")
Back to Journals » OncoTargets and Therapy » Volume 12
BRAF V600E-positive monomorphic epitheliotropic intestinal T-cell lymphoma complicating the course of hairy cell leukemia
Authors Broccoli A, Bertuzzi C, Fiorentino M, Morigi A, Stefoni V, Agostinelli C , Argnani L, Cavo M, Zinzani PL
Received 18 January 2019
Accepted for publication 3 May 2019
Published 20 June 2019 Volume 2019:12 Pages 4807—4812
DOI https://doi.org/10.2147/OTT.S202061
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr XuYu Yang
Alessandro Broccoli,1 Clara Bertuzzi,2 Michelangelo Fiorentino,3 Alice Morigi,1 Vittorio Stefoni,1 Claudio Agostinelli,2 Lisa Argnani,1 Michele Cavo,1 Pier Luigi Zinzani1
1Institute of Hematology “L. e A. Seràgnoli”, University of Bologna, Bologna, Italy; 2Hematopathology Unit, Institute of Hematology “L. e A. Seràgnoli”, University of Bologna, Bologna, Italy; 3Pathology Unit, “S.Orsola-Malpighi” Hospital, University of Bologna, Bologna, Italy
Abstract: Hairy cell leukemia (HCL) is an uncommon B-cell chronic lymphoproliferative disorder whose pathogenesis and recurrence are strictly dependent on the presence of the BRAF V600E mutant. A 65-year-old male presented a monomorphic epitheliotropic intestinal T-cell lymphoma (formerly enteropathy-associated T-cell lymphoma, type II) with HCL not responding to first-line induction with cladribine. The intestinal lymphoma bears the BRAF V600E mutant, which is the molecular hallmark of HCL, being implicated in its pathogenesis. The case is of interest, as it provides the first description of a BRAF V600E-positive intestinal T-cell lymphoma, along with immunohistochemical and molecular demonstration, occurring in concomitance with HCL. A novel digital PCR-base method for HCL disease assessment is also suggested.
Keywords: hairy-cell leukemia, BRAF V600E mutant, monomorphic epitheliotropic intestinal T-cell lymphoma, cladribine
Introduction
Hairy cell leukemia (HCL) is an uncommon B-cell chronic lymphoproliferative disorder whose pathogenesis and recurrence are strictly dependent on the presence of the BRAF V600E mutant, a pivotal player in the mitogen-activated protein (MAP) kinase pathway.1,2 The molecular landscape of HCL is rather simple: mutant BRAF is found in all cases and the same mutation persists during subsequent relapses.1 It is regarded as the sole molecular abnormality able to confer the leukemic phenotype by enhancing an autonomous cellular proliferation and inhibiting apoptosis.1 For this reason, mutated BRAF can be considered an effective therapeutic target: good responses are rapidly obtained in patients with relapsed disease treated with vemurafenib, a BRAF inhibitor.3 Although experiences with BRAF inhibitors are encouraging, first-line treatment is still based on purine analogs (cladribine and pentostatin), with up to 90% of complete responses. Even if outcomes are so favorable with induction therapy, patients tend to relapse over time and approximately 40% of them experience disease recurrence within 10 years.4,5
Despite the great biological importance of the BRAF gene mutation in this context, this seems to be quite a unique feature of HCL. Although BRAF-mutated non-Hodgkin lymphomas (NHL) have been described, mutations involving codon 600 are borne in just a minority of cases (2–16%, according to different case series) and never as a unique aberration.6,7 A few cases of BRAF V600E-positive chronic lymphocytic leukemia have been described, and some BRAF mutants have been identified in patients with Richter’s transformation.8
Here, we present an almost unique case of concomitant diagnosis of HCL and monomorphic epitheliotropic intestinal T-cell lymphoma (MEITL) in a patient who was refractory to cladribine.
Case report
A 65-year-old Caucasian man, with a history of mild thrombocytopenia (interpreted as immune) known since 2013 and never treated, was referred to our institution in August 2016 because of the recent appearance of petechiae at both lower limbs, without extension to the trunk or arms and without mucosal or major hemorrhage. Splenomegaly was detected upon physical examination.
Blood tests revealed severe neutropenia, with 1,950 leukocytes/mm3 (30% neutrophils), mild anemia (hemoglobin was 11.4 g/dL), and severe thrombocytopenia (platelets were 47,000/mm3). The biochemical profile demonstrated a mild increase of creatinine, 1.47 mg/dL, with no further abnormal findings. A bone marrow biopsy was performed, showing a marked hypocellularity (15%), along with an interstitial and diffuse infiltrate (90% of cellularity) composed of small lymphocytes with abundant pale cytoplasm and round-to-oval nuclei and a grade 1 marrow fibrosis. The immunohistochemical evaluation showed positivity for CD20 and annexin A1 (ANXA1). A droplet digital polymerase chain reaction assay performed on peripheral blood mononuclear cells demonstrated the presence of the BRAF V600E mutation, with a fractional abundance of the mutated allele (which denotes the proportion of the mutant allele frequencies) of 37.9%. A diagnosis of HCL was made.
Subcutaneous cladribine was started in September 2016 at the total dose of 10 mg to be delivered once a week for 5 consecutive weeks. The day before the second injection, the patient was admitted to the emergency room because of fever, chills, and right testis tenderness. A diagnosis of febrile neutropenia with orchiepididymitis was made. Bilateral pleural effusion, without pulmonary infiltrates, was evident at high-resolution computed tomography scan; ascites was also documented upon abdominal ultrasound. The patient rapidly became anuric. Biochemical tests showed marked hypercreatininemia (5.6 mg/dL), hyperkaliemia (6.9 mEq/L), and metabolic acidosis. Blood cultures were negative for aerobic or anaerobic bacteria and galactomannans were negative.
A wide-spectrum antibiotic treatment was started, along with furosemide and albumin supplementation to reduce the ascitic fluid. A diagnostic paracentesis demonstrated an exudate with no hairy cells. Fluid balance, diuretics, and electrolyte correction allowed a complete recovery from acute renal failure. Intravenous methylprednisolone was started at this point, with an initial clinical and laboratory improvement. Nevertheless, a sudden acute respiratory failure developed soon after due to acute pulmonary edema. Non-invasive ventilation and diuretics could restore an adequate respiratory function, and the patient was again addressed to subcutaneous cladribine administration in November 2016 (2 months after the first dose). At this point, it was decided to administer cladribine once a day for 5 consecutive days. Fifty days after treatment completion, cytopenia had not resolved, as leukocytes were still lower than 1,000/mm3 and neutrophils <500/mm3 irrespective of granulocyte colony-stimulating factor support. Thrombocytopenia also persisted (70,000/mm3), along with splenomegaly, pleural effusions, and ascites. A hairy cell marrow infiltration was confirmed (Figure 1, Panels A-B), and the BRAF V600E fractional abundance remained persistently high (10.33%), indicating a lack of response.
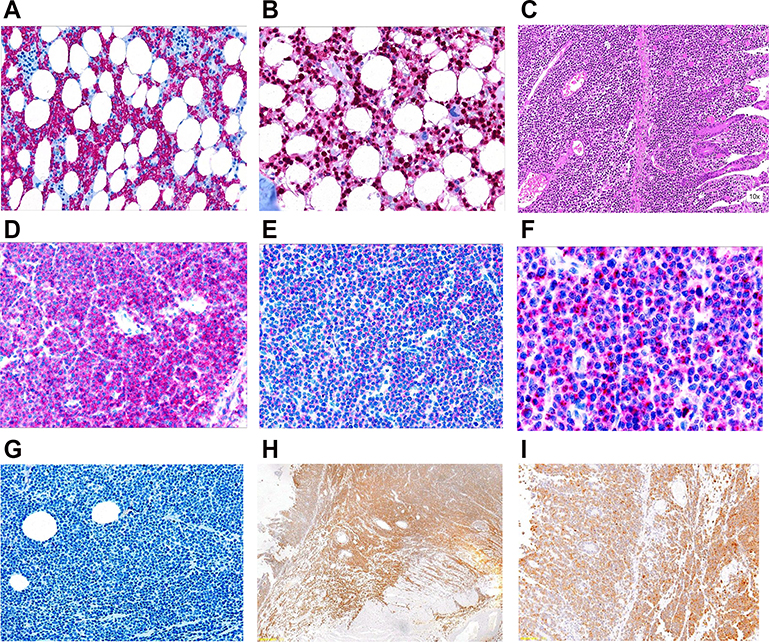 | Figure 1 Microscopic appearance of the bone marrow (panels A and B) and of the affected intestinal wall after jejunal resection (panels C–I). Bone marrow immunohistochemistry for CD20 and annexin A1, showing strong and diffuse positivity, is depicted in panels A and B, respectively. Panel C: hematoxylin-eosin staining. Panels D–F: immunohistochemistry for CD3, CD56, and TIA-1, respectively. Panel G: negative immunohistochemistry for CD20 on affected intestinal tissue (no evidence of hairy cell leukemia cells). Panels H-I: immunohistochemistry for BRAF V600E on intestinal tissue (panoramic and magnified view). Note BRAF negativity on unaffected intestinal tissue. |
Less than 1 month later, the patient was admitted again to the emergency department because of acute abdominal pain, diarrhea, and clinical signs of bowel perforation. An emergency exploratory laparotomy revealed jejunal perforation, which prompted the resection of 9 cm of affected small bowel. The resected intestinal wall was extensively infiltrated by CD20−, PAX5−, CD3+, CD4−, CD8+, and ANXA1− lymphoid elements, with a Ki-67 of 70%. CD56 and TIA-1 were also positive (Figure 1, Panels C-G). A diagnosis of MEITL was made. Interestingly, BRAF pyrosequencing on affected intestinal tissue showed a thymine to adenine substitution at position 1,799, consistently with the presence of the V600E mutant. Conversely, the mutation was absent on unaffected resection margins (Figure 2, Panel A), confirming a somatic – and not a germ-line – mutation. The expression of mutant BRAF was also confirmed by immunohistochemistry (Figure 1, Panels H-I). Molecular analysis was performed on both biopsies (bone marrow and intestine) to determine rearrangement of genes coding for immunoglobulin heavy chains (IGH) and T-cell receptor γ (TRG), trying to find out whether the two neoplasms carried rearrangements of the same size. DNA was extracted and tested by control gene PCR (100–400 bp amplicons) to verify its integrity. PCR assay was performed according to EUROCLONALITY protocols and evaluated by GeneScanning analysis to determine clonal character. Both samples presented low DNA quality and amount. Therefore, it was possible to analyze only the frameshift region 3 (FR3) of immunoglobulins, which showed the presence of a reproducible 125 bp clone in both samples (Figure 2, Panel B). The analysis of the TRG resulted partially unverifiable, even if we obtained a polyclonal pattern in both samples, the low quality of the material would have made the comparison unreliable, according to euroclonality guidelines.
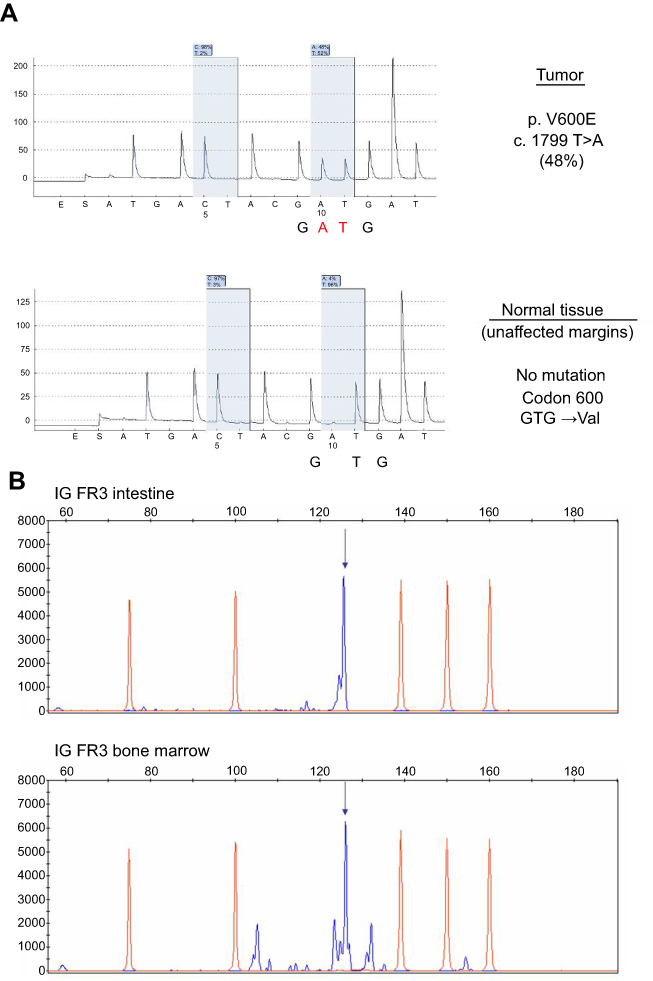 | Figure 2 Panel A: BRAF gene pyrosequencing on resected intestinal tissue, showing a T>A substitution in the affected tissue (upper diagram) and no mutation in unaffected margins (lower diagram). Panel B: analysis of the frameshift region 3 (FR3) of immunoglobulins on both marrow and intestinal samples. Proof of evidence of a reproducible 125 bp clone in both samples. |
The patient died in February 2017 due to an acute episode of intestinal occlusion. A vemurafenib-based treatment should have been taken into consideration if the patient had survived.
Discussion
The simultaneous occurrence of T- and B-cell neoplasms is exceptional and the etiologic and pathogenetic relationships between the two phenotypically different malignancies are not clear. An association between HCL and a T-cell NHL has been described in a few cases,9–11 but all the reported experiences could not provide any proof of a shared pathogenesis of the two conditions. MEITL, previously sub-classified as enteropathy-associated T-cell lymphoma type II, is a rare (<1% of NHL) and primarily gastrointestinal type of peripheral T-cell lymphoma with an aggressive clinical course and a poor prognosis. MEITL shows a unique genomic profile, characterized by recurrent mutations of the SETD2 oncogene, along with deregulated JAK/STAT pathway. The MAP-kinase pathway is also frequently affected, as up to 80% of MEITL cases bear mutations of TP53 (33%), BRAF (26%) and KRAS (20%).12
The co-occurrence of two rare malignancies leads us to hypothesize a shared pathogenetic mechanisms, as it seems unlikely a matter of chance. In other terms, a common ontogenesis of the two neoplasms can be postulated. Given that BRAF mutation and IGH-FR3 rearrangement, as detected in this case, are mutually independent, this observation strengthens the possible common ontogeny of the two neoplasms. Nevertheless, it is hard to discriminate whether they display a common origin versus one neoplasm originates from the other as a result of transdifferentiation. This is because no technique is able to reliably demonstrate this. To the best of our knowledge, this is the first time that the same underlying molecular lesion is found in MEITL concomitantly with active HCL, ie, a disease that does not respond properly to induction therapy. This reinforces the hypothesis that mutant BRAF V600E may have played a role in the development and progression of MEITL as well.
As it is known that BRAF gene mutations are found in hematopoietic stem cells in patients with HCL, the existence of a common progenitor is a possible explanation for the case we described.13 Tumoral reprogramming of an initiating HCL BRAF-mutated cell may also account for tumor cell plasticity, ending up with a change of the initial neoplastic phenotype and the development of a secondary lymphoid disease, possibly favored by the setting of profound immune suppression due to HCL itself, its treatment and complications.14 Alternatively, a B-to-T-cell dedifferentiation followed by phenotype conversion can be another possible explanation.15 The presence of a shared IGH clone in both marrow and intestinal tissue – in other words in both hairy cells and T-cell lymphoma tissue – may support these theories.
Conclusion
This is an interesting case of concurrent HCL and MEITL sharing the same genetic and phenotypic manifestation. Importantly, the T-cell lymphoma was diagnosed later than the initial HCL, when it became symptomatic. Since that moment, in fact, the patient displayed the clinical and laboratory features of HCL, although unexpected and rather unusual complications (such as ascites) occurred during treatment. The presence of the BRAF V600E mutant and consequently of a disrupted MAP kinase pathway, along with the demonstration of the same IGH rearrangement in both entities, can provide an explanation for a shared pathogenesis of the two diseases.
Ethical approval
Ethical approval was given by “S.Orsola-Malpighi” Hospital ethics committee for reporting this case.
Consent
We have obtained a written informed consent from the patient’s relatives for all of the anonymized information to be published.
Abbreviations list
HCL, hairy cell leukemia; MAP, mitogen-activated protein; NHL, non-Hodgkin lymphoma; MEITL, monomorphic epitheliotropic intestinal T-cell lymphoma; ANXA1, annexin A1; TRG, T-cell receptor γ; FR3, frameshift region 3.
Acknowledgments
The authors are grateful to Dr Anna Gazzola and Dr Claudia Mannu for their work on molecular analyses.
Author contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Tiacci E, Trifonov V, Schiavoni G, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;364(24):2305–2315. doi:10.1056/NEJMoa1014209
2. Foucar K, Falini B, Stein H, et al. Hairy cell leukaemia. In: Swerdlow SH, Campo E, Harris NL, editors WHO Classification of Tumour of Haematopoietic and Lymphoid Tissues. Lyon: IARC press; 2017:226–228.
3. Tiacci E, Park JH, De Carolis L, et al. Targeting mutant BRAF in relapsed or refractory hairy cell leukemia. N Engl J Med. 2015;373(18):1733–1747. doi:10.1056/NEJMoa1506583
4. Robak T, Matutes E, Catovsky D, et al. Hairy cell leukaemia: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26(suppl 5):100–107. doi:10.1093/annonc/mdv200
5. Else M, Dearden CE, Matutes E, et al. Long-term follow-up of 233 patients with hairy cell leukaemia, treated initially with pentostatin or cladribine, at a median of 16 years from diagnosis. Br J Haematol. 2009;145(6):733–740. doi:10.1111/j.1365-2141.2009.07668.x
6. Lee JW, Yoo NJ, Soung YH, et al. BRAF mutations in non-Hodgkin’s lymphoma. Br J Cancer. 2003;89(10):1958–1960. doi:10.1038/sj.bjc.6601371
7. Pillonel V, Juskevivius D, Ng CKY, et al. High-throughput sequencing of nodal marginal zone lymphomas identifies recurrent BRAF mutations. Leukemia. 2018;32(11):2412–2426. doi:10.1038/s41375-018-0082-4
8. Sellar RS, Fend F, Akarca A, et al. BRAF V600E mutations are found in Richter syndrome and may allow targeted therapy in a subset of patients. Br J Haematol. 2015;170(2):268–287. doi:10.1111/bjh.13279
9. Lawlor E, O’Briain DS, Flinn T, et al. The simultaneous presentation of peripheral T-cell lymphoma and hairy cell leukemia. Cancer. 1987;60(7):1537–1544.
10. Paolini R, Poletti A, Ramazzina E, et al. Co-existence of cutaneous T-cell lymphoma and B hairy cell leukemia. Am J Hematol. 2000;64(3):197–202.
11. Gasljevic G, Kloboves-Prevodnik V, Gazic B, Vovk M. Coexistent hairy cell leukemia and hepatosplenic T-cell lymphoma: a case report. Diagn Pathol. 2014;9(1):58. doi:10.1186/1746-1596-9-58
12. Roberti A, Dobay MP, Bisig B, et al. Type II enteropathy-associated T-cell lymphoma features a unique genomic profile with highly recurrent SETD2 alterations. Nat Commun. 2016;7:12602. doi:10.1038/ncomms12602
13. Chung SS, Kim E, Park JH, et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci Transl Med. 2014;6(238):238ra71. doi:10.1126/scitranslmed.3008004
14. Campos-Sánchez E, Cobaleda C. Tumoral reprogramming: plasticity takes a walk on the wild side. Biochim Biophys Acta. 2015;1849(4):436–447. doi:10.1016/j.bbagrm.2014.07.003
15. Cobaleda C, Jochum W, Busslinger M. Conversion of mature B cells into T cells by dedifferentiation to uncommitted progenitors. Nature. 2007;449(7161):473–477. doi:10.1038/nature06159
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.