")
Back to Journals » Breast Cancer: Targets and Therapy » Volume 12
Biomarkers for Inflammatory Breast Cancer: Diagnostic and Therapeutic Utility
Authors Dobiasova B, Mego M
Received 25 August 2020
Accepted for publication 3 October 2020
Published 14 October 2020 Volume 2020:12 Pages 153—163
DOI https://doi.org/10.2147/BCTT.S231502
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Pranela Rameshwar
Barbora Dobiasova, Michal Mego
2nd Department of Oncology, Comenius University, Faculty of Medicine, National Cancer Institute, Bratislava, Slovak Republic
Correspondence: Michal Mego
2 nd Department of Oncology, Comenius University, Faculty of Medicine, National Cancer Institute, Klenova 1, Bratislava 833 10, Slovak Republic
Tel +421-2-59378366
Fax +421-2-54774943
Email [email protected]
Abstract: Inflammatory breast cancer (IBC) is a rare and highly aggressive subtype of advanced breast cancer. The aggressive behavior, resistance to chemotherapy, angiogenesis, and high metastatic potential are key intrinsic characteristics of IBC caused by many specific factors. Pathogenesis and behavior of IBC are closely related to tumor surrounding inflammatory and immune cells, blood vessels, and extracellular matrix, which are all components of the tumor microenvironment (TME). The tumor microenvironment has a crucial role in the local immune r09esponse. The communication between intrinsic and extrinsic components of IBC and the abundance of cytokines and chemokines in the TME strongly contribute to the aggressiveness and high angiogenic potential of this tumor. Critical modes of interaction are cytokine-mediated communication and direct intercellular contact between cancer cells and tumor microenvironment with a variety of pathway crosstalk. This review aimed to summarize current knowledge of predictive and prognostic biomarkers in IBC.
Keywords: inflammatory breast neoplasms, biomarkers, tumor microenvironment, targeted therapy
Introduction
Inflammatory breast cancer (IBC) is rare and highly aggressive subtype of locally advanced breast cancer. The primary tumor of IBC is classified by The 2017 American Joint Committee on Cancer and the International Union for Cancer Control (AJCC-UICC) Tumor, Node, Metastasis (TNM) as T4d and characterized by the presence of many dermal tumor emboli in the papillary and reticular dermis of the skin overlying the breast, diffuse dermatologic erythema and edema (peau d’orange). IBC accounts for about 2.5% of all diagnosed breast cancers, in the United States is estimated to account for 1% to 6% cases.1,2 Actually, in some parts of the Middle East and northern Africa, this incidence can be as high as 10%.3 Younger and African-American women are especially affected by IBC. The average age is 59 years, which is less than that of patients with non-IBC breast cancer.1,2
In general, patients with non-metastatic IBC are treated similarly to those with others LABC. There are only two main differences – breast conservation therapy (BCT) and sentinel lymph node biopsy (SNB) are inappropriate for IBC.4
IBC is a very aggressive type of breast cancer. The aggressive behavior, resistance to chemotherapy, angiogenesis, and high metastatic potential are key intrinsic characteristics of IBC caused by many specific factors. Accordingly, contributions of the tumor microenvironment (TME) to the pathogenesis and aggressive behavior of IBC was declared in many studies. This review aimed to summarize current knowledge of predictive and prognostic biomarkers in IBC, including tumor tissue and tumor microenvironment associated biomarkers including blood circulation.
Tumor-Associated Biomarkers
IBC is a clinicopathologic entity. Typical clinical presentation with duration no more than six months occupying at least one-third of the breast and histology of invasive breast cancer obtained from a biopsy of the affected breast are mandatory for the diagnosis of IBC. Mammographic findings of IBC and mastitis could have a similar appearance; therefore, breast imaging could be more useful in disease monitoring.
Staging evaluations include routine laboratory tests, computed tomography of the chest, abdomen, and pelvis, a bone scan, and ultrasound-guided biopsy of the nodes in patients with suspicious lymph nodes.5
IBC cells are histopathologically similar to non-IBC cells. The tumor is not a specific histologic subtype of breast cancer, IBC is usually of the ductal type with pleomorphic cells and a high histologic grade.6 Cancer cells are typically distributed diffusely in clusters throughout the skin and breast. In IBC tumors have been reported cytokine-mediated infiltration of the lymphocytes or tumor-associated macrophages. Tumor biomarkers are in most cases evaluated in main tumor rather than in the lymphatic emboli. However, there might be a discrepancy between these two types of tumor tissue with clinical implications as non-IBC tumors can recur with IBC features and vice versa.
The classic histologic finding on biopsy is a dermal lymphatic invasion, which is found in approximately 75% of all cases, but it can also be an incidental finding in patients with non-IBC. Therefore, this finding is not necessary for the diagnosis. Within the dermal-lymphatic vessels are found formation and invasion of tumor emboli, which are responsible for the local signs and rapid metastatic potential.6–11 Several markers have been identified in preclinical tumors emboli models, such as zinc finger E-box-binding homeobox 1 (ZEB1), E-cadherin, aldehyde dehydrogenase 1 (ALDH1) and NOTCH3.12–14
The hallmark if IBC are tumor emboli (TE) formed by aggregates of tumor, immune and stromal cells, which caused blockage of dermal lymph vessels with the typical clinical presentation of “peau d’orange” appearance of the skin. Variable intercellular contact, both between cancer cells and TME and cancer cells, is responsible for the formation of a unique emboli structure. Caveolin1 and RhoC overexpression modulates these junctions and contributes to invasion of tumor emboli into the vascular structure. The emboli cells can survive despite hypoxia and direct exposure to the immune system and to form small cell clusters with high metastatic potential.15 A better understanding of the biology of tumor emboli is important to developing novel opportunities for treatment.7 Intracellular translocation of E-cadherin causes modulation of intercellular junctions and cancer cells migration into surrounding tumor microenvironment, which contributes to the special structure of IBC.7 Recent reports have linked IBC to display a hybrid epithelial/mesenchymal phenotype based on its levels of E-cadherin and its ability to migrate collectively as tumor emboli. On a molecular level, hybrid E/M cells have been shown to coexpress CD24 and CD44 (CD24hi CD44hi signature). This subpopulation resembles features of chemoresistance and have metastasis initiation properties.16,17
IBC tumors are characterized by downregulation of hormone receptors – estrogen receptor and/or progesterone receptor (ER/PR) and amplification of human epidermal growth factor receptor 2 (HER2) more frequently than non-IBC (HR-positive subtypes 30% in IBC versus 60–80% in non-IBC; HER2-positive 40% in IBC versus 25% in non-IBC). Also, incidence rates of triple-negative breast cancer (TNBC) subtypes are higher in IBC (30% versus 10–15% in non-IBC). These subtypes are generally associated with a worse prognosis and shorter disease-free survival.2,18–20
In clinical practice, the most important tumor-associated biomarkers are LE, even not present in all patients, while there is no other specific IBC-related biomarker with clinical utility, while all other biomarkers the same as in non-IBC setting.
IBC-Related Genetic Biomarkers
Due to the aggressive behavior of IBC and poor prognosis despite complex cancer treatment, recent researches have focused on investigating the molecular genetics of IBC to use targeted therapies in the treatment. Gene expression profiling attempts to characterize IBC, however, none of them have so far been able to identify a unique mutational or phenotypic profile specific to IBC. Several studies were performed that did not reveal a significant difference in gene expression between IBC and non-IBC.7 Ross et al reported the most frequently altered genes in 53 patients with IBC, which were not unique to IBC. Amplification of MYC (32%), PIK3CA (28%), HER2 (26%) and FGFR1 (17%) and mutation of p53 (62%), BRCA2 (15%) and PTEN (15%) were detected. Forty-two percent of patients with triple-negative IBC had MYC amplification, therefore, further studies of this finding are needed to improve the survival of this group of patients with a poor prognosis.21
In another study, somatic mutations in 24 patients with metastatic IBC were tested. Three major mutations were p53 (75%), PIK3CA (41,7%) and ERBB2 (16,7%). This was the first report of higher frequency of ERBB2 mutation in IBC, especially in patients with hormone-receptor positivity, which could be a potential target in treatment for HR+ IBC.22
Rana et al evaluated the results of genetic tests of 368 women with IBC. The germline mutations were found in 14.4% of patients. 7.3% had BRCA1 or BRCA2 mutations, 6.3% had other breast cancer gene mutation (PALB2, CHEK2, ATM, BARD1), and 1.6% had a non-breast cancer-associated mutation. The highest prevalence of germline mutations was among patients with triple-negative IBC (24%). The diversity of detected germline mutations suggests the need for further studies to assess the role of them in IBC.23
The role of p53 mutation in patients with IBC was evaluated in several studies. A study of 27 patients with IBC reported two mechanisms, which can subvert the normal function of p53 – cytoplasmic sequestration of the wild-type protein and direct nuclear mutations.24 A subsequent study evaluated the prognostic significance of p53. Riou et al reported an 8.6-fold higher risk of death in patients with a p53 mutation and nuclear overexpression of p53 protein compared to patients without these findings. There were also observed prognostic interactions with HR expression. ER-negative women with p53 nuclear overexpression had a 17.9-fold higher risk of death versus 2.8-fold in patients with p53 overexpression without ER-negativity.25
Small, non-coding RNAs – microRNAs (miRNAs) have also been actively investigated as molecular biomarkers for the diagnosis and prognosis of IBC tumors. MiRNAs influence tumor´s intrinsic and extrinsic components and also modulation of TME by regulation of the expression of genes by targeting mRNAs. Qi et al described 5 potential miRNAs as diagnostic molecular biomarkers in IBC (miR-301b, miR-451, miR-15a, miR-342-3p and miR-342-5p), some miRNAs associated with a better (miR-19a, miR-7, miR-324-5p) and with poorer prognosis (miR-21, miR-205).26 Other studies described the lower expression of miR-26b in IBC than in normal breast tissue and lower expression of miR-205 in IBC compared with non-IBC. Lower expression of both, miR-26b and miR-205, was associated with shorter distant metastasis-free survival and overall survival.27,28 The potential application of IBC associated miRNAs for diagnosis and prognosis of IBC requires further investigation.
The major goal of recently published studies was to identify differences in the gene expression of IBC from non-IBC breast cancers to find out targetable genomic drivers.
A novel gene called WNT1 inducible-signaling pathway protein 3 (WISP3) appears to operate as a tumor suppressor gene in IBC. About 80% of IBC is characterized by the loss of WISP3 (versus 21% in non-IBC). WISP3 is a protein able to inhibit the invasive potential of malignant cells and tumor cell growth.29
Another of the most highly overexpressed genes in IBC is the putative oncogene RhoC GTPase (90% in IBC versus 38% in non-IBC). RhoC GTPase is a member of Ras superfamily proteins and it is involved in cytoskeletal reorganization. Upregulation of the Rhoc GTPase gene leads to the release of angiogenic cytokines, enhanced invasiveness potential, and high motility of the tumors cells. RhoC GTPase is also associated with the upregulation of vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), interleukin-6 (IL-6), and interleukin-8 (IL-8), what is responsible for angiogenic stroma formation in IBC.30–32 RhoC GTPase protein, as mentioned above, plays a role in focal adhesion and invasion ability of IBC cells and in increasing metastatic capacity. RhoC activation is caused by caveolin 1, a cell membrane protein, which is upregulated in IBC and thus increases the invasiveness of cancer cells.33,34
Two studies reported high upregulation of the gene that encodes myristoylated alanine-rich C-kinase substrate (MARCKS) and the association between MARCKS overexpression and shorter metastasis-free survival (MFS) in patients with IBC. MARCKS is a substrate for protein kinase C and plays an important role in cell motility, phagocytosis, and regulation of the cell cycle. Concerning the association of MARCKS overexpression in IBC and association with poor MFS, MARCKS might represent a new potential therapeutic target in IBC.35,36
Boersma et al identified multiple pathways related to the endoplasmic reticulum stress response, that were differentially expressed in the tumor stroma of IBC versus non-IBC. Their findings suggest that the genomics of the stromal cells may play an important role in understanding the IBC phenotype.37
IBC tends to be a highly vascular tumor because of its high angiogenic and angioinvasive potential.38 Patients with IBC have greater lymphatic vessel density and higher levels of the vascular endothelial growth factor D (VEGF-D) compared to patients with non-IBC.39 VEGF-D plays an important role in angiogenesis and lymphangiogenesis of IBC. The VEGF receptor-3 is expressed in lymphatic endothelium and is activated by VEGF-C and VEGF-D ligands. The expression of VEGF-D was detected only in IBC cell lines.40–42 Thus, the efficacy of angiogenesis inhibitors in the treatment of IBC was examined. Neoadjuvant bevacizumab combined with trastuzumab and chemotherapy in HER2-positive women with IBC was efficacious and well tolerated in previously untreated IBC in Phase 2 trial (BEVERLY-2). The complete remission rate in the bevacizumab arm was markedly higher in this study than in previous works.43 In preclinical studies are evaluated platelet-derived growth factor-α (PDGFRα) inhibitors (BLU-285) and monoclonal antibodies (olaratumab). PDGFRα upregulation in tumor emboli also contributes to high angiogenesis in IBC.7
IBC cancer cells form structures that mimic the normal mammary gland, which can be the reason for high chemotherapy resistance, another intrinsic characteristic of IBC. The structures activate several signaling pathways, one of them is the epidermal growth factor receptor (EGFR) mediated pathway.7 EGFR is overexpressed in chemotherapy-resistant structures of IBC. In the study of 44 cases of IBC 30% was EGFR positive with significantly worse overall survival (OS) compared to EGFR-negative disease (P=0.01). The expression of growth factor receptors in IBC is associated with high recurrence rates and a higher risk of death; therefore, it may represent the new therapeutic targets.44
Alexander et al identified the new biomarker for therapy of IBC – Cyclin E, which plays an important role in tumors cells invasion and chemoresistance due to the regulation of many pathways important in the biology of IBC. They described a high expression of cyclin E in patients with IBC, but not correlation with cyclin E phenotype and poorer outcome as we can see in non-IBC tumors. Early targeting of this pathway may be beneficial in patients with chemotherapy resistance. We already have clinically available agents that target the cyclin E/CDK2 complex. One of these, dinaciclib, has had high toxicity in early trials,45–47 but in combination with other agents or treatment modalities and in metronomic dosing it could be a new option in therapy of IBC. CDK inhibitors could have a function in sensitization in post-mastectomy radiation, particularly in women with residual tumors, and so reduce the risk of recurrence.48
Enhancer of zeste homolog 2 (EZH2) was examined as a potential biomarker to identify patients with IBC treated with radiation with a high risk of locoregional recurrence, who may benefit from radiosensitizers. EZH2 status was tested in 62 patients with IBC who received pre- or post-operative radiation. Locoregional recurrence occurred in 16 women (25,8%), 15 of them had EZH2 expression. In addition, EZH2 positive IBC was associated significantly with negative ER status and TNR status.49
In ER-negative/HER2 positive IBC tumors, activation of the NF-κB pathway is often observed. This pathway is one of the main inflammation-mediated pathways and its activation can lead to the upregulation of antiapoptotic factors with subsequent resistance to chemotherapy. The activation was more frequently observed in IBC tumors (43% versus 4% in nonIBC).50–52
Preclinical data have demonstrated activation of the PI3K/mTOR and JAK/STAT pathways in IBC, along with the expression of inflammatory cytokines and TAMs.53 JAK/STAT pathway by promoting communication between extracellular peptide signals and cancer cell´s gene promoters supports the survival of cancer cells.54 In IBC, JAK/STAT dysregulation and subsequent isoforms pJAK2 and pSTAT3 activation and overexpression of IL-6 are observed. Jhaveri et al retrospectively analysed biomarkers expression of pJAK2, pSTAT3, IL-6, and others in IBC tumors and surrounding non-tumor tissue. Ninety-five percent of samples were pJAK2 positive, suggesting a mechanism of resistance after neoadjuvant chemotherapy. The activation of biomarkers was also demonstrated in surrounding non-tumor tissue.53,55 The combination of chemotherapy and JAK1 and JAK2 inhibitors in IBC is now being investigated in Phase I/II trial.56
Several studies have confirmed higher secretion of IL-6 in IBC tumors and significantly higher levels of serum IL-6 in IBC compared to nonIBC patients.57 The IL-6 inflammatory pathway activation is increasingly noted mainly during the lymphatic invasion of cancer cells.58 Further studies to investigate the IL-6 pathway as a therapeutic target in IBC are needed.
PIK3CA kinase, which plays an important role in the proliferation of cancer cells, is frequently mutated in breast cancer. IBC tumors are characterized by frequent genomic alterations in the HER/PI3K/mTOR pathway. PI3K may promote oncogenic signaling through mTOR activation. Marker of this activation is ribosomal S6 protein (pS6) which was positive in 100% IBC in Jhaveri´s study mentioned above.53,59 Thus, IBCs may benefit from an mTOR targeted therapy, which was demonstrated in the BOLERO-3 trial. There was the clinical benefit by mTOR inhibitor everolimus using in trastuzumab-resistant, HER2-positive patients.60
Finally, the inflammatory cyclooxygenase 2 (COX2) pathway is also well studied in IBC. COX2 is overexpressed in IBC compared to non-IBC and patients with COX2 overexpression have poorer overall survival compared to those with low COX2.61 Upregulation of COX2 and subsequently prostaglandin E2 (PGE2) overexpression is a result of chronic inflammation in TME.62 Table 1 reviews molecular biomarkers in IBC.
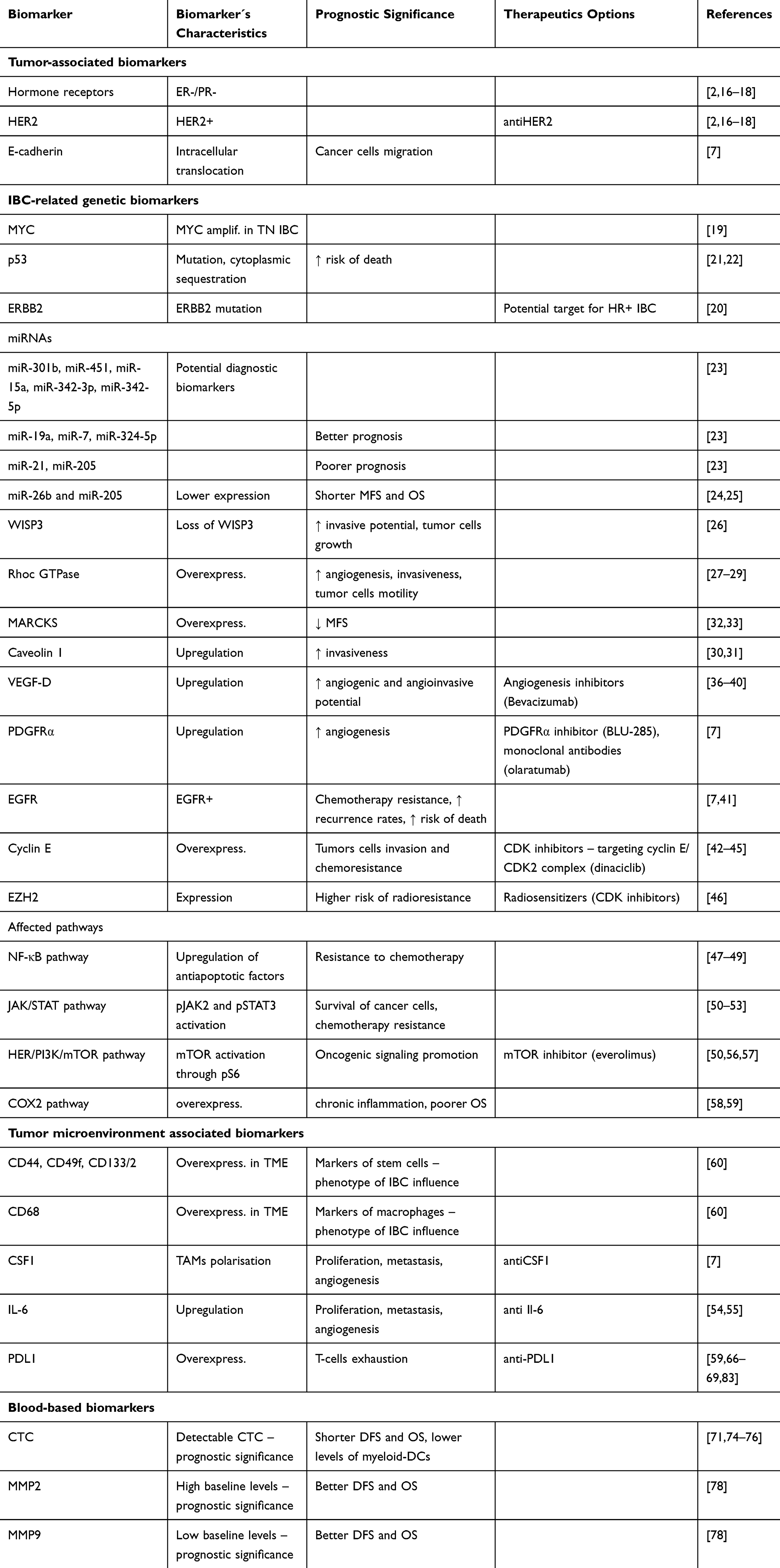 |
Table 1 Molecular Biomarkers in Inflammatory Breast Cancer |
In conclusion, while there are several signalling pathways that show different activations in IBC compared to non-IBC setting, currently, no genetic aberration and/or activation of specific signalling pathways was identified in IBC patients, that is lacking in non-IBC setting.
Tumor Microenvironment Associated Biomarkers
Pathogenesis and aggressive behavior of IBC are closely related to tumor surrounding inflammatory and immune cells, blood vessels, and extracellular matrix, which are all components of the tumor microenvironment. The tumor microenvironment has a crucial role in the local immune response.7 Term “inflammatory” breast cancer is based on clinical characteristics of IBC, that comprise inflammatory appeared breast, however, activated inflammatory pathways are not specific biological aspects of IBC. Nonetheless, accumulating data suggest specific composition of TME in IBC.7
The tumor microenvironment contains increased mammary stem cells and macrophages, which may influence the phenotype of IBC. Tumor surrounding tissue of the normal breast expresses a higher level of markers of the stem cells CD44, CD49f, CD133/2, and of the macrophages CD68 compared to non-IBC normal breast tissue.63
IBC cells induce the polarisation of macrophages into alternatively activated M2 tumor-associated macrophages (TAMs). TAMs antagonize the antitumor activity of CD8 T-cells, support survival, and proliferation of tumor cells and contribute to metastasis process and angiogenesis by the production of inflammatory mediators, immunosuppression, and by mediating tumor extracellular matrix remodeling.64–66 Macrophage colony-stimulating factor 1 is a cytokine involved in M2 macrophage polarization. Its inhibition by antibody reduced the aggressive clinical phenotype of IBC. It was shown, that TAM isolated from TME of IBC patients produce several cytokines that facilitate metastatic potential of IBC cells.
Mesenchymal stem cells (MSCs) also induce polarisation of M2 TAMs by the production of macrophage colony-stimulating factor 1 (CSF1) and interleukin family proteins. In vitro and in vivo experiments demonstrated reduction of the aggressive clinical phenotype of IBC by using an antibody antiCSF1 and modulation of the effect of interleukins on TAMs by anti-IL6 antibodies.7 Other in vitro study suggests that inhibition of M2 polarisation of macrophages by phosphopeptide mimetic PM37, targeting the SH2 domain of STAT6, can prevent radioresistance of IBC.67
T-cells infiltration and regulation of them are also an important component of TME. Infiltration of the healthy tumor-infiltrating lymphocytes (TILs) plays an important role in a good response to chemotherapy.7 Aggregates of CD8-positive T cells in the intratumoral and peritumoral desmoplastic stroma were identified in approximately half of women with IBC.59 The infiltrates of CD8 T-cells participate in immune regulation of tumor growth. TILs in IBC have often poor effector function, which is typically associated with the expression of immune checkpoint inhibitor programmed cell death ligand 1 (PDL1) on TILs and tumor cells.68,69 Recent studies describe the importance of PDL1, which was overexpressed in 38% IBC (versus 10–30% in non-IBC). High levels of PDL1 may negatively regulate the function of T-cells. Anti-PDL1 therapies may reverse, in IBC tumors with overexpression of PDL1, Tcells exhaustion. In patients with PDL1 upregulation was interestingly reported a better response to chemotherapy.70–72 PDL1 status in IBC patients remained a statistically independent predictor of OS (36,4% 5 years OS in PDL1-positive versus 47.3% in PDL1-negative IBC).73
B cells have also a critical role in the antitumor immune response. Arial-Pulido et al evaluated protein expression od PD-L1 and CD20 as prognostic biomarkers in IBC tumors. CD20+TILs/PD-L1+TILs status was an independent favorable prognostic factor for DFS and breast cancer-specific survival (BCSS) in IBC and TN IBC.74
Other important cells in TME are antigen-presenting cells – dendritic cells (DCs), which participate in T-cells activation. DCs are often suppressed in the peripheral blood of women with IBC leading in decreased tumor necrosis factor (TNF) secretion, thus novel therapeutics with DC agonistic activity may lead to a better outcome in patients with IBC by direct secretion of TNF. DCs also secrete interleukin 12 (IL12), which plays a role in T helper cells maturation, thus IL12 agonists may also be a new therapeutic option in IBC treatment.7,75–77
The communication between intrinsic and extrinsic components of IBC and the abundance of cytokines and chemokines in the TME strongly contribute to the aggressiveness and high angiogenic potential of this tumor. Critical modes of interaction are cytokine-mediated communication and direct intercellular contact between cancer cells and tumor microenvironment with a variety of pathway crosstalk.7
For clinical practice, TME biomarkers with clinical utility are the same as for non-IBC, a most useful are presence of TILs and PD-L1 expression in TNBC, while other biomarkers help us to better characterize the biology of IBC without direct effect for clinical decision making.
Blood-Based Biomarkers
Circulating tumor cells (CTC) count is an independent prognostic factor in primary and metastatic breast cancer patients.75,78 In the pooled analysis of two prospective trials in nonmetastatic IBC patients treated with neoadjuvant chemotherapy combined with neoadjuvant and adjuvant bevacizumab (BEVERLY-1, BEVERLY-2), prognostic values of circulating tumor cells (CTC) were evaluated. At baseline 39% of patients had detectable CTC, which was associated with shorter 3-year DFS (39% versus 70% in CTC negative patients) and shorter 3-year OS with a suggestion that CTC count could have a role in stratification of IBC patients.79 In a retrospective study of 147 patients with newly diagnosed IBC, patients with fewer than 5 CTCs had significantly better PFS and OS than patients with five or more CTCs detected before the start with chemotherapy. CTCs were a strong predictor of worse prognosis in these women.80 Their detection by the FDA-approved CellSearch system showed clinical validity in IBC as well as in non-IBC patients.
CTC counts also correlate with the blood dendritic cells (DC) immunophenotypes and the function of IBC. Patients with high levels of CTCs (≥5) have lower levels of myeloid-DCs (mDCs) capable of producing TNF-α and IL-12 through the toll-like receptor (TLR), which suggests general dysfunction of the immune system in IBC patients with a high number of CTCs.75 Patients with high levels of CTCs have a lower percentage of CD3+ and CD4+ T cells in peripheral blood, a lower percentage of CD8+ T cells, and a higher percentage of T-regulatory lymphocytes compared to patients without CTCs.81 Moreover, a high percentage of mDCs, which could induce a pro-inflammatory TME, is independently associated with worse OS in IBC patients.75
Also, prognostic impact of circulating levels of matrix metalloproteinases (MMP) 2 and 9 were examined in BEVERLY-2 IBC patients with HER2-positive IBC treated with the addition of bevacizumab to trastuzumab-based chemotherapy. High baseline MMP2 and low baseline MMP9 were correlated with better DFS and OS with a significant increase in MMP2 and a decrease in MMP9 levels during treatment.82
Conclusion
Despite the multimodal treatment, patients with IBC still have particularly poor prognosis and high risk of early recurrence. Their survival rate remains significantly worse compared to patients with nonIBC.
An improved biological understanding of IBC suggesting the possibility of more personalized effective targeted therapies with the improvement of the clinical outcomes in these patients. The contribution of tumor microenvironment with the abundance of cytokines and chemokines to the disease is fundamental. Thus, cytokine blockade and immunotherapy may play a crucial role in the treatment of IBC in the future.
Acknowledgments
We would like to acknowledge Denisa Manasova for her excellent technical help. We are grateful to all patients for their participation in the study.
Funding
This research was funded by the Slovak Research and Development Agency (APVV), grant number APVV-16-0010.
Disclosure
The authors declare no conflict of interest.
References
1. Hance KW, Anderson WF, Devesa SS, et al. Trends in inflammatory breast carcinoma incidence and survival: the surveillance, epidemiology, and end results program at the National Cancer Institute. J Natl Cancer Inst. 2005;97:966–975. doi:10.1093/jnci/dji172
2. Dawood S, Lei X, Dent R, et al. Survival of women with inflammatory breast cancer: a large population-based study. Ann Oncol. 2014;25:1143–1151. doi:10.1093/annonc/mdu121
3. Boussen H, Bouzaiene H, Ben Hassouna J, et al. Inflammatory breast cancer in Tunisia: epidemiological and clinical trends. Cancer. 2010;116(S11):2730–2735. doi:10.1002/cncr.25175
4. Lyman GH, Giuliano AE, Somerfield MR, et al. American Society of Clinical Oncology guideline recommendations for sentinel lymph node biopsy in early-stage breast cancer. J Clin Oncol. 2005;23:7703. doi:10.1200/JCO.2005.08.001
5. Dawood S, Merajver SD, Viens P, et al. International expert panel on inflammatory breast cancer: consensus statement for standardized diagnosis and treatment. Ann Oncol. 2011;22:515. doi:10.1093/annonc/mdq345
6. McCarthy NJ, Yang X, Linnoila IR, et al. Microvessel density, expression of estrogen receptor alpha, MIB-1, p53, and c-erbB-2 in inflammatory breast cancer. Clin Cancer Res. 2002;8:3857.
7. Lim B, Woodward WA, Wang X, et al. Inflammatory breast cancer biology: the tumour microenvironment is key. Nat Rev Cancer. 2018;18:485–499. doi:10.1038/s41568-018-0010-y
8. Robbins GF, Shah J, Rosen P, et al. Inflammatory carcinoma of the breast. Surg Clin North Am. 1974;54:801. doi:10.1016/S0039-6109(16)40383-X
9. Bonnier P, Charpin C, Lejeune C, et al. Inflammatory carcinomas of the breast: a clinical, pathological, or a clinical and pathological definition? Int J Cancer. 1995;62:382–385. doi:10.1002/ijc.2910620404
10. Manfrin E, Remo A, Pancione M, et al. Comparison between invasive breast cancer with extensive peritumoral vascular invasion and inflammatory breast carcinoma: a clinicopathologic study of 161 cases. Am J Pathol. 2014;142:299–306.
11. Charpin C, Bonnier P, Khouzami A, et al. Inflammatory breast carcinoma: an immunohistochemical study using monoclonal anti- pHER-2/neu, pS2, cathepsin, ER and PR. Anticancer Res. 1992;12:591–597.
12. Xiao Y, Ye Y, Zou X, et al. The lymphovascular embolus of inflammatory breast cancer exhibits a Notch 3 addiction. Oncogene. 2011;30(3):287–300. doi:10.1038/onc.2010.405
13. Ye Y, Gao J-X, Tian H, et al. Early to intermediate steps of tumor embolic formation involve specific proteolytic processing of E- cadherin regulated by Rab7. Mol Cancer Res. 2012;10:713–726. doi:10.1158/1541-7786.MCR-12-0009
14. Charafe-Jauffret E, Ginestier C, Iovino F, et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clin Cancer Res. 2010;16:45–55. doi:10.1158/1078-0432.CCR-09-1630
15. Silvera D, Schneider RJ. Inflammatory breast cancer cells are constitutively adapted to hypoxia. Cell Cycle. 2009;8:3091–3096. doi:10.4161/cc.8.19.9637
16. Grosse-Wilde A, Fouquier d’Hérouël A, McIntosh E, et al. Stemness of the hybrid epithelial/mesenchymal state in breast cancer and its association with poor survival. PLoS One. 2015;10:e0126522. doi:10.1371/journal.pone.0126522
17. Jolly MK, Boareto M, Debeb BG, et al. Inflammatory breast cancer: a model for investigating cluster-based dissemination. NPJ Breast Cancer. 2017;3(1):21. doi:10.1038/s41523-017-0023-9
18. Masuda H, Brewer TM, Liu DD, et al. Long- term treatment efficacy in primary inflammatory breast cancer by hormonal receptor- and HER2-defined subtypes. Ann Oncol. 2014;25:384–391. doi:10.1093/annonc/mdt525
19. Kertmen N, Babacan T, Keskin O, et al. Molecular subtypes in patients with inflammatory breast cancer; a single center experience. J BUON. 2015;20:35–39.
20. Parton M, Dowsett M, Ashley S, et al. High incidence of HER-2 positivity in inflammatory breast cancer. Breast. 2004;13(2):97–103. doi:10.1016/j.breast.2003.08.004
21. Ross JS, Ali SM, Wang K, et al. Comprehensive genomic profiling of inflammatory breast cancer cases reveals a high frequency of clinically relevant genomic alterations. Breast Cancer Res Treat. 2015;154(1):152–162. doi:10.1007/s10549-015-3592-z
22. Matsuda N, Lim B, Wang Y, et al. Identification of frequent somatic mutations in inflammatory breast cancer. Breast Cancer Res Treat. 2017;163(2):263–272. doi:10.1007/s10549-017-4165-0
23. Rana HQ, Sacca R, Drogan C, et al. Prevalence of Germline Variants in Inflammatory Breast Cancer. Cancer. 2019;125(13):2194–2202.
24. Moll UM, Riou G, Levine AJ. Two distinct mechanisms alter p53 in breast cancer: mutation and nuclear exclusion. Proc Natl Acad Sci U S A. 1992;89:7262. doi:10.1073/pnas.89.15.7262
25. Riou G, Lê MG, Travagli JP, et al. Poor prognosis of p53 gene mutation and nuclear overexpression of p53 protein in inflammatory breast carcinoma. J Natl Cancer Inst. 1993;85(21):1765. doi:10.1093/jnci/85.21.1765
26. Qi Y, Wang X, Kong X, et al. Expression signatures and roles of microRNAs in inflammatory breast cancer. Cancer Cell Int. 2019;19:23. doi:10.1186/s12935-018-0709-6
27. Ding Q, Wang Y, Zuo Z, et al. Decreased expression of microRNA-26b in locally advanced and inflammatory breast cancer. Hum Pathol. 2018;77:121–129. doi:10.1016/j.humpath.2018.04.002
28. Huo L, Wang Y, Gong Y, et al. MicroRNA expression profiling identifies decreased expression of miR-205 in inflammatory breast cancer. Mod Pathol. 2016;29(4):330–346. doi:10.1038/modpathol.2016.38
29. Kleer CG, Zhang Y, Pan Q, et al. WISP3 is a novel tumor suppressor gene of inflammatory breast cancer. Oncogene. 2002;21:3172–3180. doi:10.1038/sj.onc.1205462
30. Haga RB, Ridley AJ. Rho GTPases: regulation and roles in cancer cell biology. Small GTPases. 2016;7:207–221. doi:10.1080/21541248.2016.1232583
31. Van Golen KL, Wu ZF, Qiao XT, et al. RhoC GTPase overexpression modulates induction of angiogenic factors in breast cells. Neoplasia. 2000;2:418–425. doi:10.1038/sj.neo.7900115
32. Wu M, Wu ZF, Kumar-Sinha C, et al. RhoC induces differential expression of genes involved in invasion and metastasis in MCF10A breast cells. Breast Cancer Res Treat. 2004;84:3–12. doi:10.1023/B:BREA.0000018426.76893.21
33. Van Golen KL, Wu ZF, Qiao XT, et al. RhoC GTPase, a novel transforming oncogene for human mammary epithelial cells that partially recapitulates the inflammatory breast cancer phenotype. Cancer Res. 2000;60:5832–5838.
34. Joglekar M, Elbazanti WO, Weitzman MD, Lehman HL, van Golen KL. Caveolin-1 mediates inflammatory breast cancer cell invasion via the Akt1 pathway and RhoC GTPase. J Cell Biochem. 2015;116:923–933. doi:10.1002/jcb.25025
35. Van Laere SJ, Ueno NT, Finetti P, et al. Uncovering the molecular secrets of inflammatory breast cancer biology: an integrated analysis of three distinct Affymetrix gene expression datasets. Clin Cancer Res. 2013;19:4685–4696. doi:10.1158/1078-0432.CCR-12-2549
36. Manai M, Thomassin-Piana J, Gamoudi A, et al. MARCKS protein overexpression in inflammatory breast cancer. Oncotarget. 2017;8(4):6246–6257. doi:10.18632/oncotarget.14057
37. Boersma BJ, Reimers M, Yi M, et al. A stromal gene signature associated with inflammatory breast cancer. Int J Cancer. 2008;122(6):1324–1332. doi:10.1002/ijc.23237
38. Rosen PP. Rosen’s Breast Pathology. Philadelphia: Lippincott-Raven; 1996.
39. Levine PH, Portera CC, Hoffman HJ, et al. Evaluation of lymphangiogenic factors, vascular endothelial growth factor D and E- cadherin in distinguishing inflammatory from locally advanced breast cancer. Clin Breast Cancer. 2012;12:232–239. doi:10.1016/j.clbc.2012.04.005
40. Skobe M, Hawighorst T, Jackson DG, et al. Induction of tumor lymphangiogenesis by VEGFC promotes breast cancer metastasis. Nat Med. 2001;7:192. doi:10.1038/84643
41. Stacker SA, Caesar C, Baldwin ME, et al. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med. 2001;7:186. doi:10.1038/84635
42. Kurebayashi J, Otsuki T, Kunisue H, et al. Expression of vascular endothelial growth factor (VEGF) family members in breast cancer. Jpn J Cancer Res. 1999;90:977. doi:10.1111/j.1349-7006.1999.tb00844.x
43. Pierga JY, Petit T, Delozier T, et al. Neoadjuvant bevacizumab, trastuzumab, and chemotherapy for primary inflammatory HER2-positive breast cancer (BEVERLY-2): an open- label, single- arm phase 2 study. Lancet Oncol. 2012;13:375–384. doi:10.1016/S1470-2045(12)70049-9
44. Cabioglu N, Gong Y, Islam R, et al. Expression of growth factor and chemokine receptors: new insights in the biology of inflammatory breast cancer. Ann Oncol. 2007;18(6):1021–1029. doi:10.1093/annonc/mdm060
45. Nemunaitis JJ, Small KA, Kirschmeier P, et al. A first-in-human, Phase 1, dose-escalation study of dinaciclib, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies. J Transl Med. 2013;11:259. doi:10.1186/1479-5876-11-259
46. Mitri Z, Karakas C, Wei C, et al. A phase 1 study with dose expansion of the CDK inhibitor dinaciclib (SCH 727965) in combination with epirubicin in patients with metastatic triple negative breast cancer. Invest New Drugs. 2015;33:890–894. doi:10.1007/s10637-015-0244-4
47. Mita MM, Joy AA, Mita A, et al. Randomized Phase II trial of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin Breast Cancer. 2014;14:169–176. doi:10.1016/j.clbc.2013.10.016
48. Alexander A, Karakas C, Chen X, et al. Cyclin E overexpression as a biomarker for combination treatment strategies in inflammatory breast cancer. Oncotarget. 2017;8(9):14897–14911. doi:10.18632/oncotarget.14689
49. Debeb BG, Gong Y, Atkinson RL, et al. EZH2 expression correlates with locoregional recurrence after radiation in inflammatory breast cancer. J Exp Clin Cancer Res. 2014;33(1):58. doi:10.1186/s13046-014-0058-9
50. Shostak K, Chariot A. NF-kappaB, stem cells and breast cancer: the links get stronger. Breast Cancer Res. 2011;13:214. doi:10.1186/bcr2886
51. Xia Y, Shen S, Verma IM. NF-kappaB, an active player in human cancers. Cancer Immunol Res. 2014;2:823–830. doi:10.1158/2326-6066.CIR-14-0112
52. Wang W, Kryczek I, Dostál L, et al. Effector T cells abrogate stroma- mediated chemoresistance in ovarian cancer. Cell. 2016;165:1092–1105. doi:10.1016/j.cell.2016.04.009
53. Jhaveri K, Teplinsky E, Silvera D, et al. Hyperactivated mTOR and JAK2/STAT3 pathways: molecular drivers and potential therapeutic targets of inflammatory and invasive Ductal breast cancers after neoadjuvant chemotherapy. Clin Breast Cancer. 2016;16(2):113–122. doi:10.1016/j.clbc.2015.11.006
54. Aaronson DS, Horvath CM. A road map for those who don’t know JAK- STAT. Science. 2002;296:1653–1655. doi:10.1126/science.1071545
55. Bieche I, Lerebours F, Tozlu S, et al. Molecular profiling of inflammatory breast cancer: identification of a poor- prognosis gene expression signature. Clin Cancer Res. 2004;10:6789–6795. doi:10.1158/1078-0432.CCR-04-0306
56. US National Library of Medicine. ClinicalTrials.gov; 2017. Available from: https://www.clinicaltrials.gov/ct2/show/NCT02041429.
57. Drygin D, Ho CB, Omori M, et al. Protein kinase CK2 modulates IL-6 expression in inflammatory breast cancer. Biochem Biophys Res Commun. 2011;415:163–167. doi:10.1016/j.bbrc.2011.10.046
58. Wolfe AR, Trenton NJ, Debeb BG, et al. Mesenchymal stem cells and macrophages interact through IL-6 to promote inflammatory breast cancer in pre- clinical models. Oncotarget. 2016;7:82482–82492. doi:10.18632/oncotarget.12694
59. Hamm CA, Moran D, Rao K, et al. Genomic and immunological tumor profiling identifies targetable pathways and extensive CD8+/PDL1 +immune infiltration in inflammatory breast cancer tumors. Mol Cancer Ther. 2016;15(7):1746–1756. doi:10.1158/1535-7163.MCT-15-0353
60. Andre F, O’Regan R, Ozguroglu M, et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): a randomised, double-blind, placebo-controlled Phase 3 trial. Lancet Oncol. 2014;15:580–591. doi:10.1016/S1470-2045(14)70138-X
61. Ristimaki A, Sivula A, Lundin J, et al. Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res. 2002;62:632–635.
62. Wang X, Reyes ME, Zhang D, et al. EGFR signaling promotes inflammation and cancer stem- like activity in inflammatory breast cancer. Oncotarget. 2017;8:67904–67917. doi:10.18632/oncotarget.18958
63. Reddy JP, Atkinson RL, Larson R, et al. Mammary stem cell and macrophage markers are enriched in normal tissue adjacent to inflammatory breast cancer. Breast Cancer Res Treat. 2018;171(2):283–293. doi:10.1007/s10549-018-4835-6
64. Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi:10.1016/j.cell.2006.01.007
65. Morrow RJ, Etemadi N, Yeo B, et al. Challenging a misnomer? The role of inflammatory pathways in inflammatory breast cancer. Mediators Inflamm. 2017;2017:
66. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–1437. doi:10.1038/nm.3394
67. Rahal OM, Wolfe AR, Mandal PK, et al. Blocking Interleukin (IL)4- and IL13-mediated phosphorylation of STAT6 (Tyr641) decreases M2 polarization of macrophages and protects against macrophage-mediated radioresistance of inflammatory breast cancer. Int J Radiat Oncol Biol Phys. 2018;100(4):1034–1043. doi:10.1016/j.ijrobp.2017.11.043
68. Blank C, Mackensen A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother. 2007;56:739–745. doi:10.1007/s00262-006-0272-1
69. Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi:10.1038/nature14011
70. Bertucci F, Finetti P, Birnbaum D, et al. The PD1/PDL1 axis, a promising therapeutic target in aggressive breast cancers. Oncoimmunology. 2015;5:e1085148.
71. Bertucci F, Finetti P, Colpaert C, et al. PDL1 expression in inflammatory breast cancer is frequent and predicts for the pathological response to chemotherapy. Oncotarget. 2015;6:13506–13519. doi:10.18632/oncotarget.3642
72. Peng W, Liu C, Xu C, et al. PD-1 blockade enhances T-cell migration to tumors by elevating IFN-gamma inducible chemokines. Cancer Res. 2012;72:5209–5218. doi:10.1158/0008-5472.CAN-12-1187
73. He J, Huo L, Ma J, et al. Expression of programmed death ligand 1 (PD-L1) in posttreatment primary inflammatory breast cancers and clinical implications. Am J Clin Pathol. 2018;149(3):253–261. doi:10.1093/ajcp/aqx162
74. Arias-Pulido H, Cimino-Mathews A, Chaher N, et al. The combined presence of CD20 + B cells and PD-L1 + tumor-infiltrating lymphocytes in inflammatory breast cancer is prognostic of improved patient outcome. Breast Cancer Res Treat. 2018;171(2):273–282. doi:10.1007/s10549-018-4834-7
75. Mego M, Gao H, Cohen EN, et al. Circulating tumor cells (CTCs) are associated with abnormalities in peripheral blood dendritic cells in patients with inflammatory breast cancer. Oncotarget. 2016;8:35656–35658. doi:10.18632/oncotarget.10290
76. Fei M, Bhatia S, Oriss TB, et al. TNF- alpha from inflammatory dendritic cells (DCs) regulates lung IL-17A/IL-5 levels and neutrophilia versus eosinophilia during persistent fungal infection. Proc Natl Acad Sci USA. 2011;108:5360–5365. doi:10.1073/pnas.1015476108
77. Hilkens CM, Kalinski P, de Boer M, et al. Human dendritic cells require exogenous interleukin-12-inducing factors to direct the development of naive T- helper cells toward the Th1 phenotype. Blood. 1997;90:1920–1926. doi:10.1182/blood.V90.5.1920
78. Janni WJ, Rack B, Terstappen LW, et al. Pooled analysis of the prognostic relevance of circulating tumor cells in primary breast cancer. Clin Cancer Res. 2016;22:2583–2593. doi:10.1158/1078-0432.CCR-15-1603
79. Pierga JY, Bidard FC, Autret A, et al. Circulating tumour cells and pathological complete response: independent prognostic factors in inflammatory breast cancer in a pooled analysis of two multicentre phase II trials (BEVERLY-1 and −2) of neoadjuvant chemotherapy combined with bevacizumab. Ann Oncol. 2017;28(1):103–109. doi:10.1093/annonc/mdw535
80. Mego M, Giordano A, De Giorgi U, et al. Circulating tumor cells in newly diagnosed inflammatory breast cancer. Breast Cancer Res. 2015;17(1):2. doi:10.1186/s13058-014-0507-6
81. Mego M, Gao H, Cohen EN, et al. Circulating tumor cells (CTC) are associated with defects in adaptive immunity in patients with inflammatory breast cancer. J Cancer. 2016;7(9):1095–1104. doi:10.7150/jca.13098
82. Tabouret E, Bertucci F, Pierga JY, et al. MMP2 and MMP9 serum levels are associated with favorable outcome in patients with inflammatory breast cancer treated with bevacizumab-based neoadjuvant chemotherapy in the BEVERLY-2 study. Oncotarget. 2016;7(14):18531–18540. doi:10.18632/oncotarget.7612
83. Mohamed MM, El-Ghonaimy EA, Nouh MA, et al. Cytokines secreted by macrophages isolated from tumor microenvironment of inflammatory breast cancer patients possess chemotactic properties. Int J Biochem Cell Biol. 2014;46:138–147. doi:10.1016/j.biocel.2013.11.015
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.