")
Back to Journals » OncoTargets and Therapy » Volume 13
Biostable Double-Strand Circular Aptamers Conjugated Onto Dendrimers for Specific Capture and Inhibition of Circulating Leukemia Cells
Authors Li Y, Zhang T, Huang J, Dong H , Xie J, Jia L
Received 27 October 2020
Accepted for publication 15 December 2020
Published 5 January 2021 Volume 2020:13 Pages 13465—13477
DOI https://doi.org/10.2147/OTT.S287720
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Alberto Bongiovanni
Yu Li,1,* Ting Zhang,2,* Jing Huang,1 Haiyan Dong,1 Jingjing Xie,3 Lee Jia4
1Fujian Key Laboratory for Translational Research in Cancer and Neurodegenerative Diseases, Institute for Translational Medicine, School of Basic Medical Sciences, Fujian Medical University, Fuzhou, Fujian 350122, People’s Republic of China; 2Cancer Metastasis Alert and Prevention Center, and Pharmaceutical Photocatalysis of State Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fujian Provincial Key Laboratory of Cancer Metastasis Chemoprevention and Chemotherapy, Fuzhou University, Fuzhou 350002, People’s Republic of China; 3School of Pharmaceutical Sciences, Fujian Provincial Key Laboratory of Innovative Drug Target Research, Xiamen University, Xiamen, Fujian 361102, People’s Republic of China; 4Institute of Oceanography, Minjiang University, Fuzhou, Fujian 350108, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Haiyan Dong
Institute for Translational Medicine, School of Basic Medical Sciences, Fujian Medical University, 1 Xue Yuan Road, University Town, Fuzhou, Fujian, People’s Republic of China
Email [email protected]
Jingjing Xie
School of Pharmaceutical Sciences, Fujian Provincial Key Laboratory of Innovative Drug Target Research, Xiamen University, Xiang’an South Road, Xiamen, People’s Republic of China
Email [email protected]
Background/Objective: Circulating tumor cells (CTCs) are known as the root of cancer metastasis. Capture and inhibition of CTCs may prevent metastasis. Due to the rarity of CTCs in vivo, the current technology about CTCs capture is still challenging. The aim of our study was to conjugate the enhanced biostable double-strand (ds) circular aptamer (dApR) with dendrimers for capturing and restraining CTCs in vitro and in vivo.
Methods: CEM-targeting aptamer (Ap) was looped by ligation after phosphorylation to form circular ds aptamer dApR, which was then conjugated to dendrimers by biotin-streptavidin affinity reaction and named as G-dApR. The physicochemical properties of G-dApR were characterized by using PAGE gel electrophoresis, UV, DLS, AFM, fluorophotometer and laser confocal microscope. Biostability of G-dApR was also analyzed by gel electrophoresis. Confocal microscopy and flow cytometry were then performed to determine the binding specificity of G-dApR to CEM cells and the captured CTCs in mice and in human blood. Apoptosis of the captured cells was finally evaluated by using MTT assay, DAPI staining, AO/EB staining, cell cycle analysis and Annexin V-FITC/PI staining.
Results: Physicochemical characterization demonstrated the entity of dApR and G-dApR, and the nano-size of G-dApR (about 180 nm in aqueous phase). G-dApR exhibited the excellent biostability that confers their resistance to nuclease-mediated biodegradation in serum for at least 6 days. In our established CTCs model, we found that G-dApR could specifically and sensitively capture CTCs not non-target cells even in the presence of millions of interfering cells (108), in mice and in human blood. Finally, the activity of captured CTCs was significantly down-regulated by G-dApR, resulting in apoptosis.
Conclusion: We created the enhanced biostable dApR-coated dendrimers (G-dApR) that could specifically capture and restrain CTCs in vitro and in vivo for preventing CTC-mediated cancer metastasis.
Keywords: metastasis, CTCs, G-dApR, biostability, specificity
Introduction
Currently, aptamers have been emerged as a new powerful tool for targeting molecules since it was first discovered in 1990,1 which are being reviewed as a next-generation technology for capture and detection of biomolecules including cells and biomarkers.2,3 As an alternative to antibody, aptamers that are generated from oligonucleotides by a process termed SELEX possess many intrinsic merits, such as small size, chemically automated synthesis, flexible structure, long-term stability, lack of immunogenicity, and controllable modification. Due to these advantages, aptamers may overcome the inherent disadvantages of antibody-based biotechnology that suffers from its inherent drawbacks including instability, immunogenicity, high cost and big molecular size. Nevertheless, as the single-stranded DNA or RNA oligonucleotides, aptamers commonly face the risk of losing their functionality caused by nuclease-mediated degradation.4,5 To stabilize aptamers, helpful approaches are urgently needed, and closed circular DNA (ccDNA) may be one solution to resolve this problem because they are resistant to nuclease to some extent.6 Moreover, it is reported that nanomaterials not only can protect aptamers from enzymatic degradation and nonspecific binding,7,8 but also address the problem of insufficient efficiency in target capture owing to their unique structural and functional properties.9 These factors inspired us to construct the cyclization of the ordinary DNA aptamers, and assemble the circular aptamers onto dendrimers to improve the in vivo biostability and capture capability of ordinary aptamers. We hypothesize that assembly of double-strand circular aptamer (dApR) with PAMAM dendrimers (G-dApR) will show great biostability and specificity in capturing target.
To verify the biostability and capture specificity of G-dApR, circulating tumor cells (CTCs) was adopted as a typical target to perform the in vitro and in vivo experiments. CTCs are defined as a class of cancer cells that circulate in the bloodstream after escape from the primary tumor sites, which are considered to be the root of cancer metastasis that is the main cause for cancer-related deaths.10−12 Single-digit CTCs can even lead to cancer metastasis and lift the death rate to a higher level.13 Therefore, prevention of CTCs from metastasis is one key to suppress mortality. Although researchers have undertaken numerous efforts to isolate,14–16 enrich,17 detect,18 characterize and even ex-culture CTCs,19 since CTCs were first discovered in 1869,20 yet none has perfectly reached the final goal of CTCs-related cancer metastasis prevention. This is because CTCs are extremely rare, even appearing at rates as low as one cell per 106 or 107 leukocytes, and precisely finding CTCs among the huge number of hematologic cells and specifically inhibiting their activity are still technically challenging.21,22 Therefore, developing G-dApR for CTCs capture can provide the precise and specific tool against CTCs-related metastases.
In this work, we designed dApR to target CTCs, and then linked dApR onto the nanomaterial PAMAM dendrimer to specifically capture CTCs with enhanced biostability, namely, G-dApR. We then utilized various means to characterize G-dApR, evaluated their capability to capture CTCs in vitro and in vivo, and determined whether they can suppress CTCs activity through induction of apoptosis. Here, we provided the conceptual innovation for preventing CTCs from metastases by specific capture of the rare CTCs in blood and apoptosis-mediated inhibition of CTCs activity based on dApR-coated dendrimer with highly enhanced biostability.
Materials and Methods
Materials and Reagents
All DNA sequences were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China). PAMAM dendrimers (G4.5, theoretical MW 23438, ethylenediamine core) were purchased from Weihai CY Dendrimer Technology Co., Ltd (Shandong, China). 1-ethyl-3-(3-dimethyl-aminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS) were obtained from Aladdin Reagent Co., Ltd. T4 DNA polymerase, T4 DNA ligase, T4 polynucleotide kinase (PNK), ATP, and DNA lander (DL500) with DNA loading buffer (6×) were purchased from Takara Biotechnology Co., Ltd (Dalian, China). The RPMI 1640 medium, and fetal bovine serum (FBS) were purchased from Life Technologies GmbH (Darmstadt, Germany). SYBR Green (10,000×) and carboxyfluorescein diacetate succinimidyl ester (CFSE) were purchased from Solarbio Life Sciences (Beijing, China). Streptavidin, Yeast tRNA, 4ʹ,6-diamidino-2-phenylindole (DAPI), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Hoechst 33258, propidium iodide (PI), and other chemicals were purchased from Sigma-Aldrich.
Cell Culture
Human acute lymphatic leukemia CCRF-CEM (CEM) cell lines and Human Caucasian Burkitt’s lymphoma Ramos cell lines were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). All cell lines were authenticated and suspension-cultured in RPMI 1640 medium.
Animals
Four- to six-week-old female Balb/c mice were purchased from Fuzhou Wushi Animal Center. All mice were maintained in the plastic cages in an SPF-grade animal room with access to food and water ad libitum, and exposed to a 12 h light/dark cycle. The animal assays were conducted according to the NSFC regulation concerning the care and use of experimental animals, and approved by the Animal Care and Use Committee of Fujian Medical University.
Sequence Design of Aptamer and Confirmation of Their Capability to Target CEM Cell Lines
Our previous method23–25 was used to design the aptamer sequences and control probes. Aptamers and relevant sequences were listed in Table 1. Some sequences were modified with biotins and labeled with a fluorophore (FAM) to meet the experimental requirement. The capability of aptamers to target CEM cell lines was confirmed by flow cytometry, and the procedures were similar to what we reported previously.23–25 Briefly, cell lines were incubated with aptamers in the binding buffer at 37 °C overnight in the dark, and then determined by a FACScan cytometer (BD Biosciences).
 |
Table 1 The Sequences Designed in This Work |
Synthesis, Characterization and Biostability of dApR
The methods were similar to what we reported.24,25 Biotinylated aptamers (Ap-bio) were purchased from Sangon Biotech Co., Ltd (Shanghai, China). Firstly, we followed the manufacturer’s protocol to prepare phosphorylated aptamers and linear DNAs (L1) using ATP and PNK. Next, the ligation product of phosphorylated aptamer was obtained by hybridization with their template (T1) and ligation with T4 DNA ligase. We then utilized T4 DNA polymerase to remove the unreacted templates and aptamers followed by purification, and therefore we produced the single-strand circular aptamer and DNA, namely, ApR and C1. ApR hybridized with C1 to become the double-strand circular aptamer, namely, dApR. PAGE gel electrophoresis and flow cytometry assays were performed to characterize dApR, confirming their biostability and capability to target CEM cell lines.
Synthesis and Characterization of dApR-Coated Nanomaterials
Firstly, PAMAM dendrimer G4.5 was covalently modified with streptavidin by using cross-linking reaction with EDC and NHS, namely, G-Str. Next, biotinylated dApR was stirred with G4.5-Str for 4 h followed by purification via dialysis to generate dApR-coated G4.5, namely, G-dApR. To characterize G-dApR, various means were conducted in the present study. The PAGE gel electrophoresis was performed to determine whether dApR was conjugated onto G4.5. A laser confocal microscope was used to observe whether FAM-labeled dApR was linked to G4.5. UV spectrophotometer (Shimadzu, Japan) and a Cary Eclipse fluorescence spectrometer (Varian Ltd., USA) were applied to examine characteristic absorption of DNA at λ260 nm and fluorescence intensity of FAM at λ518 nm. The standard curves of Ap solutions for quantifying the amount of ApR on the surface of G-dApR were also obtained from fluorescent measurement. Zetasizer Nano ZS Particle Size and zeta potential Analyzer (Malvern Instruments Ltd., UK) was used to determine the mean particle size and zeta potential of G-dApR. The surface morphology and particle size of G-dApR were observed under atomic force microscope (AFM) (Agilent AFM 500, tapping probe 40 N m−1, in AC mode).
Biostability of G-dApR
Firstly, the biostability of G-dApR in buffer with different pHs (pH=9.0, 4.0 and 7.0) for 0 to 6 days was examined by a UV spectrophotometer (λuv=260 nm). Secondly, to test the biostability of G-dApR in serum, G-dApR was incubated with serum at 37 °C for 0 to 6 days, then the fluorescence intensity of treated samples was measured by a fluorescence spectrometer (FAM: λex=488 nm and λem=518 nm). The % G-dApR remaining was calculated by the following equation: % G-dApR =(S0-Sd)/S0×100%, where S0, Sd were the signal values at day 0 and thereafter. The signal value at day 0 was defined as 100%. PAGE gel assays were also conducted to confirm the biostability of G-dApR after incubation with serum at 37 °C for 0.5, 3 and 9 h-shaking under 100 rpm.
Capability of G-dApR to Capture Target Cells
The procedures were similar to what we described previously.23–25 To determine the specificity of G-dApR to bind target cells in the absence of interfering cells, G-dApR was incubated with CEM or Ramos cell lines in the binding buffer at 37 °C overnight, and then the laser confocal microscopy and flow cytometry were performed to determine specific binding between G-dApR and target cells. To confirm the capability of G-dApR to capture target cells in the presence of interfering cells, CEM cells were spiked into Ramos or RBCs at a mixture ratio of 1:106 and 1:108 followed by incubation with G-dApR for 2 h, then, we performed flow cytometry to examine the numbers of target cells captured by G-dApR. To determine the capability of G-dApR for cell capture, we also evaluated the binding affinities of G-dApR and Ap to CEM cells by following the methods in Supporting Information Methods section.
Effects of G-dApR on Cell Viability, Apoptosis and Cell Cycle of Captured Cells
The analyses were carried out as we reported.23–25 Briefly, CEM and Ramos cells were exposed to G-dApR at three concentrations (5, 20 and 45 μg/mL) for 48 h. We then performed assays of cell viability, apoptosis and cell cycle using MTT assay, DAPI staining, AO/EB staining and flow cytometry.
CTCs Capture in vivo and in the Healthy Blood
To evaluate the specificity of G-dApR in capturing CTCs, we conducted the assays of CTCs capture in vivo and in the human healthy blood by following the procedures that we reported previously.24,25 1) Assay of CTCs capture in vivo: we intravenously injected the Hoechst 33258-stained cancer cells (2×104) into the mice followed by injection of G-dApR, respectively. After that, we withdrew the mice blood and separated CEM cells using RBCs lysis followed by enrichments with centrifugation. 2) Assay of CTCs capture in the human healthy blood: 1000 CEM cells were spiked into 1, 2 or 4 mL of human healthy blood, and incubated with G-dApR for 2 h. Then, we separated CEM cells using RBCs lysis followed by enrichments with centrifugation. Finally, we used the confocal microscope and flow cytometry to analyze each sample.
Statistical Analysis
Data were expressed as means ±SD (n=3). Statistical analysis was conducted with Student’s t-test and one-way analysis of variance by SPSS statistical software (version 19.0). The symbol * and ** represent the comparison between sample and control, and NS, #, and ## represent the comparison between different groups. NS, */# and **/## indicate no significant difference, P<0.05 and P<0.01, respectively. A probability (P) value of 0.05 or less was considered significant, and P of 0.01 was highly significant.
Results and Discussion
Design, Synthesis and Characterization of Biostable dApR That Specifically Targeted CEM Cells
Generally, ordinary DNA aptamers consisted of oligonucleotides are vulnerable to nuclease-mediated biodegradation. The chemical modifications containing of the 2´-end substitutions and 3´-end modifications are often used to enhance the biostability of aptamers. However, these methods suffer from inherent defects such as unknown side-effects, low targeting capability and increased complexity of synthesis. Here, we introduced the double-strand circular DNA into aptamer to explore the enhanced stability of double-strand circular aptamers, namely dApR, aiming at resisting against nuclease-mediated degradation.
According to the method we reported before,24,25 aptamer (Ap), auxiliary sequences (L1, L1-T and T1) and control probes (probe1, probe2 and probe3) in Table 1 were designed by the mFold program (http://mfold.rna.albany.edu/). Some sequences were labeled with fluorophore FAM and/or biotin to meet experimental requirements.
We firstly designed the ordinary aptamer named as Ap to target CEM cells, and auxiliary DNA sequences for generating circular aptamer and circular DNA (Table 1). As a single-stranded (ss) oligonucleotide of 78 bases, Ap is composed of a reported aptamer that targets the protein tyrosine kinase-7 (PTK7) on the surface of CEM cell,26 a sequence for constructing circular DNA that could noticeably enhance biostability of the entire aptamer, and a fluorophore FAM that is used to visualize the binding of Ap to CEM cells. The control probes (probe1, probe2 and probe3) were correspondingly tagged with FAM (Table 1). To evaluate the capacity of Ap to target CEM cells, PTK7-negative Ramos were used as negative control cell.
Next, we tested the fluorescence responses of Ap and the controls (probe1-probe3) to CEM and Ramos cells. The fluorescence intensities resulted from binding between Ap and CEM were significantly higher than those from probe1, probe2 and probe3, but not from binding between Ap and Ramos (Figure 1A). To quantitatively compare the targeting ability of Ap to target cells, the fluorescent signal ratios of CEM and Ramos cells were calculated after incubation with aptamers and control probes (Figure 1B). We found that Ap triggered approximate 3.6-flod fluorescence increase in compare to control probes after its binding to CEM cells, indicating that Ap could specifically target CEM cells not Ramos cells.
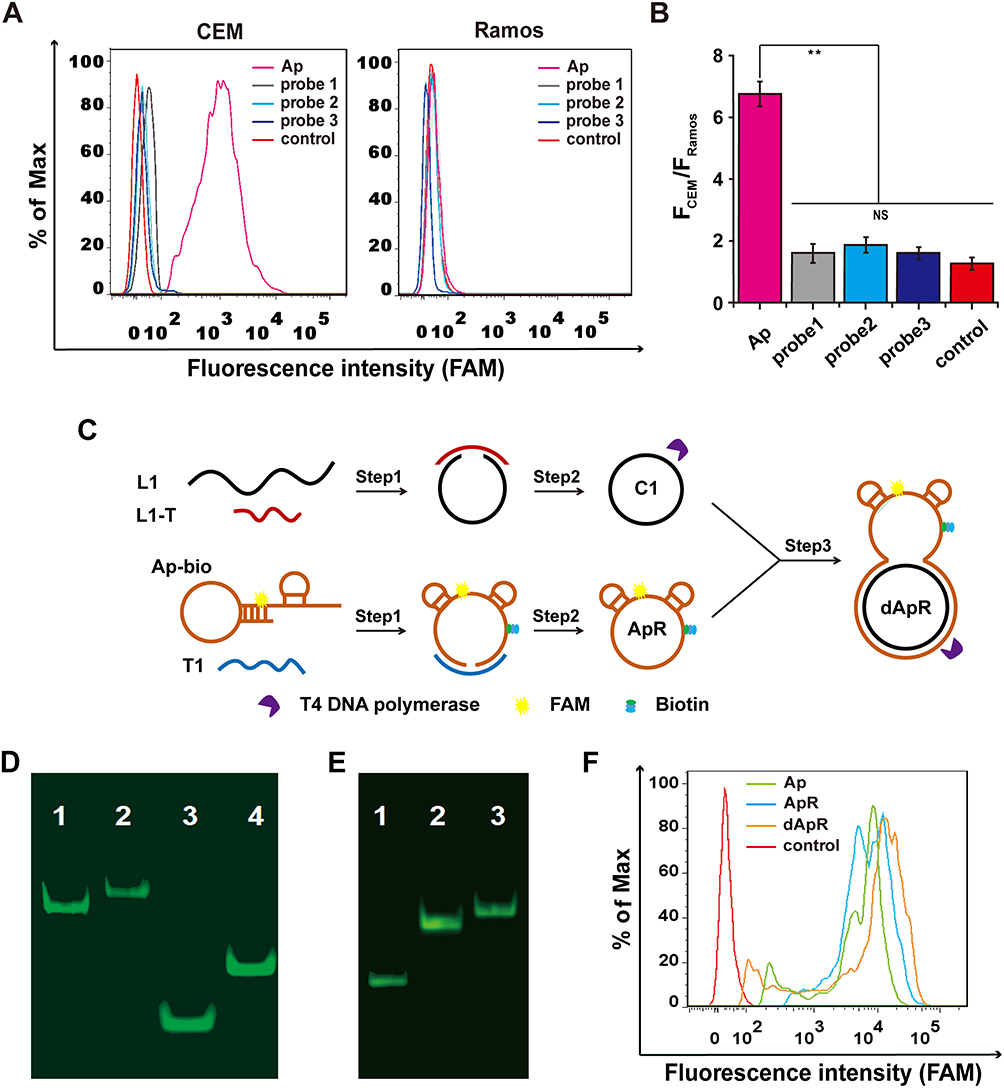 |
Figure 1 Design and synthesis of the double-strand (ds) circular aptamer (dApR) and their biostability and targeting functionality. (A) Flow cytometry assays proved that Aptamer (Ap) targeted CEM (target cells), but not the control Ramos (non-target cells), while the control probes (probe1, probe2 and probe3) did not bind to CEM and Ramos cells. (B) Quantitative fluorescence ratios of the target CEM cells to the control Ramos cells for binding Ap. (C) The procedure for preparing dApR. L1 hybridized with L1-T, while biotinylated Ap (Ap-bio) hybridized with T1, and then single strand circular DNAs (C1 and ApR) were obtained by ligation. ApR and C1 hybridized with each other to turn into the double strand circular aptamer, namely, dApR. (D) The successful synthesis of dApR was confirmed by comparison of difference in mobility between Ap and dApR, or T1 and C1 in PAGE gel. Lane 1: Ap, Lane 2: dApR, Lane 3: T1, Lane 4: C1. (E) The biostability of ApR and dApR was evaluated by PAGE gel analysis after their incubation with T4 DNA polymerase (5 μM) for 2 h, while Ap (Lane 1) degrade, but not ApR (Lane 2) and dApR (Lane 3). (F) Flow cytometry data showed that dApR bound to CEM cells more specifically than Ap and ApR did, indicating the greater binding efficiency. NS and ** respectively indicates no statistical difference and P < 0.01. |
After confirmation of the specificity of Ap, we generated double-strand circular aptamer (dApR) from ordinary aptamer to enhance its biostability by following the synthetic procedure that was shown in Figure 1C. Biotinylated Ap (Ap-bio) hybridized with template 1 (T1), meanwhile, complementary DNA (L1) hybridized with its template (L1-T) (Step 1). After DNA ligation and digestion by T4 DNA polymerase, Ap and L1 turned into single circular aptamer and single circular DNA, namely ApR and C1, respectively (Step 2). By repetition of step 1, ApR hybridized with C1 to form dApR that owned a unique capture hand carrying Ap for targeting CEM cells and a circular body for resistance to biodegradation caused by nucleases. Ordinary aptamer could be digested by nucleases, but the circular one may resist enzymatic digestion. To confirm the successful assembly of dApR, the Urea-PAGE gel electrophoresis was implemented to compare differences in mobility between dApR and Ap. Figure 1D shows that dApR (Lane 2) and C1 (Lane 4) ran slower than Ap (Lane 1) and L1 (Lane 3), suggesting the successful synthesis of dApR because circular DNAs run slower than linear ones. The result was in accordance with that we reported before.24,25 To evaluate biostability of dApR, we loaded Ap, ApR and dApR solution to PAGE gel for degradation analysis after treatment with 5 μM T4 DNA polymerase in reaction buffer at 37 °C for as long as 1 h (Figure 1E). The results demonstrated that the bands of ApR (Lane 2) and dApR (Lane 3) appeared at higher site in image, but the band of Ap (Lane 1) was seen at lower site, revealing that ApR and dApR molecules remained intact while Ap degraded (Figure 1E). We also utilized flow cytometry to analyze the targeting functionality of ApR and dApR to CEM cells. The results confirmed that ApR and dApR exhibited the identical specificity with Ap (Figure 1F).
Together, we successfully designed, synthesized and characterized dApR carrying the very biostable circular body and specific capture hand that could precisely target CEM cells.
Construction and Characterization of dApR-Coated Dendrimers
We firstly prepared dApR by following the above method (Figure 1C), and then chose carboxyl PAMAM dendrimers G4.5 (G) as a carrier for dApR because of its inherent advantages such as low cytotoxicity, good biocompatibility and large number of surface functional groups that make it easier for large numbers of dApR simultaneously linked to one G surface.27 As shown in Figure 2A, we firstly obtained G-Str by modification of G with streptavidin under standard amide coupling conditions in the presence of EDC and NHS. Then, we used the affinity interaction between streptavidin and biotin to construct dApR-coated PAMAM dendrimers, namely, G-dApR, which was able to significantly target CEM cells.
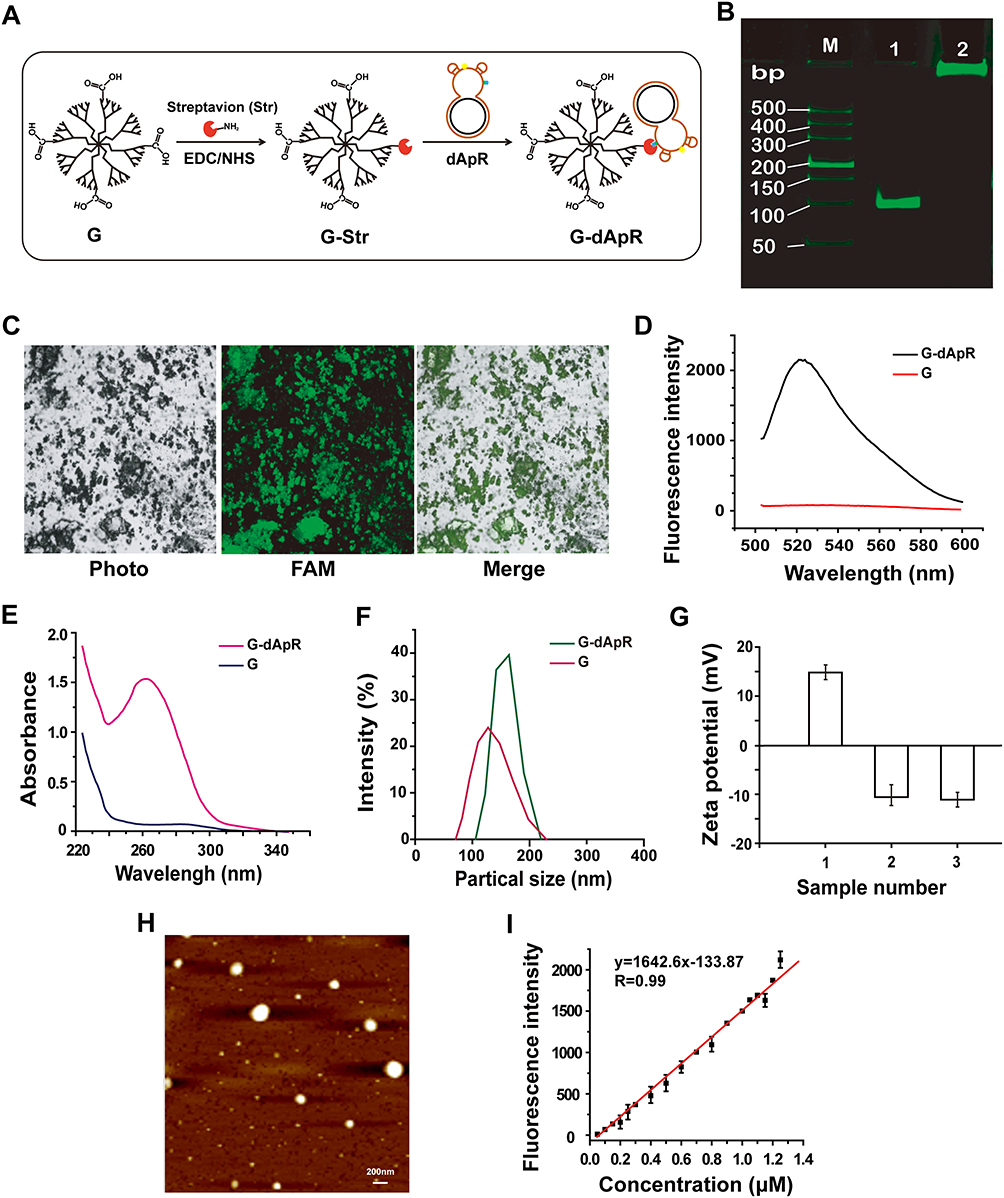 |
Figure 2 Synthesis and characterization of dApR-coated PAMAM dendrimers for targeting CEM cells. (A) Synthesis of dApR-coated G4.5 PAMAM dendrimer (G-dApR). Streptavidin reacted with carboxyl G4.5 PAMAM dendrimer (G) to obtain the intermediate product (G-Str) first, and then G-Str was conjugated with biotinylated dApR to form G-dApR. (B) PAGE gel electrophoresis verified the conjugation of dApR to G. (M) DL2000 DNA marker, Lane 1: dApR, Lane 2: G-dApR. (C) Fluorescent images of G-dApR solution by a laser confocal microscope. (D, E) Fluorescent (D) and UV (E) spectra of G and G-dApR. (F) Particle sizes of G and G-dApR in aqueous phase. (G) zeta potentials of G (1), Ap (2) and G-dApR (3). (H) AFM image of G-dApR. (I) Quantitative curves of Ap for determination of the conjugation efficiency of dApR onto PAMAM dendrimers. |
PAGE gel electrophoresis assay was conducted to confirm whether dApR was conjugated to G surface (Figure 2B). The G-dApR stayed in the sample hole without any mobility (Lane 2) while G-free entities dApR (Lane 1) ran faster on the gel, indicating that dApR was successfully linked to G. This success was also proved by fluorescent images of FAM-labeled G-dApR that were collected when it was excited at 488 nm under a laser confocal microscope. The green color in FAM channel and green-gray color in merged channel were clearly observed, providing evidence for the successful conjugation of dApR with G (Figure 2C). Moreover, the fluorescence intensity increases at λ518 nm was attributed to FAM-labeled dApR, further confirming that dApR was successfully conjugated with G (Figure 2D). The ultraviolet absorption values at λ260 nm measured by UV spectrophotometer were significantly increased as a result of the nucleic acids conjugated with G, also suggesting the successful synthesis of G-dApR (Figure 2E).
The dynamic light scattering (DLS) diameter of G-dApR in aqueous phase was measured to be about 180 nm, indicating that the size of G-dApR was slightly bigger than G (Figure 2F). The zeta potential of G became completely negative from the positive charge after Ap conjugation (Figure 2G), further suggesting the successful conjugation of dApR with G. The morphology of G-dApR was then observed under AFM (Figure 2H). It was found that the shape of G-dApR was round and its diameter was approximately 150 nm that was smaller than the DLS results possibly because of the different testing conditions, which were identical with our previous findings.24,25
We also established the fluorescent standard calibration curves of Ap with linearity R2=0.99 within the concentration range from 0.05 to 1.25 μM to quantify the amount of dApR conjugated with G (Figure 2I). After calculation, it was inferred that per mg of G carried 1.15 nmol dApR, which demonstrated that the payload capacity of G was much higher than previous achievements.28 This is probably because that affinity reaction between streptavidin and biotin is very easy to happen, and the surface area of G is enlarged by streptavidin and biotin as a linker, which makes it easier for dApR conjugated to G.
The Enhanced Biostability and Capability of G-dApR to Specifically Target CEM Cells in the Absence and Presence of Interfering Cells in vitro
The current aptamer-based technologies for CTCs detection commonly suffer from the failure in vivo and in clinical application owing to the low abundance of CTCs in the complex bloodstream, the nuclease-triggered biodegradation and the decreased specificity of ordinary aptamer.18,29 Thus, the biostability, specificity and binding affinity of G-dApR play the key role in their in vivo and clinical application. Here, we investigated the biostability, specificity and binding affinity of G-dApR for targeting CEM cells in the absence and presence of interfering cells by various means.
We evaluated the biostability of G-dApR by UV spectrophotometer. As shown in Figure 3A and B, no significant degradation was observed when they were exposed to different pH buffers (Figure 3A) and 37 °C serum (Figure 3B) reflecting the highly stable properties of G-dApR. After incubation of G-dApR with the healthy blood at 37 °C for 0.5, 3 and 9 h, no significant degradation bands were observed in PAGE gel assay (Figure 3C), further proving the enhanced biostability of G-dApR. Importantly, our finding verified previous reports that the interaction between nanomaterial dendrimers and DNA may perhaps protect DNA against degradation caused by nuclease.30,31 Because the biostability plays a key role in medical application, G-dApR could be a novel tool for capturing CTCs in vitro, particularly in vivo.
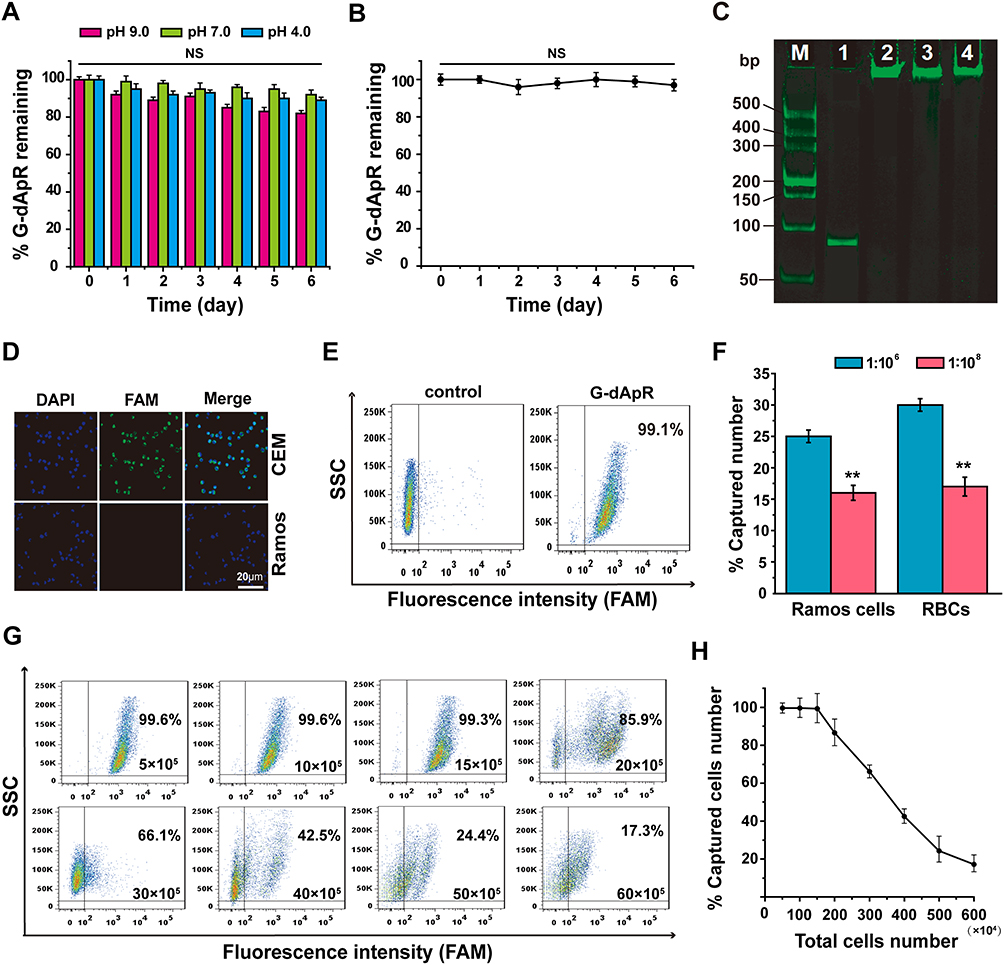 |
Figure 3 The enhanced biostability and specificity of G-dApR for targeting CEM cells. (A, B) G-dApR exhibited the enhanced biostability in buffers with different pH values (A) and in serum (B) at 37 °C for 0, 1, 2, 3, 4, 5 and 6 days. (C) In PAGE gel assay, the significant degradation of G-dApR (Lane 2, 3, 4) was not observed after incubation with the healthy blood at 37 °C for 0.5, 3 and 9 h-shaking at 100 rpm, respectively. Lane1: Ap, Lane (M) DL2000 ladder. (D) The confocal images of the bound CEM and Ramos cells after incubation with G-dApR. CEM cells incubated with G-dApR showed green-blue color for its binding, while G-dApR did not bind to Ramos cells. (E) The binding specificity of G-dApR to CEM cells was quantitatively evaluated by flow cytometry. The results showed that the percentage of captured CEM cell numbers by G-dApR was 99.1%. (F) The statistical analysis of captured target cells numbers by G-dApR from the buffer with Romas or RBCs. CEM cells were mixed with Ramos or RBCs at the ratio of 1:106 (blue pillar) or 1:108 (red pillar). The data showed that G-dApR could capture CEM cells in a cell number-dependent manner in the presence of a large number of interfering cells (Romas or RBCs). (G) Flow cytometric analysis showed that the captured number percentage of CEM cells by G-dApR maintained more than 99% accompanied with the cell amounts from 5×105 to 15×105, and then gradually decreased as more cells were added. Note: the concentration of G-dApR was the same in each sample, which was much lower than the concentration of its corresponding targets (CEM cells). (H) The curve-fitting from the flow cytometric data (G) illustrated the dynamic relationship between the captured number percentage of CEM cells (%) and the total cell amounts. Data represent mean ± standard deviation of three cell samples. NS and ** respectively indicates no statistical difference and P < 0.01. |
To assess the ability of G-dApR to target CEM cells, CEM and Ramos cells treated with G-dApR were observed by confocal microscopy. The fluorescence images were shown in Figure 3D, in which the blue color represented the nuclei of cells, green color and merged color represented the cells that were bound with G-dApR, respectively. Green and merged fluorescence were observed significantly from CEM cells, showing the specific binding between G-dApR and CEM cells, while only blue color was seen in Ramos cells. Our data clearly showed that G-dApR could specifically bind to its target CEM cell, but not negative control Ramos cell. Next, we conducted flow cytometry assays to quantitatively analyze the number of CEM cells captured by G-dApR in the absence of interfering cells. The results showed that the percentage of CEM cells captured by G-dApR was 99.1%, supporting the great binding efficiency of G-dApR to target CEM cells in the absence of interfering cells (Figure 3E). To confirm the capability of G-dApR to capture target cells in the presence of interfering cells, we mixed CEM cells with Ramos cells or RBCs at a mixture ratio of 1:106 or 1:108 to quantitatively analyze the counts of CEM cells captured from Ramos cells or RBCs by G-dApR via flow cytometry. The data showed that G-dApR could capture CEM cells in a cell density-dependent manner in the presence of a large number of interfering cells (Figure 3F). To further prove the specificity of G-dApR and discuss the binding relationship between G-dApR and CEM cells, we added the same amount of G-dApR to different samples of CEM cells solution with different numbers ranging from 5×105 to 60×105, and then tested the percentage of CEM cells captured by G-dApR per sample. In this assay, the concentration of G-dApR was much lower than its corresponding targets (CEM cells). Flow cytometry analysis showed that the percentage of captured CEM cell numbers maintained more than 99% accompanied with the cell amounts ranging from 5×105 to 15×105, and then was decreased to 17.3% with increasing numbers of CEM cells to 60×105 (Figure 3G). The dynamic relationship between %-captured cell number and total cell number was statistically analyzed and shown in Figure 3H. The data confirmed that G-dApR precisely bound with CEM cells in a one-to-one relationship. Furthermore, we calculated the dissociation coefficient (Kd) of G-dApR and Ap for confirming the binding affinity of G-dApR to CEM cells. The results showed that the Kd values of G-dApR and Ap were in the low nanomolar range, and Kd value of G-dApR was slightly lower than that of Ap, suggesting the good binding affinity of G-dApR to CEM cells (Figure S1 in Supporting Information Results section). Besides, there are reports that streptavidin-biotin linker between aptamers and nanomaterials may reduce nonspecific interplay between cell biomarkers and aptamers through permitting nucleic acid to extend out from the surface of cells.32 This is another reason why G-dApR exhibits great specificity.
Inhibition of G-dApR on Cells Activity in vitro and Associated Mechanism
Since inhibition of CTCs activity is the primary way to prevent cancer metastasis, we examined the ability of G-dApR to inhibit the viability of CEM and Ramos cells after treatment with G-dApR conjugate at various concentrations of 5, 20 and 45 μg/mL by utilizing assays of MTT, DAPI staining, AO/EB staining, cell cycle and apoptosis. MTT results showed that G-dApR significantly inhibited the viability of CEM cells under the treatment concentration of 45 μg/mL in a dose-dependent manner, while, it did not affect the activity of non-target Ramos cells (Figure 4A). As a classic morphology change of apoptotic cells even in early stage of cell death, DAPI staining images (Figure 4B) and its quantitative data (Figure 4C) showed that G-dApR resulted in nuclear condensation and chromatin margination of CEM cells more clearly than Ramos cells.
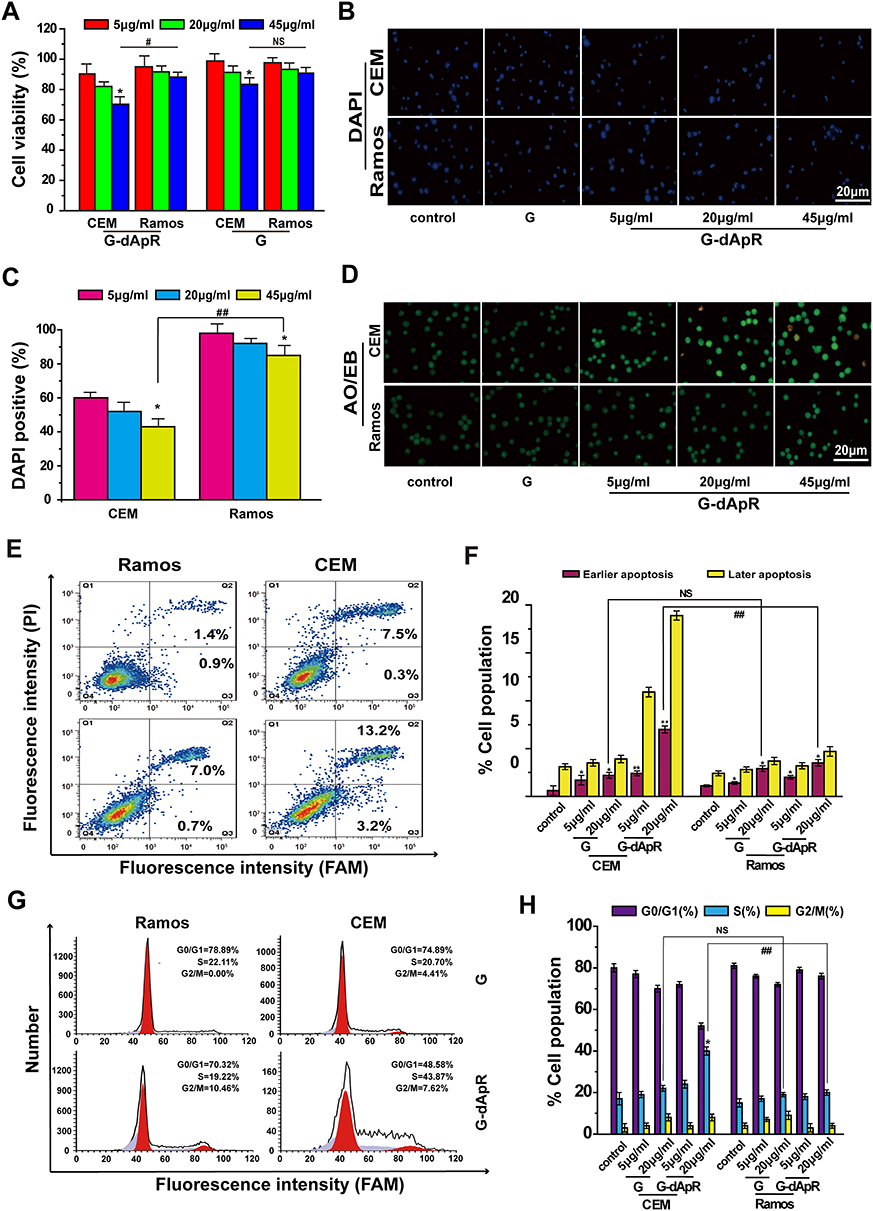 |
Figure 4 Inhibition of G-dApR on cell viability by inducing apoptosis. (A) Viability of CEM cells was decreased in a concentration-dependent manner after incubation with G-dApR (5, 20 and 45 μg/mL). (B, C) Fluorescent images (B) and quantitative analysis (C) of DAPI staining of CEM and Ramos cells after incubation with G-dApR (5, 20 and 45 μg/mL). The results showed that G-dApR induced more CEM cells into apoptosis than Ramos cells. (D) AO/EB staining of CEM and Ramos cells after incubation with G-dApR (5, 20 and 45 μg/mL). G-dApR caused more CEM cells to apoptosis (yellow dots) than Ramos cells. (E, F) Annexin V-FITC/PI apoptotic analysis confirmed the stronger dose-dependent apoptosis on CEM cells than Ramos induced by G-dApR. (G, H) Cell cycle distribution of CEM and Ramos cells after exposure to G-dApR. Flow cytometry analysis revealed that G-dApR arrested CEM cells at cell cycle S phase more than Ramos. Note: G was also measured as the control. NSIndicates no statistical difference, while *,# and **,## represents P < 0.05 and P < 0.01. |
AO/EB staining images were shown in Figure 4D, in which the color of green, yellow and orange/red, respectively, presented viable cells, early apoptosis and late apoptosis as we reported previously.24,25,33 We found that G-dApR drove more CEM cells into apoptosis than Ramos cells, and the triggered-apoptosis behavior was concentration-dependent. Furthermore, we quantitatively examined the effects of G-dApR on cell apoptosis by using flow cytometry as we conducted previously.24,25,33 The flow cytometry and statistic data were shown in Figure 4E and F. We found that G-dApR induced 16.4% CEM cells into apoptosis (Figure 4F), more cells were distributed at the later apoptotic stage, and G-dApR induced the dose-dependent apoptosis more significantly than G (the control) in CEM cells, but not Ramos cells. The cell cycle analysis showed that G-dApR significantly arrested the cell cycle of CEM cells at S phase more than G but not the control Ramos cells (Figure 4G and H). Together, these results demonstrated that G-dApR inhibited the activity of captured target CEM cells more significantly than non-target Ramos cells. This inhibition effect on cell viability seems to result from not only the capture specificity and capability of G-dApR but also the inhibition of aptamers on cell proliferation.34
Ability of G-dApR to Capture CTCs in vivo and in the Human Healthy Blood
The advanced conformation of aptamers alters by ambient conditions such as pH value, temperature and ionic strength, affecting their specific binding to the targets. This flexible nature may cause the in vitro selected aptamers to lose their binding ability in vivo because of the different conditions between in vitro and in vivo.35 Therefore, the capability evaluation of G-dApR to capture CTCs in vivo is of great significance to its medical application. Here, we evaluated the specificity of G-dApR in capturing CTCs in vivo via using the established mice model through injecting stained CEM cells into the mice followed by blood drawn and separation after treatment of G-dApR. The confocal images of captured CEM cells were shown in Figure 5A, in which the nuclei of cells was blue in Hoechst channel and WBCs were bright gray color in channels of photo and merge 2. The green color in images of merge 1 and FAM channel presented that G-dApR bound to CEM cells (Figure 5A). The data indicated the ability of G-dApR to capture CTCs in vivo. Moreover, the flow cytometry analysis further demonstrated that G-dApR captured 112 CEM cells in the collected 2×104 cells of 1 mL blood, suggesting that G-dApR could precisely capture CEM cells in vivo (Figure 5B). For the clinical application of G-dApR, we evaluated the specificity of G-dApR in capturing the target CEM cells (1000 counts) spiked into the human healthy blood (1, 2 and 4 mL) by using flow cytometry as we described previously.23–25,28 The results showed that G-dApR captured 222, 68 and 33 CEM cells from 1, 2 and 4 mL of healthy blood, exhibiting a cell density-dependent manner (Figure 5C). Hence, G-dApR has the great potential to be applied in the clinical cancer therapy.
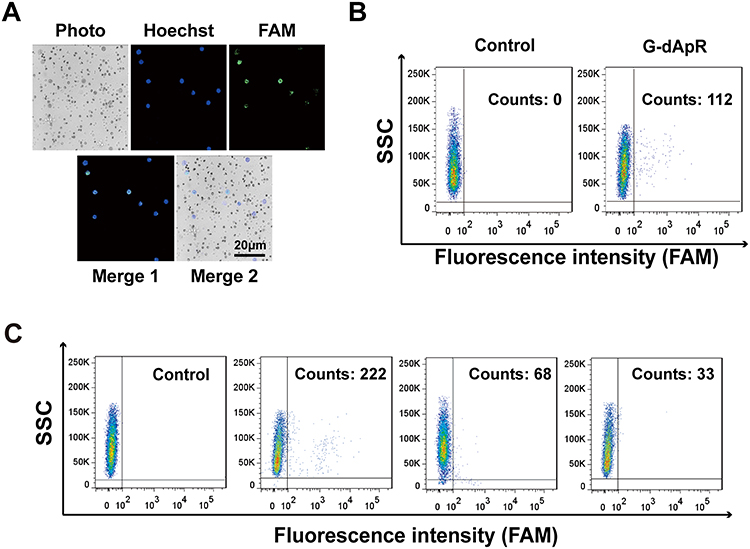 |
Figure 5 CTCs captured by G-dApR in vivo and in the human healthy blood. CEM cells (2×104/mouse) stained with Hoechst 33,258 were injected to BALB/c mice through tail vein followed by injection of G-dApR (80 μg per mouse). The in vivo captured CEM cells in mouse blood were analyzed after separation. (A) Fluorescence images of the CEM cells captured by G-dApR in vivo. The fluorescence imaging showed that the captured CEM cells by G-dApR in mouse blood exhibited FAM (λ518 nm) staining-positive color. (B) Flow cytometric analysis quantitatively showed the capture capability of G-dApR to CEM cells in vivo. (C) G-dApR could capture the rare CEM cells after 1000 CEM cells were spiked into 1, 2 or 4 mL of the human healthy blood from left to right beside the control. |
Conclusion
In the present work, for the first time, to our knowledge, we constructed the double strand circular aptamer (dApR)-coated dendrimer (G-dApR) for CTCs capture via conjugating biostable and specific dApR that targets CEM cells to PAMAM dendrimer. Our results strongly demonstrated that G-dApR could obviously resist nuclease-mediated biodegradation, specifically capture CTCs in vitro and in vivo, and significantly inhibit the captured CTCs activity by inducing apoptosis. Due to various merits including the enhanced biostability and functionality, low-cost, nano-scale, and chemical synthesis, G-dApR could be a promising aptamer nanotechnology-based platform for preventing CTC-mediated cancer metastasis through capture and suppression of CTCs.
Acknowledgments
Co-first authors: Yu Li and Ting Zhang contributed equally to this study.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC) grants 21907014, 81702988, 81961138017, 81773063 and U1505225; the grants of Fujian Medical University (XRCZX2017024 and 2017XQ1002).
Disclosure
The authors declare no conflict of interest.
References
1. Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346(6287):818–822. doi:10.1038/346818a0
2. Dickey DD, Giangrande PH. Oligonucleotide aptamers: a next-generation technology for the capture and detection of circulating tumor cells. Methods. 2016;97:94–103. doi:10.1016/j.ymeth.2015.11.020
3. Hassan EM, Willmore WG, DeRosa MC. Aptamers: promising tools for the detection of circulating tumor cells. Nucleic Acid Ther. 2016;26(6):335–347. doi:10.1089/nat.2016.0632
4. Lee JF, Stovall GM, Ellington AD. Aptamer therapeutics advance. Curr Opin Chem Biol. 2006;10(3):282–289. doi:10.1016/j.cbpa.2006.03.015
5. Wu Z, Tang LJ, Zhang XB, et al. Aptamer-modified nanodrug delivery systems. ACS Nano. 2011;5(10):7696–7699. doi:10.1021/nn2037384
6. Wu ZS, Shen Z, Tram K, et al. Engineering interlocking DNA rings with weak physical interactions. Nat Commun. 2014;5:4279. doi:10.1038/ncomms5279
7. Li F, Zhang H, Wang Z, et al. Aptamers facilitating amplified detection of biomolecules. Anal Chem. 2015;87(1):274–292. doi:10.1021/ac5037236
8. Lu DQ, He L, Zhang G, et al. Aptamer-assembled nanomaterials for fluorescent sensing and imaging. Nanophotonics. 2016;6(1):109.
9. Ming Y, Li Y, Xing H, et al. Circulating tumor cells: from theory to nanotechnology-based detection. Front Pharmacol. 2017;8:35. doi:10.3389/fphar.2017.00035
10. Chaffer CL, Weinberg RAA. Perspective on cancer cell metastasis. Science. 2011;331(6024):1559–1564. doi:10.1126/science.1203543
11. Kaiser J. Cancer’s Circulation Problem. Science. 2010;327(5969):1072–1074. doi:10.1126/science.327.5969.1072
12. Plaks V, Koopman CD, Werb Z. Circulating tumor cells. Science. 2013;341(6151):1186–1188. doi:10.1126/science.1235226
13. Cristofanilli M, Budd GT, Ellis MJ, et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004;351(8):781–791. doi:10.1056/NEJMoa040766
14. Becker FF, Wang XB, Huang Y, et al. Separation of human breast cancer cells from blood by differential dielectric affinity. Proc Natl Acad Sci U S A. 1995;92(3):860–864. doi:10.1073/pnas.92.3.860
15. Che J, Yu V, Garon EB, et al. Biophysical isolation and identification of circulating tumor cells. Lab Chip. 2017;17(8):1452–1461. doi:10.1039/c7lc00038c
16. van der Toom EE, Verdone JE, Jun C, et al. A surface tension magnetophoretic device for rare cell isolation and characterization. Med Oncol. 2017;34(2):22. doi:10.1007/s12032-016-0877-y
17. Hosokawa M, Yoshikawa T, Negishi R, et al. Microcavity array system for size-based enrichment of circulating tumor cells from the blood of patients with small-cell lung cancer. Anal Chem. 2013;85(12):5692–5698. doi:10.1021/ac400167x
18. Shen Z, Wu A, Chen X. Current detection technologies for circulating tumor cells. Chem Soc Rev. 2017;46(8):2038–2056. doi:10.1039/c6cs00803h
19. Yu M, Bardia A, Wittner BS, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339(6119):580–584. doi:10.1126/science.1228522
20. R. A T. A case of cancer in which cells similar to those in the tumors were seen in the blood after death. Aust. Med. J. 1869;14:2.
21. Alix-Panabieres C, Pantel K. Circulating tumor cells: liquid biopsy of cancer. Clin Chem. 2013;59(1):110–118. doi:10.1373/clinchem.2012.194258
22. Baccelli I, Schneeweiss A, Riethdorf S, et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol. 2013;31(6):539–544. doi:10.1038/nbt.2576
23. Dong H, Wu ZS, Xu J, et al. Novel multifunction-integrated molecular beacon for the amplification detection of DNA hybridization based on primer/template-free isothermal polymerization. Biosens Bioelectron. 2015;72:182–190. doi:10.1016/j.bios.2015.04.090
24. Dong HY, Han LY, Wang J, et al. In vivo inhibition of circulating tumor cells by two apoptosis-promoting circular aptamers with enhanced specificity. J Controlled Release. 2018;280:99–112. doi:10.1016/j.jconrel.2018.05.004
25. Dong HY, Han LY, Wu ZS, et al. Biostable aptamer rings conjugated for targeting two biomarkers on circulating tumor cells in vivo with great precision. Chem Mater. 2017;29(24):10312–10325. doi:10.1021/acs.chemmater.7b03044
26. Shangguan D, Li Y, Tang Z, et al. Aptamers evolved from live cells as effective molecular probes for cancer study. Proc Natl Acad Sci U S A. 2006;103(32):11838–11843. doi:10.1073/pnas.0602615103
27. Xie J, Gao Y, Zhao R, et al. Ex vivo and in vivo capture and deactivation of circulating tumor cells by dual-antibody-coated nanomaterials. J Control Release. 2015;209:159–169. doi:10.1016/j.jconrel.2015.04.036
28. Xie JJ, Lu YS, Dong HY, et al. Enhanced specificity in capturing and restraining circulating tumor cells with dual antibody-dendrimer conjugates. Adv Funct Mater. 2015;25(8):1304–1313. doi:10.1002/adfm.201403556
29. Zhao Y, Xu D, Tan W. Aptamer-functionalized nano/micro-materials for clinical diagnosis: isolation, release and bioanalysis of circulating tumor cells. Integr Biol (Camb). 2017;9(3):188–205. doi:10.1039/c6ib00239k
30. Seferos DS, Prigodich AE, Giljohann DA, et al. Polyvalent DNA nanoparticle conjugates stabilize nucleic acids. Nano Lett. 2009;9(1):308–311. doi:10.1021/nl802958f
31. Wu Y, Phillips JA, Liu H, et al. Carbon nanotubes protect DNA strands during cellular delivery. ACS Nano. 2008;2(10):2023–2028. doi:10.1021/nn800325a
32. Xiong X, Liu H, Zhao Z, et al. DNA aptamer-mediated cell targeting. Angew Chem Int Ed Engl. 2013;52(5):1472–1476. doi:10.1002/anie.201207063
33. Dong H, Yang X, Xie J, et al. UP12, a novel ursolic acid derivative with potential for targeting multiple signaling pathways in hepatocellular carcinoma. Biochem Pharmacol. 2015;93(2):151–162. doi:10.1016/j.bcp.2014.11.014
34. Zhou J, Tiemann K, Chomchan P, et al. Dual functional BAFF receptor aptamers inhibit ligand-induced proliferation and deliver siRNAs to NHL cells. Nucleic Acids Res. 2013;41(7):4266–4283. doi:10.1093/nar/gkt125
35. Xiao Z, Farokhzad OC. Aptamer-functionalized nanoparticles for medical applications: challenges and opportunities. ACS Nano. 2012;6(5):3670–3676. doi:10.1021/nn301869z
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.