clements D, Harrison SC. Variable surface epitopes in the crystal structure of dengue virus type 3 envelope glycoprotein. J Virol. 2005;79(2):1223–1231. | |
Falconar AK. Identification of an epitope on the dengue virus membrane (M) protein defined by cross-protective monoclonal antibodies: design of an improved epitope sequence based on common determinants present in both envelope (E and M) proteins. Arch Virol. 1999;144(12):2313–2330. | |
Schieffelin JS, Costin JM, Nicholson CO, et al. Neutralizing and non-neutralizing monoclonal antibodies against dengue virus E protein derived from a naturally infected patient. Virol J. 2010;7:28. | |
Dejnirattisai W, Wongwiwat W, Supasa S, et al. A new class of highly potent, broadly neutralizing antibodies isolated from viremic patients infected with dengue virus. Nat Immunol. 2015;16(2):170–177. | |
Rouvinski A, Guardado-Calvo P, Barba-Spaeth G, et al. Recognition determinants of broadly neutralizing human antibodies against dengue viruses. Nature. 2015;520(7545):109–113. | |
Fibriansah G, Tan JL, Smith SA, et al. A highly potent human antibody neutralizes dengue virus serotype 3 by binding across three surface proteins. Nat Commun. 2015;6:6341. | |
Robinson LN, Tharakaraman K, Rowley KJ, et al. Structure-guided design of an anti-dengue antibody directed to a non-immunodominant epitope. Cell. 2015;162(3):493–504. | |
Kwo PY, Lawitz EJ, McCone J, et al. Efficacy of boceprevir, an NS3 protease inhibitor, in combination with peginterferon alfa-2b and ribavirin in treatment-naive patients with genotype 1 hepatitis C infection (SPRINT-1): an open-label, randomised, multicentre phase 2 trial. Lancet. 2010;376(9742):705–716. | |
Gault E, Soussan P, Morice Y, et al. Evaluation of a new serotyping assay for detection of anti-hepatitis C virus type-specific antibodies in serum samples. J Clin Microbiol. 2003;41(5):2084–2087. | |
Meunier JC, Russell RS, Goossens V, et al. Isolation and characterization of broadly neutralizing human monoclonal antibodies to the e1 glycoprotein of hepatitis C virus. J Virol. 2008;82(2):966–973. | |
Lam BP, Jeffers T, Younoszai Z, Fazel Y, Younossi ZM. The changing landscape of hepatitis C virus therapy: focus on interferon-free treatment. Therap Adv Gastroenterol. 2015;8(5):298–312. | |
Cox AL. MEDICINE. Global control of hepatitis C virus. Science. 2015;349(6250):790–791. | |
Choo QL, Kuo G, Ralston R, et al. Vaccination of chimpanzees against infection by the hepatitis C virus. Proc Natl Acad Sci U S A. 1994;91(4):1294–1298. | |
Raghuraman S, Park H, Osburn WO, Winkelstein E, Edlin BR, Rehermann B. Spontaneous clearance of chronic hepatitis C virus infection is associated with appearance of neutralizing antibodies and reversal of T-cell exhaustion. J Infect Dis. 2012;205(5):763–771. | |
Ball JK, Tarr AW, McKeating JA. The past, present and future of neutralizing antibodies for hepatitis C virus. Antiviral Res. 2014;105:100–111. | |
Sautto GA, Diotti RA, Clementi M. New therapeutic options for HCV infection in the monoclonal antibody era. New Microbiol. 2012;35(4):387–397. | |
Kong L, Giang E, Nieusma T, et al. Hepatitis C virus E2 envelope glycoprotein core structure. Science. 2013;342(6162):1090–1094. | |
Kong L, Giang E, Robbins JB, et al. Structural basis of hepatitis C virus neutralization by broadly neutralizing antibody HCV1. Proc Natl Acad Sci U S A. 2012;109(24):9499–9504. | |
de Jong YP, Dorner M, Mommersteeg MC, et al. Broadly neutralizing antibodies abrogate established hepatitis C virus infection. Sci Transl Med. 2014;6(254):254ra129. | |
Keck ZY, Angus AG, Wang W, et al. Non-random escape pathways from a broadly neutralizing human monoclonal antibody map to a highly conserved region on the hepatitis C virus E2 glycoprotein encompassing amino acids 412–423. PLoS Pathog. 2014;10(8):e1004297. | |
Li Y, Pierce BG, Wang Q, et al. Structural basis for penetration of the glycan shield of hepatitis C virus e2 glycoprotein by a broadly neutralizing human antibody. J Biol Chem. 2015;290(16):10117–10125. | |
Sanchez AK, Zaki AS, Nabel SR, Ksiazek GJ, Peters TG, Filoviridae CJ. Marburg and Ebola viruses. In: Knipe DM, Howley PM, editors. Fields Virology. Philadelphia, PA: Lippincott, Williams, and Wilkins; 2001:1279–1304. | |
Towner JSS, Khristova TK, Albarino ML, et al. Newly discovered Ebola virus associated with hemorrhagic fever outbreak in Uganda. PLoS Pathog. 2008;4:e1000212. | |
Ksiazek TG, Rollin PE, Williams AJ, et al. Clinical virology of Ebola hemorrhagic fever (EHF): virus, virus antigen, and IgG and IgM antibody findings among EHF patients in Kikwit, Democratic Republic of the Congo, 1995. J Infect Dis. 1999;179(Suppl 1):S177–S187. | |
Dye JMH, Kuehne AS, Barth AI, et al. Postexposure antibody prophylaxis protects nonhuman primates from filovirus disease. Proc Natl Acad Sci U S A. 2012;109(13):5034–5039. | |
Maruyama T, Rodriguez L, Jahrling PB, et al. Ebola virus can be effectively neutralized by antibody produced in natural human infection. J Virol. 1999;73:6024–6030. | |
Parren PW, Geisbert TW, Maruyama T, Jahrling PB, Burton DR. Pre- and post-exposure prophylaxis of Ebola virus infection in an animal model by passive transfer of a neutralizing human antibody. J Virol. 2002;76:6408–6412. | |
Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature. 2008;454(7201):177–182. | |
Takada A, Ebihara H, Jones S, Feldmann H, Kawaoka Y. Protective efficacy of neutralizing antibodies against Ebola virus infection. Vaccine. 2007;25:993–999. | |
Takada A, Feldmann H, Stroeher U, et al. Identification of protective epitopes on Ebola virus glycoprotein at the single amino acid level by using recombinant vesicular stomatitis viruses. J Virol. 2003;77:1069–1074. | |
Wilson JA, Hevey M, Bakken R, et al. Epitopes involved in antibody-mediated protection from Ebola virus. Science. 2000;287:1664–1666. | |
Henao-Restrepo AM, Longini IM, Egger M, et al. Efficacy and effectiveness of an rVSV-vectored vaccine expressing Ebola surface glycoprotein: interim results from the Guinea ring vaccination cluster-randomised trial. Lancet. 2015;386(9996):857–866. | |
Marzi A, Robertson SJ, Haddock E, et al. Ebola vaccine. VSV-EBOV rapidly protects macaques against infection with the 2014/15 Ebola virus outbreak strain. Science. 2015;349(6249):739–742. | |
Pettitt J, Zeitlin L, Kim do H, et al. Therapeutic intervention of Ebola virus infection in rhesus macaques with the MB-003 monoclonal antibody cocktail. Sci Transl Med. 2013;5(199):199ra(13). | |
Qiu X, Wong G, Audet J, et al. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature. 2014;514:47–53. | |
Audet J, Wong G, Wang H, et al. Molecular characterization of the monoclonal antibodies composing ZMAb: a protective cocktail against Ebola virus. Sci Rep. 2014;4:6881. | |
WHO. Influenza (Seasonal). Available from: http://www.who.int/mediacentre/factsheets/fs211/en/. Accessed October 31, 2015. | |
Laursen NS, Wilson IA. Broadly neutralizing antibodies against influenza viruses. Antiviral Res. 2013;98(3):476–483. | |
Osterhaus AD, Rimmelzwaan GF, Martina BE, Bestebroer TM, Fouchier RA. Influenza B virus in seals. Science. 2000;288(5468):1051–1053. | |
Lambert LC, Fauci AS. Influenza vaccines for the future. N Engl J Med. 2010;363(21):2036–2044. | |
Corti D, Voss J, Gamblin SJ, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. 2011;333(6044):850–856. | |
Ekiert DC, Bhabha G, Elsliger MA, et al. Antibody recognition of a highly conserved influenza virus epitope. Science. 2009;324(5924):246–251. | |
Ekiert DC, Friesen RH, Bhabha G, et al. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science. 2011;333(6044):843–850. | |
Ekiert DC, Kashyap AK, Steel J, et al. Cross-neutralization of influenza A viruses mediated by a single antibody loop. Nature. 2012;489(7417):526–532. | |
Ekiert DC, Wilson IA. Broadly neutralizing antibodies against influenza virus and prospects for universal therapies. Curr Opin Virol. 2012;2(2):134–141. | |
Tan GS, Krammer F, Eggink D, Kongchanagul A, Moran TM, Palese P. A pan-H1 anti-hemagglutinin monoclonal antibody with potent broad-spectrum efficacy in vivo. J Virol. 2012;86(11):6179–6188. | |
Tan GS, Lee PS, Hoffman RM, et al. Characterization of a broadly neutralizing monoclonal antibody that targets the fusion domain of group 2 influenza A virus hemagglutinin. J Virol. 2014;88(23):13580–13592. | |
Pappas L, Foglierini M, Piccoli L, et al. Rapid development of broadly influenza neutralizing antibodies through redundant mutations. Nature. 2014;516(7531):418–422. | |
Benjamin E, Wang W, McAuliffe JM, et al. A broadly neutralizing human monoclonal antibody directed against a novel conserved epitope on the influenza virus H3 hemagglutinin globular head. J Virol. 2014;88(12):6743–6750. | |
Skountzou I, Satyabhama L, Stavropoulou A, et al. Influenza virus-specific neutralizing IgM antibodies persist for a lifetime. Clin Vaccine Immunol. 2014;21(11):1481–1489. | |
Casadevall A, Pirofski L. The Ebola epidemic crystallizes the potential of passive antibody therapy for infectious diseases. PLoS Pathog. 2015;11(4):e1004717. | |
Casadevall A, Scharff M. Return to the past: the case for antibody-based therapies in infectious diseases. Clin Infect Dis. 1995;21(1):150–161. | |
Hosty TS, Kissling RS, Schaeffer M, Wallace GA, Dibble EH. Human antirabies gamma globulin. Bull World Health Organ. 1959;20:1111–1119. | |
Margaret A, Keller ERS. Passive immunity in prevention and treatment of infectious diseases. Clin Microbiol Rev. 2000;13(4):602–614. | |
Lee DC, Lederman HM. Anti-tetanus toxoid antibodies in intravenous gamma globulin: an alternative to tetanus immune globulin. J Infect Dis. 1992;166:642–645. | |
McCune TR, Johnson HK, MacDonell RC Jr, et al. The effect of polyimmune gammaglobulin for prophylaxis against reactivation cytomegalovirus infection in kidney and kidney/pancreas transplant recipients. J Am Soc Nephrol. 1992;2(10):1469–1474. | |
Almansa R, Eiros JM, Fedson D, Bermejo-Martin JF. Hyperimmune serum from healthy vaccinated individuals for Ebola virus disease? Lancet. 2014;2(12):e686. | |
Rybicki EP. Plant-based vaccines against viruses. Virol J. 2014; 11(1):205. | |
Robbins JB, Schneerson R, Szu SC. Perspective: hypothesis: serum IgG antibody is sufficient to confer protection against infectious diseases by inactivating the inoculum. J Infect Dis. 1995;171(6):1387–1398. | |
Mascola JR, Lewis MG, Stiegler G, et al. Protection of macaques against pathogenic simian/human immunodeficiency virus 89.6PD by passive transfer of neutralizing antibodies. J Virol. 1999;73(5):4009–4018. | |
Mascola JR, Stiegler G, VanCott TC, et al. Protection of macaques against vaginal transmission of a pathogenic HIV-1/SIV chimeric virus by passive infusion of neutralizing antibodies. Nat Med. 2000;6(2):207–210. | |
Roben P, Moore JP, Thali M, Sodroski J, Barbas CF 3rd, Burton DR. Recognition properties of a panel of human recombinant Fab fragments to the CD4 binding site of gp120 that show differing abilities to neutralize human immunodeficiency virus type 1. J Virol. 1994;68(8):4821–4828. | |
Chen L, Kwon YD, Zhou T, et al. Structural basis of immune evasion at the site of CD4 attachment on HIV-1 gp120. Science. 2009;326(5956):1123–1127. | |
Parren PW, Ditzel H, Gulizia RJ, et al. Protection against HIV-1 infection in hu-PBL-SCID mice by passive immunization with a neutralizing human monoclonal antibody against the gp120 CD4-binding site. AIDS. 1995;9(6):F1–F6. | |
Parren PW, Marx PA, Hessell AJ, et al. Antibody protects macaques against vaginal challenge with a pathogenic R5 simian/human immunodeficiency virus at serum levels giving complete neutralization in vitro. J Virol. 2001;75(17):8340–8347. | |
Stiegler G, Armbruster C, Vcelar B, et al. Antiviral activity of the neutralizing antibodies 2F5 and 2G12 in asymptomatic HIV-1-infected humans: a phase I evaluation. AIDS. 2002;16(15):2019–2025. | |
Gardner MR, Kattenhorn LM, Kondur HR, et al. AAV-expressed eCD4-Ig provides durable protection from multiple SHIV challenges. Nature. 2015;519:87–91. | |
Butler D. Ebola raises profile of blood-based therapy. Nature. 2015;517:9–10. | |
Managed Care. Costs of biologics for inflammatory conditions vary. Available from: http://www.managedcaremag.com/archives/2014/9/costs-biologics-inflammatory-conditions-vary-0. Accessed October 31, 2015. | |
The fix. Tobacco plants hold key to Ebola virus cure ZMapp. Available from: http://www.thefix.com/content/tobacco-plants-hold-key-ebola-cure. Accessed October 31, 2015. | |
Schrand LM, Elliott JM, Ross MB, Bell EF, Mutnick AH. A cost-benefit analysis of RSV prophylaxis in high-risk infants. Ann Pharmacother. 2001;35(10):1186–1193. | |
Kamal-Bahl S, Doshi J, Campbell J. Economic analyses of respiratory syncytial virus immunoprophylaxis in high-risk infants: a systematic review. Arch Pediatr Adolesc Med. 2002;156(10):1034–1041. | |
Glennie MJ, Johnson PW. Clinical trials of antibody therapy. Immunol Today. 2000;21(8):403–410. | |
Dalle S, Thieblemont C, Thomas L, Dumontet C. Monoclonal antibodies in clinical oncology. Anticancer Agents Med Chem. 2008;8(5):523–532. | |
Schnepp BC, Johnson PR. Adeno-associated virus delivery of broadly neutralizing antibodies. Curr Opin HIV AIDS. 2014;9:250–256. | |
Haynes BF, Verkoczy L. AIDS/HIV. Host controls of HIV neutralizing antibodies. Science. 2014;344(6184):588–589. | |
Bommakanti G, Citron MP, Hepler RW, et al. Design of an HA2-based Escherichia coli expressed influenza immunogen that protects mice from pathogenic challenge. Proc Natl Acad Sci U S A. 2010;107:13701–13706. | |
Mallajosyula VV, Citron M, Ferrara F, et al. Influenza hemagglutinin stem-fragment immunogen elicits broadly neutralizing antibodies and confers heterologous protection. Proc Natl Acad Sci U S A. 2014;111:E2514–E2523. | |
Correia BE, Bates JT, Loomis RJ, et al. Proof of principle for epitope-focused vaccine design. Nature. 2014;507:201–206. | |
Li Y, O’Dell S, Walker LM, et al. Mechanism of neutralization by the broadly neutralizing HIV-1 monoclonal antibody VRC01. J Virol. 2011;85(17):8954–8967. | |
Blattner C, Lee JH, Sliepen K, et al. Structural delineation of a quaternary, cleavage-dependent epitope at the gp41-gp120 interface on intact HIV-1 Env trimers. Immunity. 2014;40(5):669–680. | |
Deng YQ, Dai JX, Ji GH, et al. A broadly flavivirus cross-neutralizing monoclonal antibody that recognizes a novel epitope within the fusion loop of E protein. PLoS One. 2011;6(1):e16059. | |
Lin HE, Tsai WY, Liu IJ, et al. Analysis of epitopes on dengue virus envelope protein recognized by monoclonal antibodies and polyclonal human sera by a high throughput assay. PLoS Negl Trop Dis. 2012;6(1):e1447. | |
Costin JM, Zaitseva E, Kahle KM, et al. Mechanistic study of broadly neutralizing human monoclonal antibodies against dengue virus that target the fusion loop. J Virol. 2013;87(1):52–66. | |
Owsianka A, Tarr AW, Juttla VS, et al. Monoclonal antibody AP33 defines a broadly neutralizing epitope on the hepatitis C virus E2 envelope glycoprotein. J Virol. 2005;79(17):11095–11104. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.