")
Back to Archived Journals » Cell Health and Cytoskeleton » Volume 7
Broader implications: biological and clinical significance of microtubule acetylation
Received 5 November 2014
Accepted for publication 3 December 2014
Published 23 January 2015 Volume 2015:7 Pages 71—82
DOI https://doi.org/10.2147/CHC.S77040
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Denis Wirtz
Sharon M Rymut,1,2 Thomas J Kelley1,2
1Department of Pharmacology, 2Department of Pediatrics, Case Western Reserve University, Cleveland, OH, USA
Abstract: Microtubule acetylation is a key posttranslational modification that enhances organelle transport, drives cell signaling, and regulates cell cycle regulation. The optimal level of microtubule acetylation is regulated by the acetyltransferase alpha-tubulin-N-acetyltransferase 1and two deacetylases, histone deacetylase 6 and sirtuin-2. Alterations in microtubule acetylation levels have been associated with the pathophysiology of a number of diseases, including various forms of neurodegenerative conditions, cancer, and even cystic fibrosis. In this review, we will highlight the biological and clinical significance of microtubule acetylation and the potential of targeting this pathway for therapeutics.
Keywords: microtubule, acetylation, HDAC6, MEC17, SIRT2, neurodegenerative disease, cancer, cystic fibrosis
Introduction
Microtubules are key structures that control intracellular transport and influence aspects of cell morphogenesis, cell signaling, and cell division, among other roles.1–3 Microtubule function is optimized by various posttranslational modifications (PTMs), including polyglutamylation, detyrosination, and acetylation (see Janke and Bulinski4 for a full review). These modifications regulate interactions with microtubule motor proteins as well as other microtubule binding proteins.3 Also, PTM levels dictate downstream protein activities contributing to the regulation of signaling cascades.2 Here, we will focus on microtubule acetylation and its role in cellular processes and disease states.
Microtubules are extensive structural networks that originate from the microtubule-organizing center and radiate to the cell periphery. Individual microtubules are composed of alpha–beta tubulin dimers that follow an alternating assembly pattern to ultimately form into hollow cylindrical structures. The external diameter has been determined to be 25 nm, while the internal diameter is approximately 12 nm. Functionally, little is known about the interior environment of the microtubule and the evolutionary relevance underlying the unique microtubule structure.
Microtubule acetylation is a dynamic PTM of both alpha- and beta-tubulin. Acetylated alpha-tubulin (Ac-tub) has long been a focus of research, while acetylated beta-tubulin has been described only recently. Ac-tub significantly aids intracellular transport by promoting kinesin-1 binding and motility as well as regulating cell cycle checkpoints.5–7 Acetylation may also be involved in microtubule stability, although it is unclear whether Ac-tub stabilizes microtubules or is simply a marker of a more stable structure.8,9 When alpha-tubulin is acetylated, the half-life of fully polymerized microtubules is hours compared with 5–10 minutes when microtubules are not acetylated.3 However, Chu et al7 demonstrate that beta-tubulin acetylation occurs at the alpha–beta dimer interface, which prevents microtubule polymerization. We will focus the rest of this review on alpha-tubulin acetylation.
The control of microtubule acetylation is dependent on the balance of acetyltransferases and deacetylases, specifically alpha-tubulin-N-acetyltransferase 1 (MEC17), histone deacetylase 6 (HDAC6), and sirtuin-2 (SIRT2). Even though much is known about the mechanism and consequences of deacetylation by HDAC6 and SIRT2, the addition of the acetyl group to tubulin by MEC17 is vastly understudied. Recently, it has been shown that the acetylation occurs in the lumen of the microtubule; however, the mechanism by which this occurs is unclear.10–12
Microtubule acetylation was first described on the axonemes of Chlamydomonas flagella; however, neurons were considered a more ideal model to pursue advanced research regarding the regulation of microtubule acetylation, due to their ease of culturing and imaging.13 With much research focused on the axonal region of neurons, the importance of microtubule regulation and acetylation on neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Rett syndrome, and Charcot–Marie–Tooth (CMT) disease, was the first to gain attention. Today, the importance of microtubule acetylation extends beyond neurodegenerative diseases. Impaired microtubule regulation has been shown to have an impact on cell signaling, inflammation, platelet activation, and other cellular processes.14–16 These impacts influence many other diseases, including cancer, cystic fibrosis (CF), and obesity, and their respective treatments.
Given the broad impact of microtubules on multiple cellular functions, regulation of microtubule acetylation is an inviting therapeutic target. Although much of the early research on HDAC6 and microtubule acetylation focuses on neuronal function and neurodegenerative diseases, therapeutic trials of HDAC inhibition as a cancer treatment are most prevalent now, and specific HDAC6 inhibitors are gaining momentum, such as ricolinostat in the treatment of non-Hodgkin lymphoma.17 As new data emerge, the application of HDAC6 inhibitors is expanding to other diseases, most with hopes to use them in combination with existing drugs. Here, we review the biological and clinical significance of microtubule acetylation, focusing on its importance in many disease states.
Regulators of microtubule acetylation
Protein acetylation is dependent on the balance of acetyltransferase function and deacetylase activity. Microtubule acetylation levels depend most on the activity levels of MEC17 and the deacetylases SIRT2 and HDAC6. Each of these proteins’ functions is highlighted in Figure 1. Further, other PTMs can regulate when and where acetylation occurs; however, there is no strong evidence describing the sequence of PTMs or how they signal to each other. Cross talk is possible, but there is great physical separation and lack of change in structure postacetylation.11,12
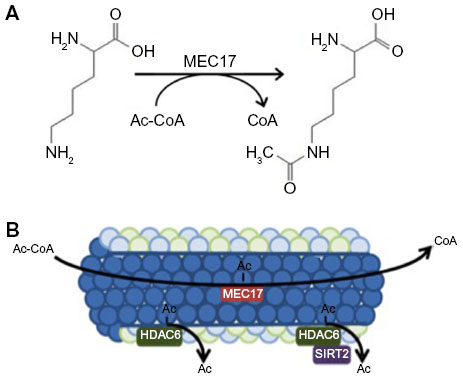 | Figure 1 Acetylation mechanisms. |
MEC17
MEC17 has been recently identified to be responsible for alpha-tubulin acetylation on lysine-40 (Figure 1A), where acetylation occurs in the lumen of the microtubule (Figure 1B). It is yet to be elucidated as to what conformation change occurs and why acetylation occurs there, however.8,10–12,18 Structurally, MEC17 is related to Gcn5 histone acetyltransferase 4.19 Mechanistically, MEC17 binds to an acetyl coenzyme A cofactor in its active site and transfers the acetyl group to tubulin at K40 (Figure 1A).20,21 It is unknown where MEC17 binds to the alpha-tubulin site or how K40 is recognized. It is suggested that Q58 creates a basic environment and is important for acetyl transfer, but extensive biochemical analysis is needed.21 Further, it is unknown how MEC17 gets to the lumen and if MEC17 requires extra cofactors for acetylation. When the microtubule becomes acetylated, no gross structural modification occurs on the exterior as determined by cryo-electron microscopy structures to hint at these solutions.12 Altering in vivo levels of MEC17 confirms the importance of MEC17, though. MEC17-knockout mice are viable; however, they have decreased advanced neurofunctions, such as learning and memory deficits, due to bulges in the dentate gyrus.22 At a cellular level, loss of MEC17 leads to impaired migration of cortical neurons, loss of focal adhesions, and inhibited proliferation through Hippo signaling.23,24 Microtubule instability through loss of MEC17 is being investigated in neuronal systems, but its role in other systems will be the subject of future research.
HDAC6
HDAC6 is accepted to be the primary regulator of microtubule deacetylation and is the solitary member of the HDAC family to be cytosolic.25 It can shuttle between the nucleus and the cytoplasm, but most of its functions occur in the cytosol.26 The list of HDAC6 targets is continually growing, and these targets can influence cellular functions, including ubiquitination, signal transduction, and transcriptional activity (see Valenzuela-Fernández et al27 for a full review). The most well-studied function of HDAC6 is its ability to interact with and deacetylate tubulin.25
HDAC6 activity can be regulated by multiple pathways, as well as be influenced by microtubule-regulating proteins such as tubulin polymerization-promoting protein (TPPP).28 TPPP has been shown to interact directly with and inhibit HDAC6 function. TPPP is phosphorylated by cyclin-dependent kinase-1, preventing its interaction with HDAC6 and resulting in increased microtubule acetylation.28 TPPP is just one example of an HDAC6 regulator that can contribute to microtubule stability.
To study the physiological importance of HDAC6, HDAC6-null mice have been generated. HDAC6-knockout mice are healthy and viable with minor delays in immune response and increases in bone mineral density.29 At a cellular level, they exhibit microtubule hyperacetylation; however, they do not show any growth defects and develop normally.29 The lack of deleterious phenotypes and the effective modulation of microtubule regulation in the HDAC6-null mice support HDAC6 as a potential therapeutic intervention for various diseases.
SIRT2
Sirtuins are a broad class of deacetylase enzymes. Of the seven family members that are described, sirtuin type 2 (SIRT2) is a nicotinamide adenine dinucleotide (NAD)+-dependent cytoplasmic enzyme that is hypothesized to either directly or indirectly deacetylate substrates, including tubulin and transcription factors FOXO1 and FOXO3a.30–32 Functionally, SIRT2 is involved in cell cycle regulation, adipocyte differentiation through peroxisome proliferator-activated receptor gamma, and oxidative stress response.30,31 SIRT2 regulation occurs by cyclin E-Cdk2, which phosphorylates SIRT2 at Ser-368, thus resulting in inactivation and altering many cellular processes, including alpha-tubulin deacetylation, cell adhesion and migration, and neurite outgrowth during neuronal differentiation.32,33
The diverse roles of SIRT2 make it difficult to distinguish between its direct and indirect influences on tubulin regulation. High expression of SIRT2 has been shown in the heart, brain, and skeletal muscle, but not everywhere, increasing skepticism about its importance in deacetylation.34,35 Expression is also increased in several malignancies compared with normal tissue, making SIRT2 a potential therapeutic target.36 Even though SIRT2 colocalizes with tubulin, no direct interactions have been reported.32 It has been proposed that SIRT2 binds to HDAC6 to facilitate its interaction with alpha-tubulin.18 Mechanistically, Nahhas et al18 demonstrate that tubulin binds only to the SIRT2–HDAC6 complex. Future research will determine the interdependence and the regulation necessary for tubulin deacetylation. What is for certain about SIRT2 is that it is a potential therapeutic target for PD and Huntington’s disease.
Importance of microtubule acetylation
Structurally, microtubules are intracellular structures that hold the shape of the cell. Functionally, microtubules facilitate intracellular transport and are key for cellular communication and cell maintenance functions such as autophagy regulation.15 The acetylation of microtubules is a key mechanism to control these and other functions, as outlined in Figure 2.
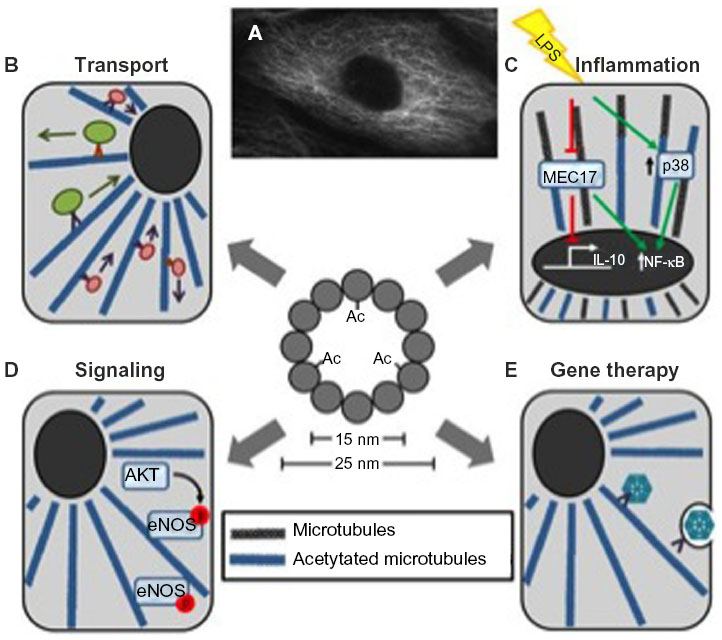 | Figure 2 Overview of the importance of microtubule acetylation. |
Intracellular transport
A key function of microtubules is to facilitate organelle transport (Figure 2B). The proper acetylation of microtubules is important to optimize this process by increasing microtubule motor affinities and improving motor function.6,37 Specifically, it has been shown that kinesin binding affinity increases proportionally when microtubules are acetylated.6 The treatment of neuronal cells with either trichostatin A (TSA), a nonspecific HDAC inhibitor, or tubacin, a specific HDAC6 inhibitor, stimulates kinesin-1-mediated anterograde transport of vesicles containing Jun-N-terminal kinase-interacting proteins at neurite tips.6,9,34
Microtubule acetylation is also known to regulate mitochondrial transport.1,38 Serotonin has been shown to increase mitochondrial movement via Akt-glycogen synthase kinase 3-beta (GSK3B) pathway.1 HDAC6 and GSK3B colocalize in hippocampal cells and regulate acetylation via HDAC6 phosphorylation.1 The implications are yet to be established, but literature shows abnormal mitochondrial transport and function in neurodegenerative diseases, including AD and PD.1,39 This may also be extended to CF, where both altered microtubule regulation and mitochondria dysfunction have been reported.40 Correcting intracellular movement is probably critical for improving these disease conditions.
Cell cycle
Microtubule acetylation is also an integral facet of cell cycle regulation.8,41 Microtubules are most highly acetylated during interphase, when much intracellular rearrangements occur, and they exhibit reduced acetylation during prophase and the transition from late telophase to interphase.42 Little is known as to why the cyclical nature of acetylation levels occurs. One hypothesis is that dynein light chain 1 LC8 promotes tubulin polymerization and stabilizes interphase microtubules.43 LC8 is a highly conserved protein that has many binding partners, including tubulin. LC8 knockdown destabilizes microtubules and inhibits bipolar spindle assembly in HeLa cells; LC8 knockdown also leads to the mitotic block in these cells.43 Mechanistic analysis suggests that LC8 binds at the alpha–beta interface, which is significantly different from where kinesin and dynein bind.43
Cell signaling
Cell signaling is a broad area, but the acetylation of microtubules is known to impact multiple cellular pathways (Figure 2C). One example is endothelial nitric oxide synthase (eNOS). eNOS plays an important role in vascular function, including the regulation of vascular tone, angiogenesis, platelet aggregation, and leukocyte–endothelial interactions.44,45 eNOS activity is regulated in part by calcium–calmodulin and also by L-arginine, NAD phosphate, S-nitrosylation, and phosphorylation.45 It is hypothesized that abnormal eNOS phosphorylation accounts for endothelial dysfunction observed in cardiovascular disease, including hypertension, hyperlipidemia, and diabetes. It has been shown that stable Ac-tub is needed for basal eNOS phosphorylation and its subcellular organization for optimal phosphorylation.44
Another example is inflammation. Inflammatory signaling has also been shown to be influenced by the control of microtubule acetylation (Figure 2B).38 Excessive inflammation can be attributed to many disease phenotypes; however, the role of Ac-tub has been controversial in both attenuating and accelerating inflammation itself, questioning its clinical implications. Excessive NLRP3 activation can cause inflammatory diseases, where microtubules mediate the assembly of the inflammasome.38 Inducers of NLRP3 cause mitochondria dysfunction, reduced NAD+ concentration, and inactivated SIRT2, which eventually leads to increased acetylation.46,47 This elevation of microtubule acetylation increases dynein trafficking and the formation and activation of NLRP3.38 Conversely, in macrophages, lipopolysaccharide-challenged cells undergo extensive remodeling of microtubule acetylation. Suppression of lipopolysaccharide-induced acetylation changes by MEC17 inactivation inhibits interleukin-10 (IL-10) induction, the anti-inflammatory cytokine.48 Increasing acetylation by HDAC6 inhibition leads to IL-10 stimulation along with blocking p38 amplification, thus attenuating inflammation.49
Microtubule acetylation in neurodegenerative diseases
The importance of intracellular transport and its control by microtubule regulation has been most thoroughly studied in neuronal models. The relationship between microtubule modifications and the impact on disease phenotypes is therefore best understood with neurological diseases. In this section, the impact of microtubule acetylation on various neurological diseases and the prospect of therapeutic interventions targeting this pathway are discussed. Highlights of where research stands are outlined in Table 1.
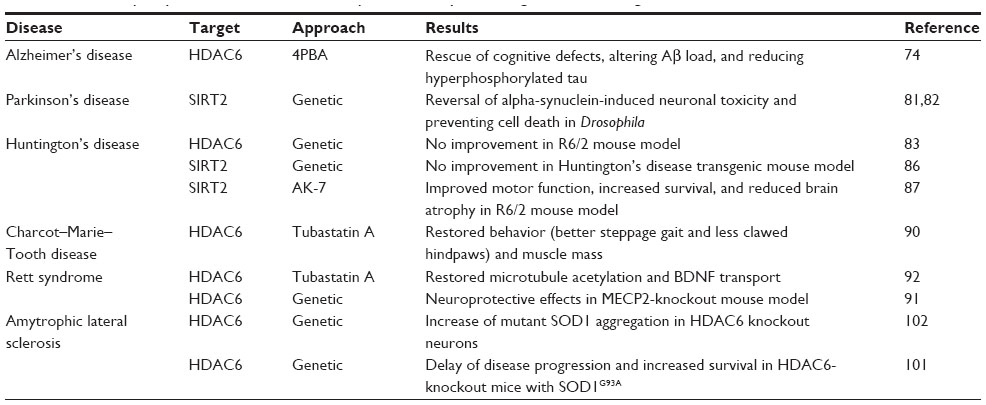 | Table 1 Summary of potential microtubule acetylation therapeutic targets in neurodegenerative diseases |
Alzheimer’s disease
AD is characterized by the presence of amyloid plaques in the brain, which mainly consist of Aβ peptide and neurofibrillary tangles.50,51 Both lead to synaptic and neuronal loss in distinct brain areas, including the neocortex and hippocampus.52,53 AD pathology is still being investigated, but atypical axonal accumulations of the amyloid precursor protein and its fragments, including Aβ, have been consistently found postmortem in AD patients.54 Aβ peptide upregulation consequently alters cytoskeletal features, including decreased Ac-tub, mitochondrial function, and F-actin polymerization.55,56 Inhibition of HDAC6 increases motility lost by the presence of Aβ.57 This functionally rescues hippocampal neurons from further Aβ deterioration.57
Also involved are neurofibrillary tangles of tau.58 The microtubule-associated protein tau is both phosphorylated and acetylated, adding to AD and taupathies complexity. In moderate to late AD, there is an excess of acetylated tau protein, but it is disputed whether this is a cause of disease progression or a consequence of tangle formations. Hempen and Brion58 have shown that these tangles inversely correlate with the extent of Ac-tub present in AD. However, Cohen et al59 report that tau acetylation promotes aggregation. Therefore, hyperacetylation is predicted to enhance tau pathologies; however, the converse is true. Treatment with HDAC6 inhibitors prevents tau aggregation and alters tau-isoform expression and subsequent phosphorylation.60 Specifically, it has been shown that KXGS motifs that are responsible for tau’s ability to polymerize are phosphorylated.60 HDAC6 inhibition results in tau acetylation and modulates tau phosphorylation and degradation in oligodendrocytes, leading to the hypothesis that HDAC6 inhibition would be beneficial in the treatment of AD.61 Recently, in a Drosophila model of taupathy, loss of HDAC6 activity rescues tau-induced microtubule defects in both muscles and neurons by increasing microtubule acetylation.62 This is the first piece of evidence that shows that HDAC6 inhibition is protective in taupathies and AD. These data also support the findings that HDAC6 knockout alleviates cognitive impairment in an AD model but does not affect its plaque burden, suggesting neuroprotection through tau.63
These data support that microtubule stability and HDAC6 inhibition can be beneficial for AD treatment. Microtubule stability through microtubule-stabilizing drugs such as paclitaxel has shown a significant increase in microtubule acetylation; therefore, these are an attractive first-generation drug class in treating AD.64 In vitro, stabilizers have been proven to be neuroprotective against tau and Aβ neurotoxicities.65,66 Consistent with these facts, paclitaxel in vivo in T44 tau transgenic (T44) mice that were affected with spinal cord tau pathology is shown to increase microtubule density and functionality in motor neurons.67 However, penetrating the blood–brain barrier has been a challenge in central nervous system therapy, making this drug unsuitable for future AD therapies. Epothilone D, which is a brain-penetrating microtubule-stabilizing agent, has been shown to prevent and ameliorate taupathies in T44 mice at low doses.68,69 However, stabilizing drugs have been shown to have significant debilitating side effects, including neutropenia and peripheral neuropathy.70,71 To avoid these side effects, more specific microtubule acetylation drugs have been tested recently. Valproic acid (VPA), a common antiepileptic agent and weak HDAC6 inhibitor, mediates some neuroprotection in AD models.72 VPA decreases Aβ production by inhibiting GSK3β cleavage of amyloid precursor protein.73 Functionally, this improves memory deficits in transgenic AD mice models and decreases neuritic plaque formations.73 Also, AD mice have shown neuroprotection with 4-phenylbutyrate, another weak HDAC6 inhibitor; spatial learning defects and memory deficits are reversed without altering beta-amyloid burdens. Mechanistically, phosphorylated tau is decreased after 4-phenylbutyrate treatment through inactivation of GSK3B.74 Specific inhibition of HDAC6 through tubastatin A and ACY-1215 has been used in preclinical trials, and results are promising.75 These results include the rescue of cognitive defects in AD mice, along with alleviating behavioral deficits, altering Aβ load, and reducing hyperphosphorylated tau.75
Parkinson’s disease
PD is characterized with loss of dopaminergic neurons in the midbrain region that controls motor function. Disease presentation includes the formation of Lewy bodies, which are insoluble ubiquitin-tagged aggregations of alpha-synuclein.76 The mechanism underlying disease pathology is unknown, but an emerging theory is that disease etiology includes disorganization of microtubules and dysfunctional regulation. Microtubules and alpha-synuclein have been reported to interact, leading to a theory that the interaction between Ac-tub and oligomeric alpha-synuclein may be increased through the inhibition of HDAC6 or SIRT2, leading to increased Lewy bodies.77,78
Outside of the realm of this review, HDAC6 inhibition in PD has been explored, but in the context of autophagy. In order for autophagy to occur, HDAC6, p62, and ubiquitin need to be bound together.79 HDAC6 inhibition also inherently enhances intracellular transport in neurons, aiding in the autophagy and lysosomal process (see Yan80 for a full review).
Unlike the rest of the neurodegenerative disorders, the therapeutic potential of SIRT2 has been explored more than HDAC6. In Drosophila, SIRT2 inhibition has been shown to reverse alpha-synuclein-induced neuronal toxicity and prevent cell death, a key observation in halting PD.81 Even though the mechanism is only hypothesized, it is suggested that SIRT2 inhibition increases intracellular movement of alpha-synuclein so that it accumulates in fewer areas, allowing for neuroprotection. It has also been shown recently that SIRT2 also deacetylates FOXO3a, which ultimately aids apoptosis through the upregulation of Bim.82 This suggests that therapeutic treatment can be achieved by SIRT2 activity outside of modulating microtubules.
Huntington’s disease
Huntington’s disease is another neurodegenerative disease marked by cognitive and motor deficits caused by abnormal polyglutamine expansion in the N-terminus of the huntingtin protein. As a consequence, transport and export of brain-derived neurotrophic factor (BDNF) become impaired, resulting in neuronal cell death.5 Dompierre et al5 show specifically that the intracellular transport process is microtubule dependent, and correct it using TSA to increase acetylation and to recruit both kinesin-1 and dynein to microtubules to reinstate bidirectional transport and release of BDNF vesicles. These findings support HDAC6-targeted therapies in Huntington’s disease; however, in vivo, genetic HDAC6 inhibition does not improve disease pathophysiology in an R6/2 mouse model of Huntington’s disease.83 Since then, SIRT2 has been investigated as a potential therapeutic target. Promising results have been shown for SIRT2 inhibition in neuroprotection in both cellular and invertebrate models.84,85 Neuroprotection is achieved in primary neuronal Huntington’s disease models, where SIRT2 inhibition leads to transcriptional repression of cholesterol biosynthesis.84 However, constitutive genetic SIRT2 inhibition in Huntington’s disease transgenic mice is neither neuroprotective nor effective against polyglutamine aggregation.86 Also, no transcriptional change in cholesterol biosynthesis in R6/2 mice is observed.86 Recently, SIRT2 inhibitor AK-7 has been developed.87 This drug is permeable to the blood–brain barrier and has been shown to be neuroprotective in vitro by reducing cholesterol levels in neurons along with polyglutamine inclusions. R6/2 mouse models using chronic treatment of AK-7 support these findings with reduction of huntingtin aggregates in Huntington’s disease mice brains.87 Improved motor function, increased survival, and reduced brain atrophy are seen as a consequence of chronic pharmacological SIRT2 inhibition in an R6/2 140CAG Htt knockin mouse model, resulting in AK-7 as a promising therapy that will go further in preclinical evaluation.87
Charcot–Marie–Tooth disease
CMT disease is a hereditary motor and sensor neuropathy that may be due to demyelinating sheaths to reduce nerve conduction velocities or decrease conduction amplitudes.88 Regardless, CMT disease patients exhibit muscle weakness and muscle mass.88 At a cellular level, CMT disease patients express mutant motor protein dynamin 2, which leads to unstable microtubules, altered Golgi apparatus structures, and decreased organelle transport.89 Mice expressing mutant heat shock p27 kDa protein beta-1 (HSPB1) exhibit phenotypes that mimic those of CMT disease patients, allowing for mechanistic evaluation.90 The decrease in Ac-tub in peripheral nerves in mutant HSPB1 mice offers an explanation for this decrease in organelle transport.90 The cause of decreased Ac-tub expression is unknown; however, it can be reestablished using HDAC6 inhibitors. Treatment of tubastatin A for mutant HSPB1 mice restores behavior, including improved steppage gait, less clawed hindpaws, and increased muscle mass, while halting and reversing other peripheral neuropathies.90 Further preclinical studies must be performed before these agents can be moved into clinical trials.
Rett syndrome
Rett syndrome is characterized by the reversal of neural development; movement, coordination, and communication are often difficult in patients, and the symptoms are often confused with autism. Primarily in girls, the postnatal neurodegenerative disease is caused by X-linked genetic mutations in the methyl-CpG-binding protein 2 (MECP2).91 Cells deficient in MECP2 and primary cells from Rett syndrome patients display an increase in HDAC6 expression, and thus increased microtubule instability and decreased Ac-tub result.92 No changes in SIRT2 expression levels are detected.92 Functional consequences include dysfunctional trafficking (both anterograde and retrograde) in MECP2 knockout neurons.91 Treatment with tubastatin A on primary patient cells restores microtubule acetylation and associated intracellular transport, specifically releasing BDNF.92 HDAC6 inhibitors have also shown neuroprotective effects in the MECP2-knockout mouse model.91
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a late-onset neurodegenerative disorder that is characterized by progressive muscle atrophy and paralysis due to death of upper and lower motoneurons. In these motoneurons and glial cells, protein inclusions have been detected as part of the diagnosis.93,94 Ninety percent of ALS is sporadic, while 10% is genetic.95 Of the familial cases, mutations are prominently located in the Cu/Zn superoxide dismutase 1 (SOD1), which is a gain of unknown toxic function.96 Mouse models that express mutated human SOD1 have the same pathological course as patients where it is hypothesized that glutamate excitotocity, oxidative stress, protein misfolding, mitochondrial defects, impaired axonal transport, and inflammation contribute to pathophysiology.95,97,98 In these models, glial cells have been identified as a potential target since they greatly contribute to motoneuron degeneration.99 Early research has indicated that TSA, a pan-HDAC inhibitor, is beneficial in ameliorating pathological characteristics and improving motor behaviors and longevity.100 However, it is unclear whether improvements in ALS are specifically due to HDAC6 activity or another HDAC. Taes et al101 have shown that disease progression is delayed in HDAC6-knockout mice that have SOD1G93A mutation. Results show that survival increases and motor axon integrity decreases in these mice. However, Gal et al102 show increased mutant SOD1 aggregation in HDAC6 knockout cells. Bruneteau et al103 show that HDAC4 is upregulated at a messenger ribonucleic acid level in subjects with rapidly progressive ALS compared with long-surviving ALS subjects, furthering the microtubule acetylation controversy in ALS.
Other conditions where microtubule acetylation is important
Cystic fibrosis
CF is a pediatric pulmonary disorder that is caused by mutations in the CF transmembrane conductance regulator (CFTR). CFTR is a cAMP-activated chloride channel that is most highly expressed in the apical membrane of epithelial cells.104 The disease is characterized by impaired transepithelial chloride transport, hyperabsorption of sodium across airway epithelium, chronic bacterial infection of the airways, heightened inflammatory responses, and metabolic changes.16,105,106 How CFTR function impacts such a wide range of cellular events is unclear. Recently, microtubules have been gaining attention in the field due to multiple MAPs and motor proteins associated with a more severe airway disease prognosis from both genome-wide association studies and exome studies.107,108 Microtubule regulation has been further implicated in CF. It has been demonstrated that there is a significant decrease in Ac-tub expression in epithelial cells and tissue.16 This decrease can be directly correlated with a decrease in previously noted dysfunctional intracellular transport marked by late endosomal/lysosomal cholesterol accumulation.109 HDAC6 inhibitors can improve endosomal transport, resulting in normal cholesterol distribution and a decrease in CF-related NF-κB activation.16 With much promise in vitro, research extending HDAC6 inhibition into CF mice models will be interesting in order to analyze its implications on multiple disease phenotypes.
Cancer
HDAC inhibitors are effective in many types of cancer, including Hodgkin lymphoma, T-cell lymphoma, and acute myeloid lymphomas. Currently, there are over 400 clinical trials using HDAC inhibitors in the treatment of cancer. Here, we will highlight the use of HDAC6 inhibitors in the treatment of acute myeloid leukemia (AML) and ovarian cancers.
In AML patients who do not qualify for traditional chemotherapy, HDAC6 inhibitors are currently being evaluated. VPA has been investigated in several AML studies, typically in conjunction with all-trans retinoic acid, especially in elderly patients or those who have relapsed.14 Overall, the toxicity levels of VPA are low, patients have complete hematological remission extending for several months, and over 30% of patients have increased peripheral blood platelet counts that last for up to 2 years.14,110
HDAC6 inhibitors have also been effective against ovarian cancer in vitro and in vivo. In previous studies, HDAC6 activity is established as a requirement for efficient oncogenic transformation in the ovarian cell line SKVO3 and breast cancer cell lines MCF7 and SKBR3.111 Without HDAC6 expression, fibroblasts are more resistant to both Ras- and ErbB2-dependent transformations.111 Mechanistically, transformation resistance is due to increased anoikis and alterations in the AKT pathway through loss of adhesion.111 More recently, tubastatin A has been suggested to increase cell elasticity without altering cytoskeletal components.112 The efficacy of HDAC6 inhibitors as an important component of combination therapies will continue to be evaluated further.
Influenza A
Most of the diseases we have focused on here have been either genetic or idiopathic. Lately, interest in HDAC6 in viral infection has been gaining attention. Specifically, influenza A infections are of interest because of HDAC6’s involvement in actin remodeling at the plasma membrane, which is the site of influenza A virus (IAV) assembly.113 Husain and Cheung113 and Husain and Harrod114 have demonstrated that IAV induces Ac-tub expression through increased Rho expression and signaling. This increase in acetylation induces the release of virions from infected cells.113,114 The contrary is true as well; when HDAC6 is overexpressed, there is a threefold decrease in releasing IAV progeny.113 Banerjee et al115 have shown mechanistically that outside of controlling the extent of acetylation, IAV infections hijack cellular machinery by mimicking misfolded protein aggregates by carrying unanchored ubiquitin chains and using the HDAC6 pathway to uncoat the virus, allowing for dissociation and viral ribonucleoproteins and matrix proteins to be dispersed in the cytosol in order to enter the nucleus. Even though much promise for HDAC6 inhibition is demonstrated in potential treatments for neurodegeneration, the effect of HDAC6 inhibition on infectious disease progression needs to be considered.
Gene therapy
Nonviral gene therapy has been a promising treatment strategy for genetic diseases. Many different strategies have been used for gene transfer with success, but efficiency is low and remains a key obstacle in bench to bedside therapeutics. Recently, mechanisms and strategies have focused on the subject of crossing the cytoplasm and evading degradation, and researchers have turned to microtubule manipulation just like that of IAV. Besides adding common transcription factors binding sites and nuclear localization sequences to plasmid gene therapy to aid delivery, hyperacetylation has been identified to increase plasmid movement using the cytoskeletal machinery in cells through greater net rates of movement and longer, continuous movement (Figure 2E).116 These results suggest using HDAC6 inhibition as a combination therapy to partially overcome the cytoplasmic intracellular transport obstacle to nonviral gene therapy.
Summary
Microtubule acetylation impacts many cellular functions, such as the regulation of various cell signaling cascades and the control of intracellular movement through the coordinated functions of the acetyltransferase MEC17 and the deacetylases HDAC6 and SIRT2. Altered regulation of microtubule acetylation has been identified in a number of disease states resulting in impaired organelle transport, loss of cellular proliferation control, and enhanced inflammatory signaling. From a number of neurodegenerative diseases to cancer to CF, the manipulation of microtubule acetylation levels through the use of SIRT2 and HDAC6 inhibitors offers a novel therapeutic target. As more is learnt about microtubules and their implications in the pathophysiology of other diseases, new therapeutic avenues will be opened and will provide new avenues for therapeutic interventions.
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
Chen S, Owens GC, Makarenkova H, Edelman DB. HDAC6 regulates mitochondrial transport in hippocampal neurons. PloS One. 2010;5(5):e10848. | |
Williams KA, Zhang M, Xiang S, et al. Extracellular signal-regulated kinase (ERK) phosphorylates histone deacetylase 6 (HDAC6) at serine 1035 to stimulate cell migration. J Bio Chem. 2013;288(46):33156–33170. | |
Bulinski JC, Gundersen GG. Stabilization of post-translational modification of microtubules during cellular morphogenesis. BioEssays News Rev Mol Cell Dev Biol. 1991;13(6):285–293. | |
Janke C, Bulinski JC. Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol. 2011;12(12):773–786. | |
Dompierre JP, Godin JD, Charrin BC, et al. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J Neurosci. 2007;27(13):3571–3583. | |
Reed NA, Cai D, Blasius TL, et al. Microtubule acetylation promotes kinesin-1 binding and transport. Curr Biol. 2006;16(21):2166–2172. | |
Chu C-W, Hou F, Zhang J, et al. A novel acetylation of β-tubulin by San modulates microtubule polymerization via down-regulating tubulin incorporation. Mol Biol Cell. 2011;22(4):448–456. | |
Piperno G, LeDizet M, Chang XJ. Microtubules containing acetylated alpha-tubulin in mammalian cells in culture. J Cell Biol. 1987;104(2):289–302. | |
Takemura R, Okabe S, Umeyama T, Kanai Y, Cowan NJ, Hirokawa N. Increased microtubule stability and alpha tubulin acetylation in cells transfected with microtubule-associated proteins MAP1B, MAP2 or tau. J Cell Sci. 1992;103(Pt 4):953–964. | |
Nogales E, Whittaker M, Milligan RA, Downing KH. High-resolution model of the microtubule. Cell. 1999;96(1):79–88. | |
Howes SC, Alushin GM, Shida T, Nachury MV, Nogales E. Effects of tubulin acetylation and tubulin acetyltransferase binding on microtubule structure. Mol Biol Cell. 2014;25(2):257–266. | |
Soppina V, Herbstman JF, Skiniotis G, Verhey KJ. Luminal localization of α-tubulin K40 acetylation by cryo-EM analysis of fab-labeled microtubules. PloS One. 2012;7(10):e48204. | |
Greer K, Maruta H, L’Hernault SW, Rosenbaum JL. Alpha-tubulin acetylase activity in isolated Chlamydomonas flagella. J Cell Biol. 1985;101(6):2081–2084. | |
Tassara M, Döhner K, Brossart P, et al. Valproic acid in combination with all-trans retinoic acid and intensive therapy for acute myeloid leukemia in older patients. Blood. 2014;123(26):4027–4036. | |
Monastyrska I, Rieter E, Klionsky DJ, Reggiori F. Multiple roles of the cytoskeleton in autophagy. Biol Rev Camb Philos Soc. 2009;84(3):431–448. | |
Rymut SM, Harker A, Corey DA, et al. Reduced microtubule acetylation in cystic fibrosis epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2013;305(6):L419–L431. | |
Dasmahapatra G, Patel H, Friedberg J, Quayle SN, Jones SS, Grant S. In vitro and in vivo interactions between the HDAC6 inhibitor ricolinostat (ACY1215) and the irreversible proteasome inhibitor carfilzomib in non-hodgkin lymphoma cells. Mol Cancer Ther. 2014;13(12):2886–2897. | |
Nahhas F, Dryden SC, Abrams J, Tainsky MA. Mutations in SIRT2 deacetylase which regulate enzymatic activity but not its interaction with HDAC6 and tubulin. Mol Cel. Biochem. 2007;303(1–2):221–230. | |
Steczkiewicz K, Kinch L, Grishin NV, Rychlewski L, Ginalski K. Eukaryotic domain of unknown function DUF738 belongs to Gcn5-related N-acetyltransferase superfamily. Cell Cycle. 2006;5(24):2927–2930. | |
Friedmann DR, Aguilar A, Fan J, Nachury MV, Marmorstein R. Structure of the α-tubulin acetyltransferase, αTAT1, and implications for tubulin-specific acetylation. Proc Natl Acad Sci U S A. 2012;109(48):19655–19660. | |
Taschner M, Vetter M, Lorentzen E. Atomic resolution structure of human α-tubulin acetyltransferase bound to acetyl-CoA. Proc Natl Acad Sci U S A. 2012;109(48):19649–19654. | |
Kim G-W, Li L, Gorbani M, You L, Yang X-J. Mice lacking α-tubulin acetyltransferase 1 are viable but display α-tubulin acetylation deficiency and dentate gyrus distortion. J Biol Chem. 2013;288(28):20334–20350. | |
Aguilar A, Becker L, Tedeschi T, Heller S, Iomini C, Nachury MV. α-tubulin K40 acetylation is required for contact inhibition of proliferation and cell-substrate adhesion. Mol Biol Cell. 2014;25(12):1854–1866. | |
Li L, Wei D, Wang Q, et al. MEC-17 deficiency leads to reduced α-tubulin acetylation and impaired migration of cortical neurons. J Neurosci. 2012;32(37):12673–12683. | |
Hubbert C, Guardiola A, Shao R, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417(6887):455–458. | |
Liu Y, Peng L, Seto E, Huang S, Qiu Y. Modulation of histone deacetylase 6 (HDAC6) nuclear import and tubulin deacetylase activity through acetylation. J Biol Chem. 2012;287(34):29168–29174. | |
Valenzuela-Fernández A, Cabrero JR, Serrador JM, Sánchez-Madrid F. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008;18(6):291–297. | |
Schofield AV, Gamell C, Suryadinata R, Sarcevic B, Bernard O. Tubulin polymerization promoting protein 1 (Tppp1) phosphorylation by Rho-associated coiled-coil kinase (rock) and cyclin-dependent kinase 1 (Cdk1) inhibits microtubule dynamics to increase cell proliferation. J Biol Chem. 2013;288(11):7907–7917. | |
Zhang Y, Kwon S, Yamaguchi T, et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol Cell Biol. 2008;28(5):1688–1701. | |
Jing E, Gesta S, Kahn CR. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 2007;6(2):105–114. | |
Wang F, Nguyen M, Qin FX-F, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007;6(4):505–514. | |
North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11(2):437–444. | |
Pandithage R, Lilischkis R, Harting K, et al. The regulation of SIRT2 function by cyclin-dependent kinases affects cell motility. J Cell Biol. 2008;180(5):915–929. | |
Inoue T, Hiratsuka M, Osaki M, et al. SIRT2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene. 2007;26(7):945–957. | |
Maxwell MM, Tomkinson EM, Nobles J, et al. The sirtuin 2 microtubule deacetylase is an abundant neuronal protein that accumulates in the aging CNS. Hum Mol Genet. 2011;20(20):3986–3996. | |
Kim H-S, Vassilopoulos A, Wang R-H, et al. SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell. 2011;20(4):487–499. | |
Bulinski JC. Microtubule modification: acetylation speeds anterograde traffic flow. Curr Biol. 2007;17(1):R18–R20. | |
Misawa T, Takahama M, Kozaki T, et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol. 2013;14(5):454–460. | |
Winklhofer KF, Haass C. Mitochondrial dysfunction in Parkinson’s disease. Biochim Biophys Acta. 2010;1802(1):29–44. | |
Valdivieso AG, Clauzure M, Marín MC, et al. The mitochondrial complex I activity is reduced in cells with impaired cystic fibrosis transmembrane conductance regulator (CFTR) function. PloS One. 2012;7(11):e48059. | |
Gundersen GG, Bulinski JC. Distribution of tyrosinated and nontyrosinated alpha-tubulin during mitosis. J Cell Biol. 1986;102(3):1118–1126. | |
Wolf KW, Spanel-Borowski K. Acetylation of alpha-tubilin in different bovine cell types: implications for microtubule dynamics in interphase and mitosis. Cell Biol Int. 1995;19(1):43–52. | |
Asthana J, Kuchibhatla A, Jana SC, Ray K, Panda D. Dynein light chain 1 (LC8) association enhances microtubule stability and promotes microtubule bundling. J Biol Chem. 2012;287(48):40793–40805. | |
Giustiniani J, Couloubaly S, Baillet A, et al. Basal endothelial nitric oxide synthase (eNOS) phosphorylation on Ser(1177) occurs in a stable microtubule- and tubulin acetylation-dependent manner. Exp Cell Res. 2009;315(20):3509–3520. | |
Mattila JT, Thomas AC. Nitric oxide synthase: non-canonical expression patterns. Front Immunol. 2014;5:478. | |
Kim SR, Kim DI, Kim SH, et al. NLRP3 inflammasome activation by mitochondrial ROS in bronchial epithelial cells is required for allergic inflammation. Cell Death Dis. 2014;5:e1498. | |
McGettrick AF, O’Neill LAJ. How metabolism generates signals during innate immunity and inflammation. J Biol Chem. 2013;288(32):22893–22898. | |
Shida T, Cueva JG, Xu Z, Goodman MB, Nachury MV. The major alpha-tubulin K40 acetyltransferase alphaTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc Natl Acad Sci U S A. 2010;107(50):21517–21522. | |
Wang B, Rao Y-H, Inoue M, et al. Microtubule acetylation amplifies p38 kinase signalling and anti-inflammatory IL-10 production. Nat Commun. 2014;5:3479. | |
Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120(3):885–890. | |
Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8(1):159–168. | |
Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL. Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science. 1984;225(4667):1168–1170. | |
Masliah E, Hansen L, Albright T, Mallory M, Terry RD. Immunoelectron microscopic study of synaptic pathology in Alzheimer’s disease. Acta Neuropathol (Berl). 1991;81(4):428–433. | |
Sennvik K, Bogdanovic N, Volkmann I, Fastbom J, Benedikz E. Beta-secretase-cleaved amyloid precursor protein in Alzheimer brain: a morphologic study. J Cell Mol Med. 2004;8(1):127–134. | |
Henriques AG, Vieira SI, da Cruz E Silva EF, da Cruz E Silva OAB. Abeta promotes Alzheimer’s disease-like cytoskeleton abnormalities with consequences to APP processing in neurons. J Neurochem. 2010;113(3):761–771. | |
Chen JX, Yan SS. Role of mitochondrial amyloid-beta in Alzheimer’s disease. J Alzheimers Dis. 2010;20 Suppl 2:S569–S578. | |
Kim C, Choi H, Jung ES, et al. HDAC6 inhibitor blocks amyloid beta-induced impairment of mitochondrial transport in hippocampal neurons. PloS One. 2012;7(8):e42983. | |
Hempen B, Brion JP. Reduction of acetylated alpha-tubulin immunoreactivity in neurofibrillary tangle-bearing neurons in Alzheimer’s disease. J Neuropathol Exp Neurol. 1996;55(9):964–972. | |
Cohen TJ, Guo JL, Hurtado DE, et al. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2011;2:252. | |
Cook C, Carlomagno Y, Gendron TF, et al. Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum Mol Genet. 2014;23(1):104–116. | |
Noack M, Leyk J, Richter-Landsberg C. HDAC6 inhibition results in tau acetylation and modulates tau phosphorylation and degradation in oligodendrocytes. Glia. 2014;62(4):535–547. | |
Xiong Y, Zhao K, Wu J, Xu Z, Jin S, Zhang YQ. HDAC6 mutations rescue human tau-induced microtubule defects in Drosophila. Proc Natl Acad Sci U S A. 2013;110(12):4604–4609. | |
Govindarajan N, Rao P, Burkhardt S, et al. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol Med. 2013;5(1):52–63. | |
Schiff PB, Horwitz SB. Taxol stabilizes microtubules in mouse fibroblast cells. Proc Natl Acad Sci U S A. 1980;77(3):1561–1565. | |
Shemesh OA, Spira ME. Rescue of neurons from undergoing hallmark tau-induced Alzheimer’s disease cell pathologies by the antimitotic drug paclitaxel. Neurobiol Dis. 2011;43(1):163–175. | |
Michaelis ML, Ranciat N, Chen Y, et al. Protection against beta-amyloid toxicity in primary neurons by paclitaxel (Taxol). J Neurochem. 1998;70(4):1623–1627. | |
Trojanowski JQ, Smith AB, Huryn D, Lee VM-Y. Microtubule-stabilising drugs for therapy of Alzheimer’s disease and other neurodegenerative disorders with axonal transport impairments. Expert Opin Pharmacother. 2005;6(5):683–686. | |
Brunden KR, Zhang B, Carroll J, et al. Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy. J Neurosci. 2010;30(41):13861–13866. | |
Zhang B, Carroll J, Trojanowski JQ, et al. The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J Neurosci. 2012;32(11):3601–3611. | |
Lee JJ, Swain SM. Peripheral neuropathy induced by microtubule-stabilizing agents. J Clin Oncol. 2006;24(10):1633–1642. | |
Bedard PL, Di Leo A, Piccart-Gebhart MJ. Taxanes: optimizing adjuvant chemotherapy for early-stage breast cancer. Nat Rev Clin Oncol. 2010;7(1):22–36. | |
Noh H, Seo H. Age-dependent effects of valproic acid in Alzheimer’s disease (AD) mice are associated with nerve growth factor (NGF) regulation. Neuroscience. 2014;266:255–265. | |
Qing H, He G, Ly PTT, et al. Valproic acid inhibits Abeta production, neuritic plaque formation, and behavioral deficits in Alzheimer’s disease mouse models. J Exp Med. 2008;205(12):2781–2789. | |
Ricobaraza A, Cuadrado-Tejedor M, Pérez-Mediavilla A, Frechilla D, Del Río J, García-Osta A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer’s disease mouse model. Neuropsychopharmacol. 2009;34(7):1721–1732. | |
Zhang L, Liu C, Wu J, et al. Tubastatin A/ACY–1215 improves cognition in Alzheimer’s disease transgenic mice. J Alzheimers Dis. 2014; 41(4):1193–1205. | |
Wakabayashi K, Tanji K, Mori F, Takahashi H. The Lewy body in Parkinson’s disease: molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathol. 2007;27(5):494–506. | |
Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115(6):727–738. | |
Donmez G, Outeiro TF. SIRT1 and SIRT2: emerging targets in neurodegeneration. EMBO Mol Med. 2013;5(3):344–352. | |
Fusco C, Micale L, Augello B, et al. HDAC6 mediates the acetylation of TRIM50. Cell Signal. 2014;26(2):363–369. | |
Yan J. Interplay between HDAC6 and its interacting partners: essential roles in the aggresome-autophagy pathway and neurodegenerative diseases. DNA Cell Biol. 2014;33(9):567–580. | |
Outeiro TF, Kontopoulos E, Altmann SM, et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science. 2007;317(5837):516–519. | |
Liu L, Arun A, Ellis L, Peritore C, Donmez G. SIRT2 enhances 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced nigrostriatal damage via apoptotic pathway. Front Aging Neurosci. 2014;6:184. | |
Bobrowska A, Paganetti P, Matthias P, Bates GP. Hdac6 knock-out increases tubulin acetylation but does not modify disease progression in the R6/2 mouse model of Huntington’s disease. PloS One. 2011;6(6):e20696. | |
Luthi-Carter R, Taylor DM, Pallos J, et al. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc Natl Acad Sci U S A. 2010;107(17):7927–7932. | |
Ma Y, Chen H, He X, et al. NAD+ metabolism and NAD(+)-dependent enzymes: promising therapeutic targets for neurological diseases. Curr Drug Targets. 2012;13(2):222–229. | |
Bobrowska A, Donmez G, Weiss A, Guarente L, Bates G. SIRT2 ablation has no effect on tubulin acetylation in brain, cholesterol biosynthesis or the progression of Huntington’s disease phenotypes in vivo. PloS One. 2012;7(4):e34805. | |
Chopra V, Quinti L, Kim J, et al. The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington’s disease mouse models. Cell Rep. 2012; 2(6):1492–1497. | |
Suter U, Scherer SS. Disease mechanisms in inherited neuropathies. Nat Rev Neurosci. 2003;4(9):714–726. | |
Tanabe K, Takei K. Dynamic instability of microtubules requires dynamin 2 and is impaired in a Charcot-Marie-Tooth mutant. J Cell Biol. 2009;185(6):939–948. | |
D’ Ydewalle C, Krishnan J, Chiheb DM, et al. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med. 2011;17(8):968–974. | |
Xu X, Kozikowski AP, Pozzo-Miller L. A selective histone deacetylase-6 inhibitor improves BDNF trafficking in hippocampal neurons from Mecp2 knockout mice: implications for Rett syndrome. Front Cell Neurosci. 2014;8:68. | |
Gold WA, Lacina TA, Cantrill LC, Christodoulou J. MeCP2 deficiency is associated with reduced levels of tubulin acetylation and can be restored using HDAC6 inhibitors. J Mol Med (Berl). Epub September 12, 2014. | |
Piao Y-S, Wakabayashi K, Kakita A, et al. Neuropathology with clinical correlations of sporadic amyotrophic lateral sclerosis: 102 autopsy cases examined between 1962 and 2000. Brain Pathol. 2003;13(1):10–22. | |
Watanabe M, Dykes-Hoberg M, Culotta VC, Price DL, Wong PC, Rothstein JD. Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis. 2001;8(6):933–941. | |
Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7(9):710–723. | |
Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. | |
Chiu AY, Zhai P, Dal Canto MC, et al. Age-dependent penetrance of disease in a transgenic mouse model of familial amyotrophic lateral sclerosis. Mol Cell Neurosci. 1995;6(4):349–362. | |
Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264(5166):1772–1775. | |
Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52(1):39–59. | |
Yoo Y-E, Ko C-P. Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2011;231(1):147–159. | |
Taes I, Timmers M, Hersmus N, et al. Hdac6 deletion delays disease progression in the SOD1G93A mouse model of ALS. Hum Mol Genet. 2013;22(9):1783–1790. | |
Gal J, Chen J, Barnett KR, Yang L, Brumley E, Zhu H. HDAC6 regulates mutant SOD1 aggregation through two SMIR motifs and tubulin acetylation. J Biol Chem. 2013;288(21):15035–15045. | |
Bruneteau G, Simonet T, Bauché S, et al. Muscle histone deacetylase 4 upregulation in amyotrophic lateral sclerosis: potential role in reinnervation ability and disease progression. Brain J Neurol. 2013;136(Pt 8):2359–2368. | |
Rich DP, Anderson MP, Gregory RJ, et al. Expression of cystic fibrosis transmembrane conductance regulator corrects defective chloride channel regulation in cystic fibrosis airway epithelial cells. Nature. 1990;347(6291):358–363. | |
Ciofu O, Hansen CR, Høiby N. Respiratory bacterial infections in cystic fibrosis. Curr Opin Pulm Med. 2013;19(3):251–258. | |
Hodges CA, Grady BR, Mishra K, Cotton CU, Drumm ML. Cystic fibrosis growth retardation is not correlated with loss of Cftr in the intestinal epithelium. Am J Physiol Gastrointest Liver Physiol. 2011; 301(3):G528–G536. | |
Wright FA, Strug LJ, Doshi VK, et al. Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat Genet. 2011;43(6):539–546. | |
Emond MJ, Louie T, Emerson J, et al. Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat Genet. 2012;44(8):886–889. | |
White NM, Jiang D, Burgess JD, Bederman IR, Previs SF, Kelley TJ. Altered cholesterol homeostasis in cultured and in vivo models of cystic fibrosis. Am J Physiol Lung Cell Mol Physiol. 2007;292(2):L476–L486. | |
Fredly H, Gjertsen BT, Bruserud O. Histone deacetylase inhibition in the treatment of acute myeloid leukemia: the effects of valproic acid on leukemic cells, and the clinical and experimental evidence for combining valproic acid with other antileukemic agents. Clin Epigenetics. 2013;5(1):12. | |
Lee Y-S, Lim K-H, Guo X, et al. The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res. 2008;68(18):7561–7569. | |
Ketene AN, Roberts PC, Shea AA, Schmelz EM, Agah M. Actin filaments play a primary role for structural integrity and viscoelastic response in cells. Integr Biol Quant Biosci Nano Macro. 2012;4(5):540–549. | |
Husain M, Cheung C-Y. Histone deacetylase 6 inhibits influenza A virus release by downregulating the trafficking of viral components to the plasma membrane via its substrate, acetylated microtubules. J Virol. 2014;88(19):11229–11239. | |
Husain M, Harrod KS. Enhanced acetylation of alpha-tubulin in influenza A virus infected epithelial cells. FEBS Lett. 2011;585(1):128–132. | |
Banerjee I, Miyake Y, Nobs SP, et al. Influenza A virus uses the aggresome processing machinery for host cell entry. Science. 2014; 346(6208):473–477. | |
Badding MA, Dean DA. Highly acetylated tubulin permits enhanced interactions with and trafficking of plasmids along microtubules. Gene Ther. 2013;20(6):616–624. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.