")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Brigatinib for ALK-positive metastatic non-small-cell lung cancer: design, development and place in therapy
Authors Ali R, Arshad J, Palacio S, Mudad R
Received 25 August 2018
Accepted for publication 9 January 2019
Published 8 February 2019 Volume 2019:13 Pages 569—580
DOI https://doi.org/10.2147/DDDT.S147499
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Manfred Ogris
Robert Ali,1 Junaid Arshad,1 Sofia Palacio,1 Raja Mudad2
1Department of Medicine, Division of Oncology, Jackson Memorial Hospital, University of Miami, Miller School of Medicine, Sylvester Comprehensive Cancer Centre, Miami, FL 33131, USA; 2Department of Medicine, Division of Oncology, University of Miami, Miller School of Medicine, Sylvester Comprehensive Cancer Centre, Miami, FL 33136, USA
Abstract: Despite the benefits of first and second generation anaplastic lymphoma kinase (ALK) inhibitors in the management of ALK-rearranged advanced non-small-cell lung cancer (NSCLC), the development of acquired resistance poses an ongoing dilemma. Brigatinib has demonstrated a wider spectrum of preclinical activity against crizotinib-resistant ALK mutant advanced NSCLC. The current review narrates a brief history of tyrosine kinases, the development and clinical background of brigatinib (including its pharmacology and molecular structure) and its use in ALK-positive NSCLC.
Keywords: non-small cell lung cancer, ALK positive, ALK inhibitors, brigatinib, TKI
Plain language summary
Discovery of the anaplastic lymphoma kinase along with EML4 translocation in 2007 was the first important step toward the development of the ALK tyrosine kinase inhibitors. Ceritinib and alectinib, two second generation ALK inhibitors, have been approved to treat patients with ALK-positive NSCLC who have progressed on crizotinib. However, the indication for alectinib has been expanded to the first line setting in the management of those patients with advanced ALK-positive NSCLC. An open-label, Phase I/II trial evaluated the role of brigatinib in the treatment of advanced malignancies, particularly ALK-rearranged NSCLC, which were refractory to available therapies. The ALTA (open-label Phase II randomized) trial showed improvement in PFS in patients with ALK-positive NSCLC. Brigatinib demonstrates an improved CNS PFS in patients with intracranial metastasis in both Phase I/II and Phase II (ALTA trial). Brigatinib acts as a multi-kinase inhibitor with a broad-spectrum activity against ALK, ROS1, FLT3, mutant variants of FLT3 and T790M-mutant EGFR. Pulmonary toxicity is a serious and dose-limiting side effect.
Introduction
According to the most recent Surveillance, Epidemiology, and End Results (SEER) database update, lung cancer accounts for 13.5% of all new cancer cases, with an estimated 234,030 newly diagnosed patients in 2018 among the US population. Conversely, lung cancer accounts for 25.3% of all cancer deaths and 5-year survival averages 18.6%.1
Traditionally, lung cancer has been categorized based on histopathology and immunohistochemical stains, being designated as either small-cell lung cancer or non-small-cell lung cancer (NSCLC). NSCLC accounts for about 80% of all lung cancer cases, and often presents at an advanced stage. Historically, cytotoxic chemotherapy has been the backbone of systemic therapy, with only a marginal improvement in the median overall survival (OS) of less than 1 year from diagnosis.2 However, the discovery of subsets of NSCLC with specific genetic alterations, and the development of targeted therapy for those subsets, has dramatically changed this treatment paradigm.
The identification of the anaplastic lymphoma kinase (ALK) gene rearrangement has been one of the more pivotal breakthroughs in the management of NSCLC over the past decade. It is present in 3%–7% of NSCLCs, and affords susceptibility to targeted therapy with ALK tyrosine kinase inhibitors (TKIs).3,4 Notably, preclinical work has also demonstrated that the patient population harboring epidermal growth factor receptor (EGFR) mutations did not overlap with those harboring the ALK gene rearrangement, recognizing ALK-positive NSCLC as a distinct, novel subclass within NSCLC.3
In this paper, we will delve into the details of the clinical development and management of this disease, focusing on the place of brigatinib in therapy.
Clinical manifestations of ALK-positive disease
Patients with ALK-positive rearrangement are diagnosed at a median age of 52 years, which is typically younger compared to the general NSCLC population regardless of subtype, and also among those harboring EGFR mutations. Whereas EGFR mutations are more common among females, ALK rearrangements have a greater predilection among males.5,6 Consistent among both subgroups, however, is that patients are never-smokers or light smokers (<10 pack-years). Tumors generally tend to be more centrally located, and patients often present with advanced disease. Cerebral and hepatic metastases are not uncommon, nor are pleural and pericardial effusions. This pattern seems to underscore the inherent aggressive nature of this disease.7,8
History of drug development for ALK-positive disease
Crizotinib was the first US Food and Drug Administration (FDA)-approved ALK TKI. Based on results from the Phase III clinical trial PROFILE 1014, it demonstrated superiority to chemotherapy, thus cementing its role as standard-of-care first line therapy in patients newly diagnosed with ALK-rearranged NSCLC. Among those treated with crizotinib in the first line, 74% had an objective response with a progression-free survival (PFS) of 11 months, compared to 45% and 7 months, respectively, in the platinum-doublet first line arm.9
Nonetheless, the acquisition of mutations that confer resistance seems almost inevitable, with progression usually occurring around 1 year following the initiation of therapy.9 Multiple mechanisms have been implicated in the development of resistance, and are generally stratified as either ALK dominant or ALK non-dominant.10,11 Furthermore, among patients treated with crizotinib, the first site of progression is usually the central nervous system (CNS) (25%–50%), and is believed to be due to inadequate CNS penetration of this drug.12,13 Similar to other TKIs, crizotinib appears to be a substrate for ABC transporters such as the ATP-dependent P-glycoprotein, which are able to actively restrict the passage of the drug through the blood–brain barrier.14 Consequently, this prompted the development of newer generation ALK TKIs to overcome these resistance patterns, and these include ceritinib, alectinib, brigatinib, ensartinib and lorlatinib.
The FDA granted accelerated approval of ceritinib in April 2014, for patients who progressed while receiving crizotinib.15 Alectinib received a similar approval for the same population in December 2015,16 followed by brigatinib in April 2017.17 Other ALK TKIs, such as lorlatinib, have been granted priority review or orphan drug status by the FDA for patients who have ALK TKI resistance. Approval of these agents has relegated traditional cytotoxic chemotherapy, and even immune checkpoint inhibitors, to the third line setting and beyond.
The J-ALEX study was a randomized, Phase III study comparing alectinib to crizotinib among patients with ALK-positive NSCLC, who were either chemotherapy-naïve or had received one prior chemotherapy regimen. Alectinib demonstrated superiority in terms of PFS and side-effect tolerance, with fewer patients discontinuing the drug compared to those in the crizotinib arm.18 Based on these results, alectinib is currently approved in both the USA and Europe in the first line setting. Nonetheless, for those communities where alectinib is not available or easily accessible, many treatment guidelines continue to recommend crizotinib upfront, with a switch to an alternative TKI upon progression or an intolerable side-effect profile. The choice of agent thereafter is often dependent on drug approval patterns, overlapping toxicity and, ultimately, concerns over cost.
Clinical background
Cellular homeostasis and mitogenesis is regulated by the interaction of growth factors with specific receptors present on the cell surface. One of the most important classes of receptors is the tyrosine kinase-associated receptor. Tyrosine kinase-associated cellular pathways are involved in altering important functions such as transcription and translation, and if dysregulated can lead to oncogenesis.19
There are at least 20 different classes of receptor tyrosine kinases (RTKs), including epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR), ALK and ROS receptor families.20 EGFR was one of the first RTKs to be discovered, in 1960.21
Honegger et al reported that the tyrosine kinase function of EGFR is related to the ATP binding pocket, which can interfere with the receptor signaling.22 Further studies led to the development of an EGFR inhibitor, gefitinib, which was later approved for the treatment of NSCLC in the USA in 2003.23
The development of other TKI molecules continued to be a hot topic for research and drug development. Although the ALK gene was initially discovered in 1994 in anaplastic large-cell lymphoma, it then led to the discovery of the EML4-ALK fusion gene in 2007 in a (5%) subset of pulmonary adenocarcinomas with the inversion (2)(p21;p23) rearrangement. Both ALK and EML4 genes are located on the short arm of chromosome 2. EML4-ALK translocation with chromosome 2 p inversion leads to a driver mutation with potent oncogenic potential. This translocation leads to the formation of a protein translated by the EML4 gene. As a result of the fusion with its partners, the new ALK protein migrates from the cell membrane to the cytoplasm and becomes more stable (increased half-life), which in turn results in ALK overexpression and activation.
Crizotinib was the first available TKI targeting the cMET and ALK fusion protein. There were two randomized controlled trials that led to the accelerated approval of crizotinib in ALK-positive NSCLC patients in 2010.24,51 Crizotinib was found to be effective only in patients with the ALK translocation. Some of the patients with NSCLC developed gatekeeper mutations L1196M within the kinase domain, making it unresponsive to crizotinib.25 One-third of ALK-positive NSCLC patients develop secondary mutations and approximately 40% have a primary refractory disease.3 This led to the development of second generation inhibitors of ALK fusion protein, namely ceritinib4 and alectinib.26 Although some of the second generation ALK inhibitors were able to overcome crizotinib-resistant mutations, novel mutations resistant to each of these agents quickly arose.27–29
This prompted the development of a newer generation TKI which would target these emerging mutations, namely brigatinib.
Pharmacology
Brigatinib is composed of a dimethylphosphine oxide (DMPO) group constructed in a U-shaped confirmation around a bis-anilinopyrimidine scaffold. It differs from crizotinib, which is developed around an aminopyridine group. The C2 and C4 positions in the pyrimidine ring bear two aniline groups, whereas C5 holds a chlorine atom. There is a methoxy group on the aniline ring at C2 which binds to a pocket under the ALK L1198 residue, thus filling the ribose binding pocket and providing interaction sites for more residues. The C5 chlorine atom interacts with the ALK L1196 gatekeeper residue. The DMPO group is incorporated as a hydrogen bond acceptor at the C4 aniline. These features impart important properties to the molecules, including increased hydrophilicity, decreased lipid solubility and limited protein binding.30 The route of administration is oral. After oral absorption, 66% of the drug is bound to the plasma proteins with an elimination half-life of 25 hours. The recommended doses include an initial dose of 90 mg/day for 7 days followed by an increase in the dose to 180 mg/day afterwards, if tolerable.17
Brigatinib acts as a multi-kinase inhibitor with a broad-spectrum activity against ALK, ROS1, FLT3, mutant variants of FLT3, T790M-mutant EGFR (deletions and point mutations) and IGFR-1R, with minimal activity against wild-type EGFR or MET.30 Brigatinib also has in vitro kinase activity against all mutations resistant to the first and second generation inhibitors, including ALK C1156Y, I1171S/T, V1180L, L1196M, L1152R/P, E1210K and G1269A mutations.30
The use of brigatinib is associated with a number of side effects. With grade 1–2 toxicities, the drug can be resumed at a lower dose after returning to the baseline. However, grade 4 toxicities require the complete discontinuation of the drug.
The most common side effects include nausea, vomiting and diarrhea, along with headaches. The most serious side effects include pneumonitis/interstitial lung disease, as per the ALK in Lung Cancer Trial of AP26113 (ALTA).17 In the trial, severe pulmonary adverse events were present in 3.7% of patients in the 90 mg group; however, there was an increase in the occurrence of events in patients with a dose increment to 180 mg. The pulmonary side effects manifest as worsening dyspnea and cough, especially in the first week. A high degree of clinical suspicion is required. Hypertension is another important side effect in all TKIs. As per the ALTA trial, grade 3 hypertension was found in 5.9% of the overall patient population. Blood pressure monitoring starting after 2 weeks of intake followed by monthly monitoring of the blood pressure is advised. Bradycardia, visual disturbances, creatinine phosphokinase (CPK) elevation, increased amylase/lipase and hyperglycemia are some of the less common side effects.17
Preclinical data and resistance patterns
The emergence of resistance to first line crizotinib occurs through two basic pathways: ALK-dependent mechanisms (accounting for 30% of patients) and ALK-independent mechanisms. The ALK-dependent model involves the gain of secondary mutations in ALK that interfere with crizotinib binding and/or amplification of the ALK fusion gene, with more than 10 secondary mutations having been identified. The most common mutations are L1196M and G1269A.10,11 Furthermore, the CNS as the first site of progression occurs in about 50% of patients, alluding to inadequate CNS penetration of crizotinib as the basis for resistance.12,31 Alternatively, ALK-independent mechanisms involve the emergence of a second mutated, overexpressed or amplified oncogene relative to the pretreated NSCLC, such as EGFR, KRAS, BRAF, MET, HER2 and KIT.10
Ceritinib and alectinib, two second generation ALK inhibitors, have been approved to treat patients with ALK-positive NSCLC who have progressed on crizotinib. Preclinical data have shown that these agents inhibit ALK more potently than crizotinib, and also maintain activity against many of the secondary mutations associated with crizotinib resistance.32,33 This has been corroborated clinically, with response rates of 49% for ceritinib and 58% for alectinib among previously treated patients. Furthermore, both of these agents have demonstrated activity in untreated CNS disease.15,34 That being said, progression is invariable, and a shorter time to progression has been demonstrated compared to first line crizotinib (median PFS =6.9–8.9 months).15,35 Furthermore, ALK secondary mutations related to resistance have been identified: F1174c/V specific for ceritinib, I1171N/T/S for alectinib and G1202R common to both.10,31,36
Brigatinib was subsequently developed as a potent, selective inhibitor of ALK, capable of overcoming resistance mechanisms associated with crizotinib. It is able to achieve levels of exposure in patients that substantially exceed those required to inhibit native ALK. An extensive characterization of the preclinical properties of brigatinib was outlined by Zhang et al.37 The experimental design was such that a kinase screen was performed to evaluate the selectivity profile of brigatinib, and the cellular and in vivo activities of ALK TKIs were compared using engineered and cancer-derived cell lines.
Across a panel of eight ALK-positive tumor-derived and engineered cell lines, brigatinib (median inhibitory concentration [IC50] =10 nmol/L) inhibits ALK with 12-fold greater potency than crizotinib. Total steady-state plasma levels (Cave) of brigatinib in patients dosed at 90 mg (582 nmol/L) and 180 mg (1,447 nmol/L) exceed the IC90 for native ALK inhibition by 15–38-fold, compared to the 2-fold increment of the crizotinib plasma steady state. These findings may be attributed to the higher selectivity of brigatinib for ALK, and imply a lesser propensity for pharmacological failure. Furthermore, the brigatinib doses coincide with those employed in the pivotal Phase II study.
In addition, brigatinib is less susceptible to inducing secondary resistance mutations in ALK, compared to crizotinib, ceritinib and alectinib. Various concentrations of the four ALK TKIs were analyzed in an in vitro mutagenesis screen using Ba/F3 cells expressing native EML4-ALK. In the side-to-side comparison, only brigatinib was able to suppress emergence of any ALK secondary mutant at 500 nmol/L, whereas higher concentrations of the other ALK TKIs (up to 1,000 nmol/L) were needed. Another panel of Ba/F3 cell lines was generated containing native EML4-ALK or 17 different secondary ALK mutations. The inhibitory profile of brigatinib was found to be superior to that of the other three ALK TKIs in both analyses. The IC50 of brigatinib against native EML4-ALK was 14 nmol/L, vs 107 nmol/L, 37 nmol/L and 25 nmol/L of crizotinib, ceritinib and alectinib, respectively. In addition, brigatinib was active against all resistant mutations tested (IC50 <200 nmol/L), specifically those reported in patients who progressed on ceritinib and alectinib (F1174C/V, I1171N and G1202R mutations). Brigatinib also inhibited nine different mutants with 3–54-fold greater potency compared to ceritinib and/or alectinib. Save for L1198F, brigatinib demonstrated greater activity than crizotinib for all mutants. Despite the overall superior selectivity over parental cells and for all mutants, G1202R remained relatively the most resistant to treatment (IC50 =184 nmol/L).
These promising in vitro data were corroborated with in vivo-based experiments. ALK-positive Karpas-299 (ALCL) and H2228 (NSCLC) xenograft mouse models were treated with escalated doses of oral brigatinib: 10 mg/kg, 25 mg/kg or 50 mg/kg daily. This led to a dose-dependent inhibition of tumor growth in both models, with deeper and more sustained remissions coinciding with increased plasma levels of brigatinib. Specifically, H2228-derived tumors demonstrated greater sensitivity to ALK inhibition than Karpas-299-derived tumors when exposed to concordant drug doses, with substantial tumor regression being maintained for greater than 28 days after treatment. Moreover, a 100 mg/kg/day dose of crizotinib inhibited tumor growth and ALK signaling to a similar degree to 10 mg/kg brigatinib.37
CNS activity of brigatinib was also evaluated using an orthotopic brain tumor model. ALK-positive H2228 (NSCLC) cells were injected intracranially to form brain tumors, and the mice were subsequently treated daily with vehicle, crizotinib (100 mg/kg) or brigatinib (25 or 50 mg/kg). Median survival for vehicle, crizotinib, brigatinib 25 mg/kg and brigatinib 50 mg/kg was 28 days, 47.5 days, 62 days and >64 days (study termination at day 64), respectively. There was also a significant reduction in tumor burden in the mice treated with 50 mg/kg brigatinib vs crizotinib.37
It should also be noted that the percentage of brigatinib not bound to human plasma protein in vitro is 4–100-fold greater than that of crizotinib, ceritinib and alectinib. Consequently, cellular assays performed at physiological levels of human plasma protein demonstrated a 2-fold reduction in brigatinib potency compared with a 2.7–4-fold reduction with crizotinib, ceritinib and alectinib.37 This may reflect an even greater potency of brigatinib in the clinical experience.
Clinical development
Despite the benefit of first and second generation ALK inhibitors in the management of ALK-rearranged advanced NSCLC, the development of acquired resistance poses an ongoing dilemma. Brigatinib (AP26113; ARIAD Pharmaceuticals, Cambridge, MA, USA) has demonstrated a wider spectrum of preclinical activity against crizotinib-resistant ALK mutants compared to both ceritinib and alectinib, adding to the ever-expanding treatment armamentarium.37
These findings were subsequently corroborated in two published clinical trials. The first was a Phase I/II study by Gettinger et al.39 In the post-hoc analysis, an objective response was clearly evident among 51 of the 71 patients who were previously treated with crizotinib. A reasonable PFS was also demonstrated, and the drug was fairly well tolerated. Notably, an idiosyncratic pulmonary toxicity was observed, but this was curtailed following the introduction of an antecedent dose-reduced loading phase.
The follow-up Phase II trial, ALTA, explored optimal dosing between a 90 mg/day regimen and a 180 mg/day regimen, each with a 1-week 90 mg/day pretreatment phase. Those patients treated with the 180 mg dose demonstrated a higher response, which was particularly notable among those with brain metastases.17
Several such studies are currently underway to evaluate the benefit of the newer generation ALK inhibitors over crizotinib. These include brigatinib (ALTA-1L, ALK in Lung Cancer Trial of Brigatinib in 1st Line [NCT02737501]), lorlatinib (NCT03052608) and ensartinib (eXalt3 trial [NCT027670804]).
Phase I/II trial
The trial was a Phase I/II design, and was single armed and open labeled. The aim was to evaluate the role of brigatinib in the treatment of advanced malignancies, particularly ALK-rearranged NSCLC, which were refractory to available therapies and for which no standard or available curative treatments existed.
In the Phase I dose-escalation stage of the trial, patients received oral brigatinib at total daily doses ranging from 30 to 300 mg, with an endpoint of establishing the recommended Phase II dose. Dose-limiting toxicities, specifically one grade 3 ALT elevation and one grade 4 dyspnea, were observed at the 240 mg/day and 300 mg/day dose, respectively. This led to the designation of 180 mg/day as the optimal Phase II dose.
In the Phase II expansion stage, three regimens were specified: 90 mg/day, 180 mg/day and 180 mg/day with a 7-day lead-in at 90 mg/day. The 7-day lead-in was stipulated in an attempt to offset the accumulating early pulmonary toxicity observed with upfront 180 mg/day dosing. Five cohorts were defined: 1) ALK inhibitor-naïve ALK-rearranged NSCLC, 2) crizotinib-treated ALK-rearranged NSCLC, 3) EGFR
A total of 137 patients were enrolled in the Phase I dose-escalation and the Phase II dose-expansion cohorts, all of whom were treated. Seventy-nine patients (58%) had ALK-rearranged NSCLC, and of these, 71 patients had previously received crizotinib. The median duration of treatment was 7.5 months (IQR 1.8–18.6 months) for all patients and 15.4 months (IQR 7.1–20.9 months) for patients with ALK-rearranged NSCLC. The median follow-up was 15.7 months (IQR 6.8–21.0 months) for all patients and 17.0 months (IQR 11.4–22.1 months) for patients with ALK-rearranged NSCLC.
Of the five patient groups, cohort 1 demonstrated an objective response rate of 100%, with all four patients responding to treatment with brigatinib (95% CI 40%–100%). The results were similarly impressive for cohort 2, with 31/42 patients showing a response (overall response rate [ORR] 74%, 95% CI 58%–86%). Notably, none of the three patients in cohort 3 demonstrated any response, and only 3/18 patients in cohort 4 responded (ORR 17%, 95% CI 4%–41%). The Phase II primary outcome for cohort 5 was 83%, with 5/6 patients demonstrating a CNS response.
Post-hoc analysis revealed that 51/71 patients (72%, 95% CI 60%–82%) with ALK-rearranged NSCLC who had previously been treated with crizotinib had an objective response. Furthermore, four (6%) of these patients were found to have a complete response (CR). Moreover, the median duration of response was 11.2 months (95% CI 7.6–29.7 months) in the crizotinib-treated group, but was not reached (NR) among crizotinib-naïve patients (95% CI 5.6 months to NR). When stratified according to various dosing schema, the impact on response was as follows: for 90 mg/day, 180 mg/day with the 7-day lead-in at 90 mg, and 180 mg/daily total dosing, the ORRs were 77% (13/10 patients, 95% CI 46%–95%), 80% (20/25, 95% CI 59%–93%) and 65% (15/23, 95% CI 43%–84%), respectively.
Median PFS was most robust in cohorts 1 and 2, not being reached in the ALK-inhibitor-naïve group (95% CI 7.4 months to NR) and 14.5 months (95% CI 9.2 months to NR) among those previously treated with crizotinib. Less impressively, cohort 4 had a median PFS of 1.8 months (95% CI 1.7–3.7 months) and one patient in cohort 3 progressed at 7.4 months. Finally, no PFS events (progression or death) were reported in cohort 5. In addition, when ALK-rearranged NSCLC patients were analyzed based on prior crizotinib therapy, those previously treated showed a median PFS of 13.2 months, but this was not reached in the crizotinib-naïve group.
ALTA trial
Based on the results of the prior study, an open-label, randomized Phase II trial was conducted to evaluate two different brigatinib dosing regimens among patients with locally advanced or metastatic ALK-positive NSCLC, who were refractory to crizotinib. It was titled the ALTA trial: ALK in Lung Cancer Trial of AP26113 (NCT02094573). Patients were randomized 1:1 to either brigatinib 90 mg/day (arm A) or 180 mg/day with a 7-day lead-in at 90 mg (arm B). Patients were also stratified based on brain metastases and best response to crizotinib. ORR according to RECIST v1.1 was set as the primary endpoint, with secondary endpoints including CNS response, duration of response, PFS, OS and safety and tolerability.17
A total of 222 patients were enrolled into the study, with 112 being assigned to arm A and 110 to arm B. Patients were followed for an average of 8 months. Investigator-assessed confirmed ORR was 45% (97.5% CI 43%–56%) in arm A, including one CR, and 54% (97.5% CI 43%–65%) in arm B, including four CRs. Notably, responses included one patient in arm B with the recalcitrant G1202R mutation who achieved a confirmed partial response (PR). The median time to response approached 2 months in both arms, and median PFS was 9.2 months (97.5% CI 7.4–15.6 months) in arm A and 12.9 months (97.5% CI 11.1 months to NR) in arm B.
An update of the ALTA trial, presented at the 2017 International Association for the Study of Lung Cancer (IASLC) World Conference on Lung Cancer, corroborated the benefit of brigatinib in this setting, particularly highlighting the advantage of the 180 mg dosing. Median follow-up was now more than twice that of the initially published study, and demonstrated ORRs of 46% and 55%, and median PFS of 9.2 months and 15.6 months in arms A and B, respectively. The median OS was not reached in arm A, and was 27.6 months in arm B.52
See Table 1 for a comparison between the pivotal Phase I/II trial and the randomized Phase II (ALTA) trial.
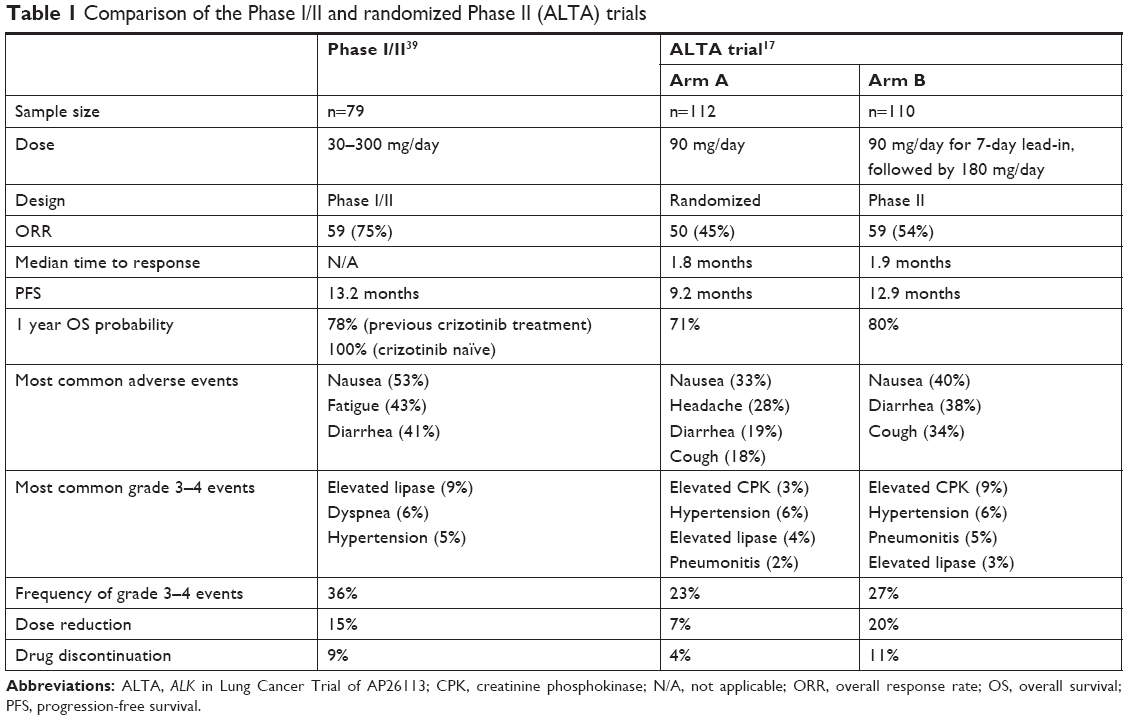 | Table 1 Comparison of the Phase I/II and randomized Phase II (ALTA) trials |
CNS activity
In the Phase I/II trial, 50 (63%) of the 79 patients with ALK-rearranged NSCLC had brain metastases at baseline, and 23 (46%) of these were naïve to cranial irradiation. Post-hoc analysis revealed that 46 of these 50 patients (92%) had had baseline imaging prior to starting brigatinib and at least one follow-up scan, allowing for evaluation of CNS response, with a median follow-up of 17.5 months. Fifteen of these 46 assessable patients (33%) had measurable brain metastases, with an intracranial response observed in 8/15 (53%). However, among the 31 (67%) patients with non-measurable disease, 11/31 (35%) had complete disappearance of CNS lesions on follow-up imaging. For all 19 patients who responded, the estimated median duration of intracranial response was 18.9 months (95% CI 5.5 months to NR), whereas for the entire group of 46 evaluable patients (responders and non-responders), the median intracranial PFS was 15.6 months (95% CI 13 months to NR).
Furthermore, 21/46 patients (47%) had not received prior cranial irradiation. Of the nine patients with measurable disease, five (56%) had a response, and similarly for those with non-measurable lesions, 7/12 (58%) had complete resolution of their brain lesions. The median intracranial PFS for this group of 21 patients without previous brain irradiation and treated with brigatinib was 22.3 months (95% CI 8–22.3 months).
In the ALTA trial, 154 patients (69%) had baseline brain metastases, with 44 of these patients having measurable lesions. Active brain metastases were defined as lesions without prior radiotherapy or with investigator-assessed progression after prior radiotherapy. Among patients with measurable baseline brain metastases, the independent review committee-assessed intracranial ORR was 42% (11/26 patients, 95% CI 23%–63%) in arm A and 67% (12/18 patients, 95% CI 41%–87%) in arm B. For those patients who demonstrated an intracranial response, the median duration of response was not reached in either arm. Furthermore, the median intracranial PFS was 15.6 months (95% CI 7.3–15.7 months) and 12.8 months (95% CI 11.0 months to NR) in arms A and B, respectively.
A follow-up analysis by Camidge et al which was published in May 2018 provides updates on the CNS response of patients enrolled in each trial.38 These results are summarized in Table 2, which compares baseline CNS characteristics and responses between the pivotal Phase I/II trial and the randomized Phase II (ALTA) trial. Of note, three patients in arm A of the ALTA trial had progression in the brain while receiving brigatinib 90 mg/day, owing to either increased size of the target brain lesion or the appearance of a new brain lesion. Following escalation of the dose to 180 mg/day, serial imaging revealed the best reductions in the sum of diameters of measurable lesions to be 19%, 38% and 100%. By extension, those patients in arm B had a numerically lower incidence of first disease progression both intracranially and extracranially compared to those in arm A. The whole-body efficacy among ALK-positive NSCLC patients with baseline brain metastases was quite reasonable, with ORRs of 74% (34/46), 40% (32/80) and 59% (43/73), and median systemic PFS of 14.5 months, 8.8 months and 12.9 months in the Phase I/II, ALTA arm A and ALTA arm B trials, respectively.38
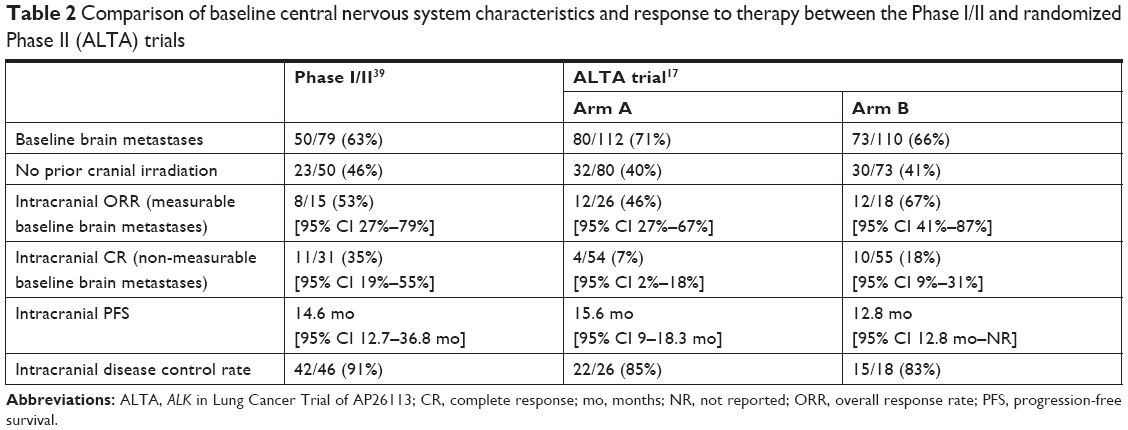 | Table 2 Comparison of baseline central nervous system characteristics and response to therapy between the Phase I/II and randomized Phase II (ALTA) trials |
In both trials, brigatinib has consistently yielded an impressive intracranial efficacy, with both high and durable responses. Furthermore, compared to both ceritinib and alectinib, ORRs were superior with brigatinib 180 mg (with lead-in) among patients with crizotinib-refractory ALK-positive NSCLC with measurable baseline brain metastases: 38% and 64% vs 67%, respectively.38,41 That being said, the limitation of both studies is that brain imaging was not performed and collected in a systematic, protocolized manner. This issue will be addressed in the upcoming randomized, Phase III trial of brigatinib 180 mg (with lead-in) vs crizotinib in ALK inhibitor-naïve patients with advanced ALK-positive NSCLC (ALTA-1L; Clinical trials identifier: NCT02737501), where intracranial disease will be followed longitudinally and prior CNS therapy will be fully detailed.
Safety and tolerability
ALK-specific TKIs offer an effective treatment strategy for NSCLC patients who are ALK rearranged. Implicit to the durable responses observed, as well as sequential therapy with the advent of newer ALK inhibitors, patients will foreseeably have a lengthy exposure to TKI therapy. As a result, it is paramount that the drugs have an acceptable safety and tolerability profile.
The Phase I/II trial was the first to explore this metric in a clinical setting, with doses ranging from 30 mg to 300 mg. In the dose-escalation phase, two dose-limiting toxicities were observed: one grade 3 increase in alanine aminotransferase and one grade 4 dyspnea, at 240 mg and 300 mg, respectively. This led to the selection of 180 mg as the initial recommended Phase II dose. This was subsequently modified, introducing an antecedent 7-day lead-in of 90 mg followed by the established 180 mg/day, in an attempt to circumvent the pulmonary toxicities observed with upfront higher dosing.
The most frequent adverse events included nausea (53%), fatigue (43%) and diarrhea (41%), which were primarily grades 1–2. Grade 3–4 adverse events included mainly elevated lipase (9%), dyspnea (6%) and hypertension (5%). Notably, there was a group of pulmonary events that categorically occurred within 7 days of starting treatment, but especially within the first 48 hours. Events were characterized by dyspnea, hypoxia, cough, pneumonia and pneumonitis, with an increasing frequency of events mirroring higher starting doses of brigatinib. This phenomenon seemed to have been mitigated with the 7-day lead-in dose of 90 mg, as none of the 32 patients treated in this sequence experienced pulmonary issues.
Dose reduction was mandated in 15/98 patients (15%) owing to adverse events in the Phase II expansion. Sixteen patients (12%) died during treatment or within 31 days of the last dose of brigatinib. Eight cases were attributed to progression of the underlying neoplasm. However, the cause of death among each of the other eight cases was mainly pulmonary related, and included pneumonia, acute respiratory distress syndrome, dyspnea, hypoxia and respiratory failure. One patient experienced sudden death and another patient died from unknown causes. Three of these deaths were, at least in part, believed to be secondary to treatment.
The spectrum of adverse events observed in the ALTA trial closely paralleled that in the Phase I/II study. Nausea (33%/40%, arm A/B), diarrhea (19%/38%, arm A/B), headache (28%/27%, arm A/B) and cough (18%/34%, arm A/B) were the most common events, and were mainly grade 1–2. Likewise, grade 3 and higher events were hypertension, pneumonia and increased lipase, all generally occurring in 3%–6% of cases.
Also akin to the Phase I/II trial, there was a subset of early pulmonary events of similar character occurring with a median time to onset of 2 days (range 1–9 days). Fourteen (6%) events all occurred at the 90 mg dose; however, no further episodes were observed despite escalation to 180 mg. Six patients required dose interruption and reintroduction, while seven patients discontinued treatment entirely.
Dose reduction secondary to any adverse event was required in 8/109 patients (7%) in arm A and 22/110 (20%) patients in arm B. Dose interruption was necessary in 20 patients (18%) and 40 patients (36%) in arms A and B, respectively. Eight patients (4%) died within 30 days of the last dose.
In the 2017 trial update, the most common grade 3 or higher events increased blood CPK (3%/11%), hypertension (4%/4%), increased lipase (4%/4%), rash (1%/4%) and pneumonitis (2%/4%) in arms A and B, respectively. Also, for arms A and B, dose-reduction rates were 9% and 30%, respectively, while treatment was discontinued in 4% and 11% of patients.
Tolerability was also gaged from the patient’s perspective, utilizing the European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire Core 30 (QLQ-C30). Questionnaires were completed at baseline and at the beginning of each cycle. Although no statistically significant difference was observed between the two arms, 80% of patients reported either an improvement or at least no change in their quality of life scores. Furthermore, up to 30% experienced a clinically meaningful reduction in symptoms.
What is the place of brigatinib in therapy?
The preceding two trials have clearly demonstrated the benefit of brigatinib among patients with ALK-positive NSCLC who have progressed on crizotinib (see Table 1). Brigatinib also shows promising activity among those patients who are ALK inhibitor naïve, as well as those with brain metastases. Furthermore, compared to the other two second generation ALK inhibitors, ceritinib and alectinib, the objective response rates and median PFS of brigatinib are comparable, and may even be superior. Nonetheless, cross-trial comparisons should be interpreted with a fair amount of scrutiny, bearing in mind the differences among the patient populations being analyzed.
The real question now arises as to how to appropriately sequence therapy to obtain the proverbial “biggest bang for one’s buck”. Alectinib has replaced crizotinib as the approved first line therapy among patients with ALK-rearranged NSCLC, with ceritinib and brigatinib being relegated to second line therapy following progression. But which of these should be used in preference? It is evident from preclinical data that brigatinib has the broadest in vitro coverage against resistance patterns as defined by the acquisition of ALK mutations. This was subsequently corroborated in the clinical realm when a patient with the recalcitrant G1202R mutation had a confirmed PR in the ALTA trial.
The J-ALEX study introduced new insight into upfront treatment with second generation ALK inhibitors, demonstrating an improved PFS favoring alectinib (not estimable) over crizotinib (10.2 months) in the first line setting (HR 0.34, P<0.0001).18 A similar study conducted by Peters et al further cemented the benefit of alectinib vs crizotinib in previously untreated, advanced ALK-positive NSCLC. The results of this study mirrored those of the J-ALEX trial, with PFS not being reached in the alectinib arm and being 11.1 months in the crizotinib group.40
In July 2018, Takeda Pharmaceuticals, the maker of brigatinib, issued a press release confirming that the ALTA-1L trial, a pivotal Phase III study investigating the activity of brigatinib in treatment-naïve patients, had reached its primary endpoint of superior PFS for brigatinib compared to crizotinib in the first line setting. Furthermore, in updates of the ALTA Phase II trial, brigatinib has shown an ORR of 56% and an unprecedented median PFS of 16.7 months and OS of 34.1 months. This is the longest PFS of any ALK inhibitor to be reported in the second line setting for patients who have progressed on crizotinib. Once brigatinib receives FDA approval, it will become another option for the first line treatment of ALK-positive NSCLC. In addition, a subsequent trial (NCT02706626) is being planned to elucidate the role of brigatinib following progression on second generation ALK inhibitors.
In terms of safety profiles, alectinib seems to be generally well tolerated, while ceritinib has a higher incidence of clinically significant drug-related adverse events. Brigatinib has also been shown to be reasonably safe, with the most concerning adverse event being early-onset pulmonary toxicity. This presented mainly as either pneumonitis or interstitial lung disease, and was associated with the 90 mg dose, regardless of whether this was the baseline dosing regimen or the 7-day lead-in to 180 mg. Notably, no events occurred with escalation to 180 mg.
Based on the efficacy and safety observed in both the Phase I/II trial and the ALTA study, the FDA granted accelerated approval to brigatinib for the treatment of metastatic crizotinib-resistant, ALK-positive NSCLC patients on April 28, 2017. The European Committee for Medicinal Products for Human Use has also recently mirrored this outlook, giving a positive opinion about brigatinib use in patients previously treated with crizotinib who have progressed or become intolerant to the drug. European marketing approval was eventually granted in November 2018.53
That being said, there are multiple other agents currently in development for this ALK/ROS1 subpopulation. Lorlatinib, another new generation ALK inhibitor, has proven efficacy in both in vitro and in vivo experiments, inhibiting cell growth in crizotinib- or alectinib-resistant ALK mutant lung cancer cell lines, as well as demonstrating a systemic and intracranial response in mouse models leading to prolonged survival.42,43 In the Phase I study conducted by Shaw et al, 54 patients with advanced ALK-positive or ROS1-positive NSCLC were enrolled to receive various doses of lorlatinib.44 The major dose-limiting toxicity was a grade 2 CNS effect which manifested as slowed speech and mentation, as well as difficulty with finding words. This was observed at doses of 200 mg/day, whereas a dose of 100 mg/day was found to be both efficacious and well tolerated, and was eventually adopted as the recommended dose in the subsequent Phase II study.45 Notably, Shaw et al detected a double mutation (ALK C1156Y/L1198F) in patients resistant to lorlatinib, which conferred resensitization to crizotinib.46
In the Phase II trial (NCT01970865), 275 ALK- or ROS1-positive patients received lorlatinib at 100 mg/day. Results were reported in six expansion cohorts according to prior treatments, with patients from four of these cohorts (197/257) having received prior therapy with ALK inhibitors. Among these, the ORR was 62.4% (95% CI 33%–74%) and intracranial ORR was 54.9% (95% CI 39%–75%). Moreover, among the 30 patients who received lorlatinib as first line therapy, 27 (90%) had a confirmed ORR. The median duration of response was 12.5 months (95% CI 8.4–23.7 months). As of November 2018, lorlatinib has been granted accelerated approval by the FDA for use in patients with ALK-positive metastatic NSCLC who have progressed on one or more ALK TKIs, based on this study. The Phase III CROWN study (NCT03052608), an open-label, randomized, double-blind, two-arm trial, which aims to compare lorlatinib with crizotinib as a first line treatment in patients with advanced ALK-positive NSCLC, is still recruiting.
Yet another agent with the potential to revolutionize treatment of ROS1-positive advanced NSCLC is entrectinib. It is a small-molecule TKI that targets oncogenic rearrangements in NTRK, ROS1 and ALK.47 At the IASLC 18th World Conference on Lung Cancer, Doebele’s group presented results of an integrated analysis of 53 patients with ROS1-positive NSCLC from three different clinical trials of entrectinib: Phase II STARTRK-2, Phase I STARTRK-1 and Phase I ALKA-372-001. The ORR was 77.4%, with a median duration of response of 24.6 months. Among the 23 patients with evaluable brain metastases, the intracranial response was 55%, with a duration of intracranial response of 12.9 months. Median PFS was 19 months, and with a median follow-up of 15.5 months, nine patients (17%) have died. Tolerance was reasonable, with only 3.9% of patients discontinuing the drug because of adverse events. Given that crizotinib is the only drug approved for ROS1-positive locally advanced/metastatic NSCLC, entrectinib may now be a viable first line option, especially for those patients with CNS involvement.48
Future directions
The discovery of the ALK gene rearrangement has revolutionized the treatment paradigm for NSCLC patients with this subtype. Multiple ALK inhibitors have already been trialed and tested in established niches, and, with several other agents on the horizon, the question now becomes: what is the optimal sequencing to offer the greatest longevity to our patients? Likewise, the durability of each TKI is variable, and suboptimal target inhibition may explain pharmacological failure and the emergence of various resistance mechanisms. Current and upcoming trials will inform choices for optimal first line therapy and how to effectively maneuver ALK TKI therapy, especially given that many tumors remain ALK dependent even beyond progression on first line ALK TKI.
Moreover, the advent of genomic profiling and next generation sequencing is leading the way for “patient-specific” therapy, now appropriately labeled precision medicine. Combination therapies involving ALK inhibitors may become the new trend. Indeed, data from recent trials such as KeyNote 04249 and IMpower 15050 have very encouragingly demonstrated the benefit of anti-programmed cell death protein-1 (PD-1) drugs among patients with advanced NSCLC. However, these drugs have traditionally not been as effective among never-smokers, who constitute a large proportion of the ALK-positive faction. Determining whether ALK inhibitors can potentiate the immune system and thereby magnify the effect of immunotherapy may be a worthwhile pursuit.
Ultimately, the true benefit of these targeted agents may be in elucidating an effective “add-on” strategy, which is both tolerable and safe. Incumbent on this premise would be to transform ALK-rearranged NSCLC into a chronic disease. Targeted therapies have indeed heralded a new era in the treatment of oncogene-driven malignancies.
Disclosure
Dr Raja Mudad has attended one advisory board for brigatinib by Takeda Pharmaceuticals, and received an honorarium for that activity in March 2018. The authors report no other conflicts of interest in this work.
References
SEER. Cancer stat facts: lung and bronchus cancer. Bethesda, MD: National Cancer Institute. Available from: https://seer.cancer.gov/statfacts/html/lungb.html. Accessed January 21, 2019. | ||
Schiller JH, Harrington D, Belani CP, et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med Overseas Ed. 2002;346(2):92–98. | ||
Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. | ||
Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13(10):1011–1019. | ||
Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer Who Harbor EML4-ALK. J Clin Oncol. 2009;27(26):4247–4253. | ||
Shaw AT, Solomon B. Targeting anaplastic lymphoma kinase in lung cancer. Clin Cancer Res. 2011;17(8):2081–2086. | ||
Doebele RC, Lu X, Sumey C, et al. Oncogene status predicts patterns of metastatic spread in treatment-naive nonsmall cell lung cancer. Cancer. 2012;118(18):4502–4511. | ||
Kang HJ, Lim HJ, Park JS, et al. Comparison of clinical characteristics between patients with ALK-positive and EGFR-positive lung adenocarcinoma. Respir Med. 2014;108(2):388–394. | ||
Solomon BJ, Mok T, Kim DW, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–2177. | ||
Katayama R, Lovly CM, Shaw AT. Therapeutic targeting of anaplastic lymphoma kinase in lung cancer: a paradigm for precision cancer medicine. Clin Cancer Res. 2015;21(10):2227–2235. | ||
Toyokawa G, Seto T. Updated evidence on the mechanisms of resistance to ALK inhibitors and strategies to overcome such resistance: clinical and preclinical data. Oncol Res Treat. 2015;38(6):291–298. | ||
Costa DB, Shaw AT, Ou SH, et al. Clinical experience with crizotinib in patients with advanced ALK-rearranged non-small-cell lung cancer and brain metastases. J Clin Oncol. 2015;33(17):1881–1888. | ||
Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol. 2011;29(15):e443–e445. | ||
Tang SC, Nguyen LN, Sparidans RW, Wagenaar E, Beijnen JH, Schinkel AH. Increased oral availability and brain accumulation of the ALK inhibitor crizotinib by coadministration of the p-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridar. Int J Cancer. 2014;134(6):1484–1494. | ||
Shaw AT, Kim DW, Mehra R, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370(13):1189–1197. | ||
Shaw AT, Gandhi L, Gadgeel S, et al; Study investigators. Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer: a single-group, multicentre, phase 2 trial. Lancet Oncol. 2016;17(2):234–242. | ||
Kim DW, Tiseo M, Ahn MJ, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase-positive non-small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol. 2017;35(22):2490–2498. | ||
Hida T, Nokihara H, Kondo M, et al. Alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer (J-ALEX): an open-label, randomised phase 3 trial. Lancet. 2017;390(10089):29–39. | ||
Perona R. Cell signalling: growth factors and tyrosine kinase receptors. Clin Transl Oncol. 2006;8(2):77–82. | ||
Ségaliny AI, Tellez-Gabriel M, Heymann MF, Heymann D. Receptor tyrosine kinases: characterisation, mechanism of action and therapeutic interests for bone cancers. J Bone Oncol. 2015;4(1):1–12. | ||
Schlessinger J. Receptor tyrosine kinases: legacy of the first two decades. Cold Spring Harb Perspect Biol. 2014;6(3):a008912. | ||
Honegger AM, Szapary D, Schmidt A, et al. A mutant epidermal growth factor receptor with defective protein tyrosine kinase is unable to stimulate proto-oncogene expression and DNA synthesis. Mol Cell Biol. 1987;7(12):4568–4571. | ||
Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4(5):361–370. | ||
Camidge DR, Bang YJ, Kwak EL, et al. Progression-free survival (PFS) from a phase I study of crizotinib (PF-02341066) in patients with ALK-positive non-small cell lung cancer (NSCLC). J Clin Oncol. 2011;29(suppl;abstr 2501). | ||
Katayama R, Khan TM, Benes C, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A. 2011;108(18):7535–7540. | ||
Kinoshita K, Asoh K, Furuichi N, et al. Design and synthesis of a highly selective, orally active and potent anaplastic lymphoma kinase inhibitor (CH5424802). Bioorg Med Chem. 2012;20(3):1271–1280. | ||
Huang WS, Liu S, Zou D, et al. Discovery of Brigatinib (AP26113), a phosphine oxide-containing, potent, orally active inhibitor of anaplastic lymphoma kinase. J Med Chem. 2016;59(10):4948–4964. | ||
Ignatius Ou SH, Azada M, Hsiang DJ, et al. Next-generation sequencing reveals a novel NSCLC ALK F1174V mutation and confirms ALK G1202R mutation confers high-level resistance to alectinib (CH5424802/RO5424802) in ALK-rearranged NSCLC patients who progressed on crizotinib. J Thorac Oncol. 2014;9(4):549–553. | ||
Katayama R, Friboulet L, Koike S, et al. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin Cancer Res. 2014;20(22):5686–5696. | ||
Sabari JK, Santini FC, Schram AM, et al. The activity, safety, and evolving role of brigatinib in patients with ALK-rearranged non-small cell lung cancers. Onco Targets Ther. 2017;10:1983–1992. | ||
Weickhardt AJ, Scheier B, Burke JM, et al. Local ablative therapy of oligoprogressive disease prolongs disease control by tyrosine kinase inhibitors in oncogene-addicted non-small-cell lung cancer. J Thorac Oncol. 2012;7(12):1807–1814. | ||
Friboulet L, Li N, Katayama R, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4(6):662–673. | ||
Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011;19(5):679–690. | ||
Gadgeel SM, Gandhi L, Riely GJ, et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014;15(10):1119–1128. | ||
Ou SH, Ahn JS, De Petris L, et al. Alectinib in crizotinib-refractory ALK-rearranged non-small-cell lung cancer: a phase II global study. J Clin Oncol. 2016;34(7):661–668. | ||
Ou SH, Greenbowe J, Khan ZU, et al. I1171 missense mutation (particularly I1171N) is a common resistance mutation in ALK-positive NSCLC patients who have progressive disease while on alectinib and is sensitive to ceritinib. Lung Cancer. 2015;88(2):231–234. | ||
Zhang S, Anjum R, Squillace R, et al. The potent ALK inhibitor Brigatinib (AP26113) overcomes mechanisms of resistance to first- and second-generation ALK inhibitors in preclinical models. Clin Cancer Res. 2016;22(22):5527–5538. | ||
Camidge DR, Kim DW, Tiseo M, et al. Exploratory analysis of Brigatinib activity in patients with anaplastic lymphoma kinase-positive non-small-cell lung cancer and brain metastases in two clinical trials. J Clin Oncol. 2018;36(26):2693–2701. | ||
Gettinger SN, Bazhenova LA, Langer CJ, et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: a single-arm, open-label, phase 1/2 trial. Lancet Oncol. 2016;17(12):1683–1696. | ||
Peters S, Camidge DR, Shaw AT, et al; ALEX Trial Investigators. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N Engl J Med. 2017;377(9):829–838. | ||
Gadgeel SM, Shaw AT, Govindan R, et al; ALEX Trial Investigators. Pooled analysis of CNS response to alectinib in two studies of pretreated patients with ALK-positive non-small-cell lung cancer. J Clin Oncol. 2016;34(34):4079–4085. | ||
Zou HY, Friboulet L, Kodack DP, et al. PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical models. Cancer Cell. 2015;28(1):70–81. | ||
Collier TL, Normandin MD, Stephenson NA, et al. Synthesis and preliminary PET imaging of 11C and 18F isotopologues of the ROS1/ALK inhibitor lorlatinib. Nat Commun. 2017;8:15761. | ||
Shaw AT, Felip E, Bauer TM, et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017;18(12):1590–1599. | ||
Solomon B, Shaw A, Ou S, et al. OA 05.06 phase 2 study of lorlatinib in patients with advanced ALK+/ROS1+ non-small-cell lung cancer. J Thorac Oncol. 2017;12(11):S1756. | ||
Shaw AT, Friboulet L, Leshchiner I, et al. Resensitization to Crizotinib by the Lorlatinib ALK Resistance Mutation L1198F. N Engl J Med. 2016;374(1):54–61. | ||
Liu D, Offin M, Harnicar S, Li BT, Drilon A. Entrectinib: an orally available, selective tyrosine kinase inhibitor for the treatment of NTRK, ROS1, and ALK fusion-positive solid tumors. Ther Clin Risk Manag. 2018;14:1247–1252. | ||
Ahn MJ, Cho BC, Siena S, et al. Entrectinib in patients with locally advanced or metastatic ROS1 fusion-positive non-small cell lung cancer (NSCLC). Presented at: IASLC 18th World Conference on Lung Cancer; 2017;15–18. Yokohama, Japan. Abstract 8564. | ||
De Lima Lopes G, Wu YL, Sadowski S, et al. P2.43: pembrolizumab vs platinum-based chemotherapy for PD-L1+ NSCLC: phase 3, randomized, open-label KEYNOTE-042 (NCT02220894): track: immunotherapy. J Thorac Oncol. 2016;11(10S):S244–S245. | ||
Socinski MA, Jotte RM, Cappuzzo F, et al. Atezolizumab for first-line treatment of metastatic Nonsquamous NSCLC. N Engl J Med. 2018;378(24):2288–2301. | ||
Crino LKD, Kim D, Riley GJ, et al. Initial phase II results with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC): PROFILE 1005. J Clin Oncol. 2011;29(suppl;abstr 7514). | ||
Ahn M, Camidge DR, Tiseo M, et al. Brigatinib in crizotinib refractory ALK+ NSCLC: updated efficacy and safety results from ALTA, a randomized phase 2 trial. J Thorac Oncol. 2017;12 No 11S2:S1755–1756. | ||
European Commission Approves ALUNBRIG® (brigatinib) for ALK+ Non-Small Cell Lung Cancer in Patients Previously Treated with Crizotinib, Advancing Treatment Paradigm in Europe. November 27, 2018. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.