")
Back to Journals » Infection and Drug Resistance » Volume 11
Biofilm infections between Scylla and Charybdis: interplay of host antimicrobial peptides and antibiotics
Authors Chernysh S , Gordya N , Tulin D , Yakovlev A
Received 22 November 2017
Accepted for publication 11 January 2018
Published 9 April 2018 Volume 2018:11 Pages 501—514
DOI https://doi.org/10.2147/IDR.S157847
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Sergey Chernysh,* Natalia Gordya,* Dmitry Tulin, Andrey Yakovlev
Laboratory of Insect Biopharmacology and Immunology, Faculty of Biology, St. Petersburg State University, St. Petersburg, Russia
*These authors contributed equally to this work
Purpose: The aim of this study is to improve the anti-biofilm activity of antibiotics. We hypothesized that the antimicrobial peptide (AMP) complex of the host’s immune system can be used for this purpose and examined the assumption on model biofilms.
Methods: FLIP7, the AMP complex of the blowfly Calliphora vicina containing a combination of defensins, cecropins, diptericins and proline-rich peptides was isolated from the hemolymph of bacteria-challenged maggots. The complex interaction with antibiotics of various classes was studied in biofilm and planktonic cultures of Staphylococcus aureus, Escherichia coli, Pseudomonas aeruginosa, Klebsiella pneumoniae and Acinetobacter baumannii by the checkerboard method using trimethyl tetrazolium chloride cell viability and crystal violet biofilm eradication assays supplemented with microscopic analysis.
Results: We found that FLIP7 demonstrated: high synergy (fractional inhibitory concentration index <0.25) with meropenem, amikacin, kanamycin, ampicillin, vancomycin and cefotaxime; synergy with clindamycin, erythromycin and chloramphenicol; additive interaction with oxacillin, tetracycline, ciprofloxacin and gentamicin; and no interaction with polymyxin B. The interaction in planktonic cell models was significantly weaker than in biofilms of the same strains. The analysis of the dose–effect curves pointed to persister cells as a likely target of FLIP7 synergistic effect. The biofilm eradication assay showed that the effect also caused total destruction of S. aureus and E. coli biofilm materials. The effect allowed reducing the effective anti-biofilm concentration of the antibiotic to a level well below the one clinically achievable (2–3 orders of magnitude in the case of meropenem, ampicillin, cefotaxime and oxacillin).
Conclusion: FLIP7 is a highly efficient host antimicrobial system helping antibiotics to overcome biofilm barriers through persisters’ sensitization and biofilm material destruction. It is promising for the treatment of biofilm infections as an adjuvant of various small-molecule antibiotics.
Keywords: insect antimicrobial peptides, antibiotics, synergy, biofilms, persisters, Calliphora vicina
Introduction
Most bacteria form biofilms in order to survive in the host organism. These biofilms are sedimented multicellular communities embedded in a biopolymer matrix. Biofilms account for over 80% of microbial infections that cause nonhealing wounds, ulcers and lesions of the skin, mucosa and internal organs.1 A growing body of data also points to the important role of biofilms in the occurrence of diseases that are usually not considered to be infectious, such as cancer2–4 and cardiovascular disorders.5,6 Moreover, biofilms, which invade the surfaces of catheters, implants and other medical devices, make a significant contribution to morbidity and economic losses from bacterial infections.7 The transition of planktonic bacteria to biofilms gives them multifactorial protection against the host’s immune system and allows persistence in the body for an unlimited time.8,9 Enhanced resistance to antibiotics and antiseptics is another clinically important factor in biofilm infections.10–14 The escalating prevalence of antibiotic-resistant strains makes the task of treating biofilms even more difficult. Thus, the search for new ways of treating biofilm infections is one of the important areas of health care progress.
Over the last three decades, animal antimicrobial peptides (AMPs) have attracted attention as a promising platform for the development of new antibacterial drugs. More than 2600 sequences of AMPs toxic to planktonic bacteria have been identified in animals, plants, fungi and microorganisms.15 Much less is known about anti-biofilm AMPs, even though some of them exhibit strong cell-killing and/or matrix-destroying activity against bacterial biofilms.16–19 Using the checkerboard method, it was possible to detect two-component AMP–AMP20,21 and AMP–antibiotic17,19,22 combinations with a synergistic anti-biofilm effect. However, further progress in the development of such combinations encounters significant technical difficulties, since not only the type of interaction (synergism, additivity, antagonism) but also other parameters like antibacterial activity spectrum, toxicity, the rate of resistance development and so on should be estimated. The search for combinations comprising three or more components seems to be an almost insoluble problem from this point of view.
At the same time, the immune systems of multicellular animals exploit multicomponent AMP systems optimized by natural selection in all the parameters important for the survival of the host organism. Although the idea of using natural AMP complexes as a platform for the development of antimicrobial drugs seems attractive from various points of view, it has so far been neglected. Only recently, the key benefits of this approach have received experimental confirmation. Particularly, experimental evolution studies revealed that none of the Gram-negative bacteria which were tested were able to acquire resistance to the host AMP complexes of blowfly Calliphora vicina and other Calliphoridae in contrast to conventional antibiotics.23 A further study has shown that a C. vicina complex containing a combination of four AMP families (defensins, cecropins, diptericins and proline-rich peptides) is active against not only planktonic bacteria but also their biofilms.24 Thus, the natural AMP complex is superior to conventional antibiotics in at least two respects: the ability to prevent the development of acquired resistance and the effectiveness against biofilms. The question arises as to whether it is possible to combine the merits of host AMP complexes and antibiotics and thus create a more effective approach to the treatment of biofilm infections.
In this study, we tried to find an answer to this question by investigating the interaction of a C. vicina AMP complex referred to as FLIP7 (Fly Larvae Immune Peptides) and antibiotics of the main classes using the checkerboard method and in vitro models of biofilms formed by clinically relevant pathogens Staphylococcus aureus, Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa and Acinetobacter baumannii.
Materials and methods
C. vicina AMP complex
The sample of FLIP7 was isolated from the hemolymph of blue blowfly C. vicina diapausing maggots. The maggots were inoculated with E. coli M17 strain (Microgen, Moscow, Russia) to induce an immune response and left overnight at 25°C. The hemolymph was collected in ice-cold tubes through a cuticle puncture, frozen and kept at −70°C until use. The thawed hemolymph was acidified with 0.1% trifluoroacetic acid (TFA) to a final concentration of 0.05%, and the insoluble particles were removed by centrifugation (30 minutes at 8000 × g at 4°C). The supernatant was applied to reversed-phase Sep-Pak C18 cartridges (Waters Corporation, Milford, MA, USA) stabilized by 0.05% TFA in the amount of 5 mL/g of sorbent. Highly hydrophilic compounds were removed by cartridge washing with 0.05% TFA. The compounds absorbed in the cartridge were eluted with 50% acetonitrile solution acidified with 0.05% TFA, lyophilized (FreeZone; Labconco, Kansas City, MO, USA) and stored at −70°C. Prior to use, the lyophilized sample was dissolved in deionized water (50 mg/mL), sterilized by filtration through a membrane with a pore size of 0.22 μm (Milliex-GS; Merck Millipore, Billerica, MA, USA) and frozen at −70°C.
The sample used in this work was structurally characterized in our previous paper using a combination of liquid chromatography/mass spectrometry and transcriptome methods.24 Ten peptides belonging to four AMP families (defensins, diptericins, cecropins and proline-rich peptides) of the insects were deciphered, although some additional AMPs found in the complex remained uncharacterized. The database of mRNAs isolated from bacteria-challenged C. vicina maggots and available at https://www.ncbi.nlm.nih.gov/sra/SRX2523753 (submission number SRR5210297) predicts the existence of additional AMP isoforms especially diverse in the family of diptericins.
Antibiotics
The following commercial preparations were used in the experiments: aminoglycosides amikacin sulfate (Sintez, Kurgan, Russia), kanamycin sulfate (Sintez) and gentamicin sulfate (AppliChem, Darmstadt, Germany), natural penicillin oxacillin sodium (Sintez), aminopenicillin ampicillin sodium (Sintez), carbopenem meropenem trihydrate (AstraZeneca, Cambridge, UK), third-generation cephalosporin cefotaxime sodium (Abolmed, Moscow, Russia), naturally occurring polypeptide polymyxin B sulfate (AppliChem), glycopeptide vancomycin (Kraspharma, Krasnoyarsk, Russia), lincosamid clindamycin phosphate (Hemopharm, Vršac, Serbia), macrolide erythromycin phosphate (Sintez), second-generation fluoroquinolone ciprofloxacin hydrochloride (AppliChem), chloramphenicol sodium succinate (AppliChem), tetracycline hydrochloride (AppliChem) and antiseptic benzalkonium chloride (Fluka, Buchs, Switzerland). The antibiotics were dissolved in sterile deionized water at a concentration of 10, 1 and 0.1 mg/mL, aliquoted in 0.05 mL volumes and kept at −70°C until use. Chloramphenicol and tetracycline were dissolved in 50% ethanol in sterile deionized water.
Bacteria
E. coli ATCC 25922 and P. aeruginosa ATCC 27583 strains as well as clinical strains of S. aureus 203, K. pneumoniae 145 and A. baumannii 28 were used as model biofilm-forming bacteria. The annotated genome assembly of E. coli ATCC 25922 and P. aeruginosa ATCC 27583 is available at the links http://www.ncbi.nlm.nih.gov/nuccore/CP009072 and http://www.pseudomonas.com/strain/show/3104, respectively. The A. baumannii 28 genome is available at https://www.ncbi.nlm.nih.gov/nuccore/NZ_MAFT00000000. Strain genome sequencing predicts resistance to aminoglycosides, beta-lactams and chloramphenicol. The clinical strain of K. pneumoniae 145 originates from the surgery clinic of the Kirov Military Medical Academy (St. Petersburg, Russia), and the strains of A. baumannii 28 and S. aureus 203 originate from the collection of The Northwestern State Medical Mechnikov University and Institute of Experimental Medicine (St. Petersburg, Russia), respectively. The profiles of the clinical strains’ antibiotic resistance were determined as recommended.47 The strains were classified as susceptible, intermediate or resistant to the antibiotic in testing with disc diffusion method. To test the resistance profiles of K. pneumoniae 145, the following antibiotics were used: amikacin, amoxicillin/clavulanic acid, gentamicin, imipenem, meropenem, cefepime, cefoperazone, cefoperazone/sulbactam, cefotaxime, ceftazidime, ciprofloxacin, piperacillin/tazobactam and tigecycline (Oxoid Limited, Basingstoke, UK). The strain was found to be resistant to all the antibiotics that were tested except imipenem, meropenem and tigecycline. The S. aureus 203 strain was found to be sensitive to all the antibiotics tested.
Biofilm formation
The preparation of 24-hour-old biofilms corresponded to the previously described procedure ensuring the formation of a dense biofilm by the strains used in experiments.24 The biofilm formation was confirmed by the staining of 24-hour biofilm with crystal violet dye. The bacteria were cultured in 5 mL of LB liquid nutrient medium (Thermo Fisher Scientific, Waltham, MA, USA) for 18–20 hours at 37°C. Overnight cultures were adjusted to 5 × 105 CFU/mL measured by optical density (OD). One-hundred-microliter aliquots of the diluted bacterial suspension were inoculated into each well of a 96-well flat-bottomed polystyrene plate (Sarstedt AG & Co., Nümbrecht, Germany) and incubated in a humidified incubator for 24 hours at 37°C. The negative control was an LB liquid nutrient medium.
TTC cell-killing assay
The effect of antimicrobial agents on the viability of biofilm bacteria was assessed using a standard trimethyl tetrazolium chloride (TTC) test identical to that described previously.24 The 24-hour biofilms, formed in a 96-well tissue culture microtiter plate, were washed three times with 200 µL of sterile physiological buffered saline (PBS) to remove unattached bacteria and then air-dried. An antimicrobial agent dissolved in 100 µL LB liquid nutrient medium (Thermo Fisher Scientific) was added to each corresponding well, and the plates were incubated for 24 hours at 37°C in a humidified incubator. Then, 11 µL of 0.2% TTC (Lenreactiv, St. Petersburg, Russia) was added to a final concentration of 0.02%. After 1 hour of incubation at 37°C, the OD540 was measured on the Epoch microplate reader (BioTek Instruments, Winooski, VT, USA). The mean OD540 value of 48-hour biofilm cells without antimicrobial agent treatment was set as the control. Each experiment was performed in two independent assays. The minimum biofilm-inhibiting concentration (MBIC) was assessed with MBIC90, the concentration decreasing TTC staining by 90% compared to the control.
Crystal violet biofilm eradication assay
A standard crystal violet biofilm eradication assay was performed with minor modifications as described previously.24 This method gives the total amount of the biofilm material binding to the dye, including extracellular and cellular components. The 24-hour biofilms, formed in a 96-well tissue culture microtiter plate, were washed three times with 200 µL of sterile PBS solution and air-dried. One hundred microliters of antimicrobial composition dissolved in LB liquid nutrient medium (Invitrogen, Carlsbad, CA, USA) was added to each corresponding well, and the plates were incubated for 24 hours at 37°C. Then, the waste media was removed, and the plates were washed three times with 200 µL PBS solution, air-dried and stained with crystal violet 0.1% (in water) (Lenreactiv, St. Petersburg, Russia) for exactly 2 minutes. The stained biofilms were washed three times with 200 µL PBS solution, air-dried and solubilized with 200 µL of 95% ethanol for 1 hour. Then, the biofilm-associated dye was measured at OD570 using the Epoch reader (BioTek Instruments). Each experiment was performed in two independent assays. The minimum biofilm eradication concentration (MBEC) was assessed with MBEC90, which is the concentration of antimicrobials decreasing crystal violet binding in preformed biofilms by 90% compared to untreated control.
Planktonic cells’ viability assessment
The standard microdilution method was carried out for the determination of planktonic cells minimum inhibiting concentration (MIC), as recommended.48 The initial inoculum was grown on a solid LB agar nutritive medium (Invitrogen, Carlsbad, CA, USA). Individual colonies were collected, transferred into liquid medium (Luria broth base, 25 g/L) and incubated overnight at 37°C. Individual wells of a 96-well tissue culture plate (Sarstedt, Newton, NC, USA) containing 100 μL of LB liquid nutrient medium (Invitrogen, Carlsbad, CA, USA) with doubling antimicrobial dilutions were inoculated with approximately 5 × 105 CFU/mL of test bacteria. Microtiter plates were incubated for 20 hours at 37°C. The cells’ viability was determined with the TTC method as described above. The MIC was assessed with MIC90, the concentration decreasing TTC staining by 90% compared to the untreated control.
The AMP complex and antibiotics interaction studies
To establish the type and quantitative characteristics of the interaction of C. vicina AMP complex and antibiotics of different classes, the checkerboard titration method with some modifications was used.25 The 24-hour biofilms formed in a 96-well tissue culture microtiter plate were washed three times with 200 µL of sterile PBS and air-dried. In another 96-well microplate, combinations of FLIP7 and antibiotics were prepared in such a way that two-fold dilutions of FLIP7 were placed in horizontal rows of wells, and two-fold dilutions of antibiotics were placed in vertical rows. Then, 100 μL of the contents of each well from this microplate was transferred to a biofilm microplate and incubated for 24 hours at 37°C. The biofilm state was evaluated by the TTC and crystal violet methods as described above. All the experiments were performed in two independent replicates. In experiments with planktonic cultures, dilutions of FLIP7 and an antibiotic were pooled at 25 μL in wells of the microplate according to the scheme, and 50 μL of bacterial suspension with a cell concentration of 106 CFU/mL was added to each well. The microplate was incubated for 24 hours at 37°C, and then the contents of the wells were stained with TTC as described above.
The effects of the combinations were examined by calculating the fractional inhibitory concentration index (FICI) of each combination as follows: [(MIC of drug A, tested in combination)/(MIC of drug A, tested alone)] + [(MIC of drug B, tested in combination)/(MIC of drug B, tested alone)]. In the TTC test and the crystal violet test, the values of MBIC90 and MBEC90 were used instead of MIC. The profile of the combination was defined as synergistic if the FICI was ≤0.5, indifferent if the FICI was >0.5 but ≤4.0 and antagonistic if the FICI was >4. The ratio of the biofilm MBIC90 to the planktonic culture MIC90 determined in the TTC cell viability assay was used as a quantitative characteristic of the biofilm-related resistance (BRR).
Biofilm microscopic visualization
A coverslip of 24 × 24 mm (Thermo Fisher Scientific Gerhard Menzel B.V. & Co. KG, Braunschweig, Germany) was placed in each well of six-well tissue culture microtiter plate (Falcon; Becton Dickinson Labware, Franklin Lakes, NJ, USA). The wells were filled with 2.8 mL of S. aureus 203 suspension, prepared as mentioned in the “Biofilm formation” section, and the plates were incubated for 24 hours at 37°C. Then, the 24-hour biofilms were washed three times with 3 mL of sterile PBS to remove unattached bacteria and filled with fresh culture medium (control) or a medium supplemented with FLIP7, meropenem or a combination of both. The plates were incubated for another 24 hours at 37°C, and then coverslips were removed from the wells, washed in PBS, placed on a glass slide upside down and photographed through a Leica DMI 2500 microscope (Leica Microsystems, Wetzlar, Germany) with Nomarski optics at ×400 and ×1000 magnification.
Statistical methods
Statistical processing of data was performed using the Primer of Biostatistics software, version 4.03. Statistical significance of the differences was analyzed with ANOVA and Z-test considering P-values <0.05 as significant.
Results
BRR profiles quantification
For the analysis of model biofilms’ recalcitrance to antibiotics, the ratio of biofilm MBIC90 and planktonic cells MIC90 was used as a quantitative characteristic referred here to as BRR. The obtained values of BRR (Table 1) show that bacteria in the state of biofilm have increased recalcitrance to the majority of antibiotics studied compared to planktonic cells of the same strain. However, the level of BRR varies widely, depending on the type of antibiotic and the type of bacteria. The maximum BRR growth was registered in S. aureus, particularly to meropenem (over 2000-fold), ampicillin, amikacin and kanamycin. A significant BRR increase was also noted for vancomycin, benzalkonium chloride and erythromycin (10- to 84-fold). Resistance to oxacillin and chloramphenicol only slightly exceeded the level of planktonic cells (1.25- to 2.4-fold).
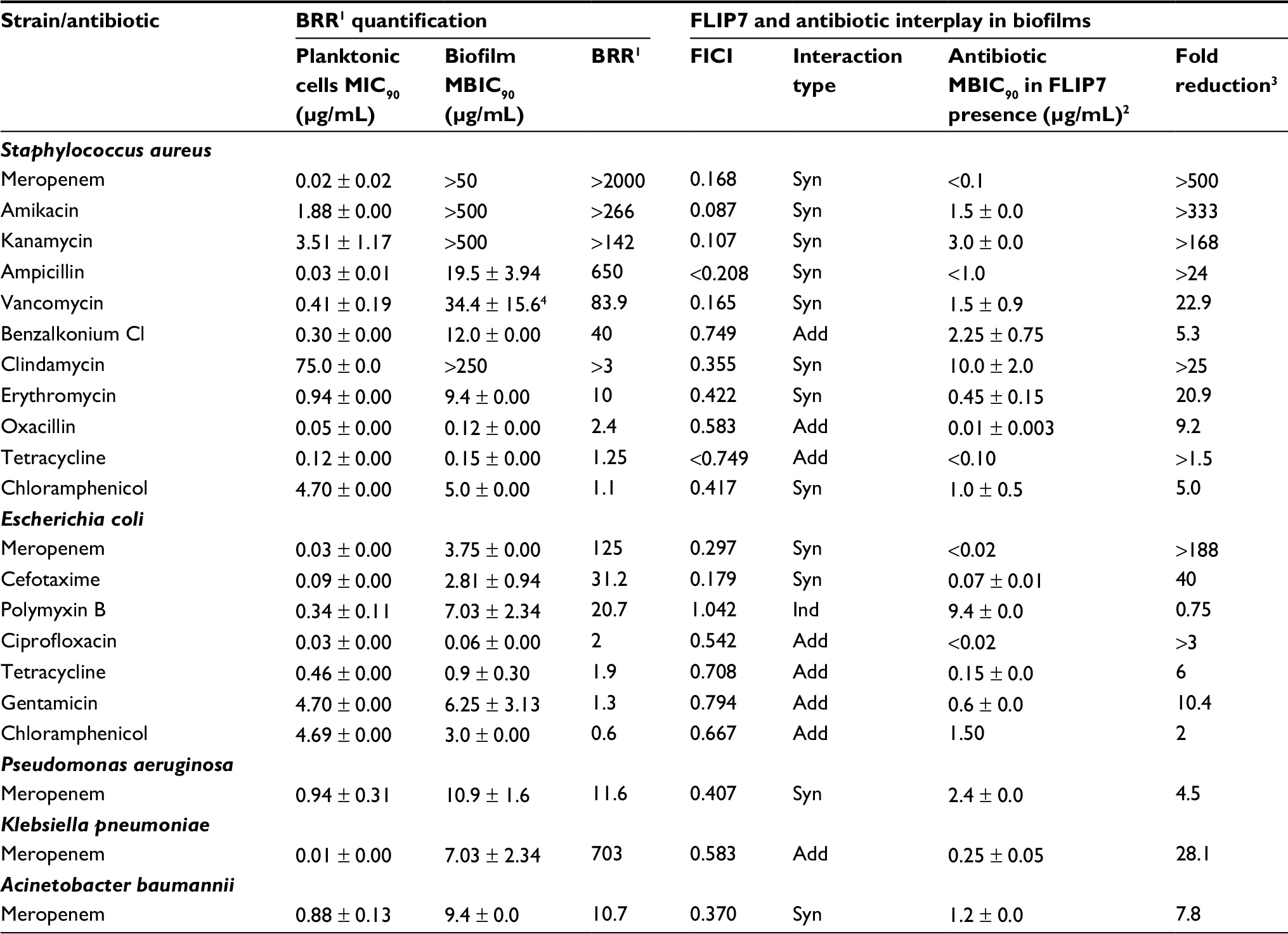 | Table 1 Biofilm-related resistance and FLIP7–antibiotic interaction in biofilms (TTC assay) Notes: 1Biofilm-related resistance determined as antibiotic MBIC90 to planktonic cells MIC90 ratio. 2MBIC90 of antibiotic in combination with FLIP7 concentration causing maximum fold reduction. 3Ratio of antibiotic MBIC90 in the absence and presence of FLIP7 concentration causing maximum antibiotic fold reduction. 4The difference from the planktonic culture MIC90 was statistically unreliable (P = 0.161). Data presented as mean ± SE. Abbreviations: TTC, trimethyl tetrazolium chloride; BRR, biofilm-related resistance; MIC, minimum inhibiting concentration; MBIC, minimum biofilm-inhibiting concentration; FICI, fractional inhibitory concentration index; Syn, synergy; Add, additivity; Ind, independence. |
The E. coli biofilm exhibited maximum resistance growth to meropenem (125-fold), and a less significant resistance growth to cefotaxime and polymyxin B (21- to 31-fold), whereas resistance to ciprofloxacin, tetracycline, gentamicin and chloramphenicol was only slightly different from the planktonic cells level.
Meropenem resistance changes were also tested in P. aeruginosa, A. baumannii and K. pneumoniae. Biofilms of P. aeruginosa and A. baumannii demonstrated intermediate 11- to 20-fold resistance growth, whereas in K. pneumoniae it was 703-fold.
Thus, the model biofilms showed an increase in resistance compared to planktonic cells of the same strain, but the level of resistance to different antibiotics varied greatly. The maximum BRR increase was found with meropenem, which at the same time proved to be highly effective against planktonic cells.
FLIP7 and antibiotics interaction (TTC cell viability assay)
To study the interaction of FLIP7 and antibiotics, we tested the antimicrobial activity of 1827 combinations varying in the type of antibiotic and concentrations of the components in accordance with the standard checkerboard method. The test results are summarized in Table 1 (columns 5–8). FICI values of the S. aureus biofilm demonstrated high synergy (FICI <0.25) of FLIP7 with meropenem, amikacin, kanamycin, ampicillin and vancomycin. All these antibiotics were also characterized by a high level of BRR. At the same time, antibiotics with a low BRR level and antiseptic benzalkonium chloride showed much higher FICI values, and correspondingly, weaker interaction with FLIP7.
In E. coli, the highest synergy was found for meropenem and cefotaxime, which showed the maximum BRR level, whereas antibiotics with low BRR demonstrated a lack of synergy. Polymyxin B with its evidential BRR growth and lack of interaction with FLIP7 stood alone among the other antimicrobials tested. Meropenem and FLIP7 synergy was also significant in experiments with P. aeruginosa and A. baumannii biofilms, although in K. pneumoniae they interacted at the level of additivity.
Antibiotic fold reduction in the presence of FLIP7 effective concentration (column 8 of Table 1) is another important characteristic of the antimicrobials interaction. The most significant fold reduction was found in S. aureus biofilm treated with meropenem, amikacin or kanamycin (over 142–500 times). The fold reduction of other antibiotics and antiseptic benzalkonium chloride varied, ranging from 5 to over 25 times. In E. coli, meropenem showed the most pronounced fold reduction (over 188 times). The reduction of MBIC90 of other antibiotics was reliable as well, except for polymyxin B. FLIP7 also reduced the effective concentration of meropenem in P. aeruginosa, K. pneumoniae and A. baumannii biofilms, although not as significantly as in S. aureus and E. coli.
In parallel with experiments in biofilms, the interaction of FLIP7 and antibiotics was studied in planktonic cultures of the same strains (Table 2). In general, the interaction of FLIP7 and antibiotics in planktonic cells was significantly weaker than in biofilms and mainly additive. Decline of the interaction was particularly noticeable in high-BRR antibiotics. The fold reduction analysis demonstrated a similar trend with some exceptions. Thus, polymyxin B showed no interaction with FLIP7 in the E. coli biofilm, while with the planktonic culture the interaction was well defined. A more significant level of fold reduction in E. coli planktonic cells compared to biofilms was also noted in tetracycline and chloramphenicol. A. baumannii exhibited a similar synergy rate in biofilm and planktonic states and an even higher fold reduction level in planktonic cells.
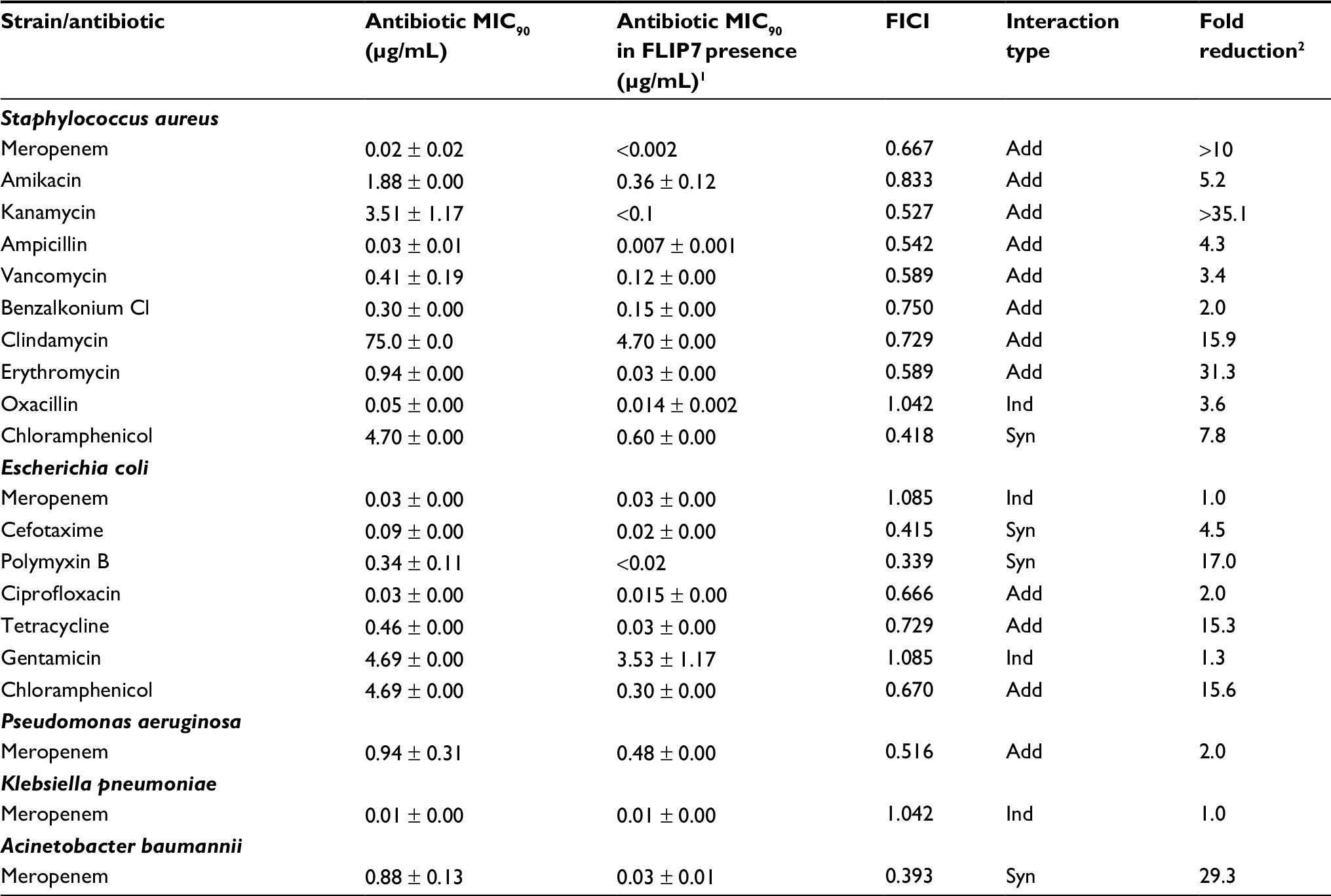 | Table 2 FLIP7 and antibiotics interaction on planktonic cells (TTC assay) Notes: 1MIC90 of antibiotic in combination with FLIP7 concentration causing maximum antibiotic fold reduction. 2Ratio of antibiotic MIC90 in the absence and presence of FLIP7 concentration causing maximum antibiotic fold reduction. Data presented as mean ± SE. Abbreviations: TTC, trimethyl tetrazolium chloride; MIC, minimum inhibiting concentration; FICI, fractional inhibitory concentration index; Syn, synergy; Add, additivity; Ind, independence. |
Dose–effect curves demonstrate another important feature of FLIP7 and antibiotics interaction – the ability to eliminate antibiotic-insensitive persister cells, which is especially pronounced in S. aureus biofilm (Figure 1). Meropenem caused a dose-dependent decrease in cell survival in the concentration range from 0.1 to 0.8 μg/mL, and further increase in concentration did not lead to a decrease in survival. In accordance with the generally accepted interpretation,26 this indicates the presence in the population of antibiotic-insensitive persistent cells. The combination of meropenem with an effective concentration of FLIP7 completely eliminated (or reduced to an undetectable level) metabolically active cells, starting with an antibiotic concentration of 0.1 μg/mL. The same effect was observed in experiments with other high-BRR antibiotics: amikacin, ampicillin and kanamycin. With antibiotics with low or zero BRR (oxacillin, chloramphenicol), persistent bacteria were not detected in the biofilm. Antibiotics with an intermediate (10- to 84-fold) level showed the presence in the population of cells with intermediate resistance to vancomycin, benzalkonium chloride and erythromycin (data not shown). E. coli dose–effect curves of high-BRR antibiotics meropenem and cefotaxime also revealed the presence of persister cells eliminated by the antibiotic combination with FLIP7, whereas those of the low-BRR antibiotics ciprofloxacin and chloramphenicol did not reveal persister cells (Figure 2). The persisters were not clearly detected in meropenem-treated P. aeruginosa, K. pneumoniae and A. baumannii biofilms (data not shown).
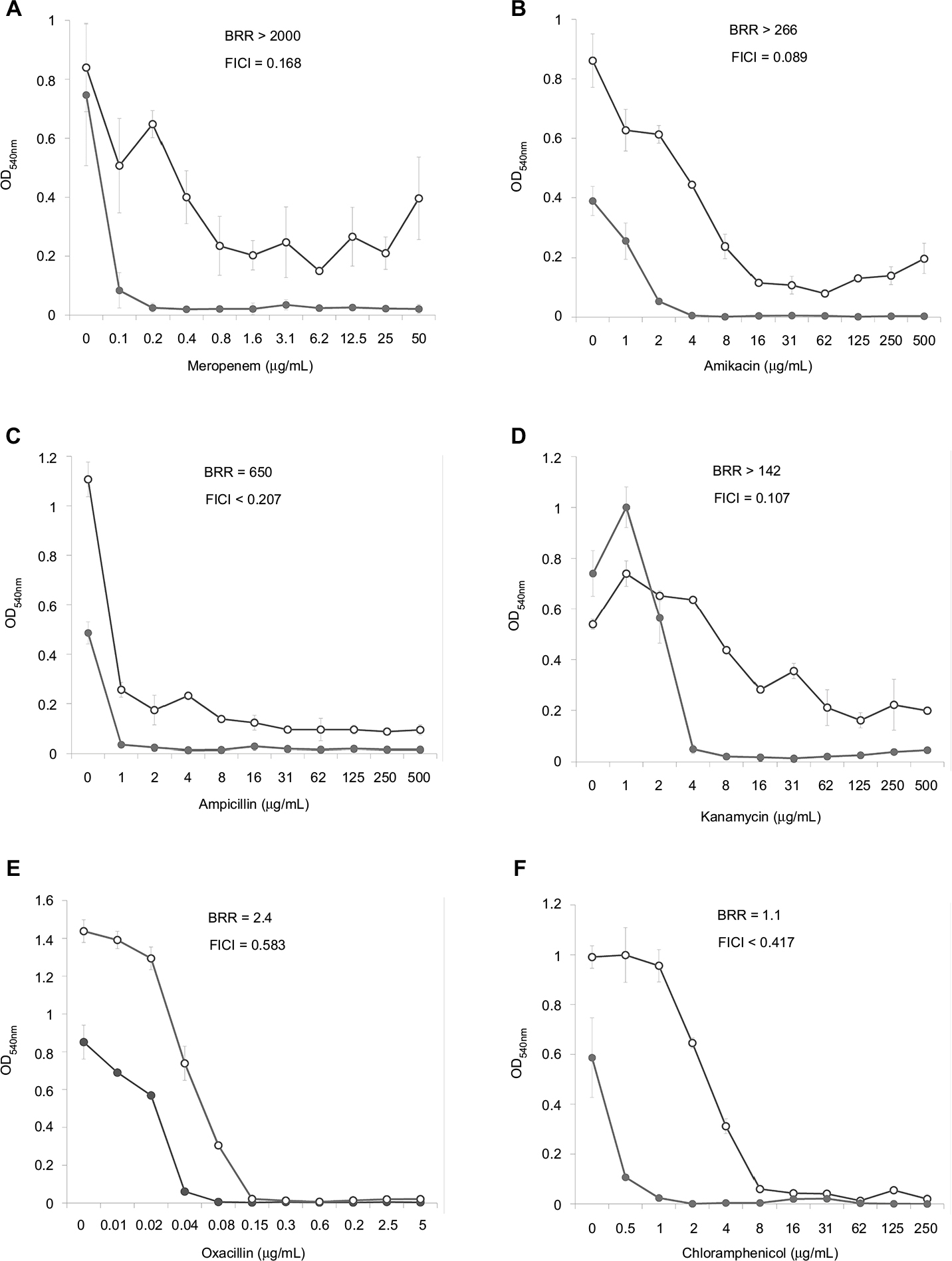 | Figure 1 Doze–effect curves of antibiotics alone and in combination with FLIP7 against Staphylococcus aureus biofilm (TTC assay). Notes: (A) Meropenem versus meropenem + FLIP7 125 µg/mL. (B) Amikacin versus amikacin + FLIP7 250 µg/mL. (C) Ampicillin versus ampicillin + FLIP7 125 µg/mL. (D) Kanamycin versus kanamycin + FLIP7 250 µg/mL. (E) Oxacillin versus oxacillin + FLIP7 250 µg/mL. (F) Chloramphenicol versus chloramphenicol + FLIP7 125 µg/mL. Open circles – antibiotic, closed circles – the antibiotic + FLIP7; Y-axis – optical density (mean ± standard error). Effective concentrations of FLIP7 were individually determined for each antibiotic based on the checkerboard experiments. Abbreviations: TTC, trimethyl tetrazolium chloride; OD, optical density; BRR, biofilm-related resistance; FICI, fractional inhibitory concentration index. |
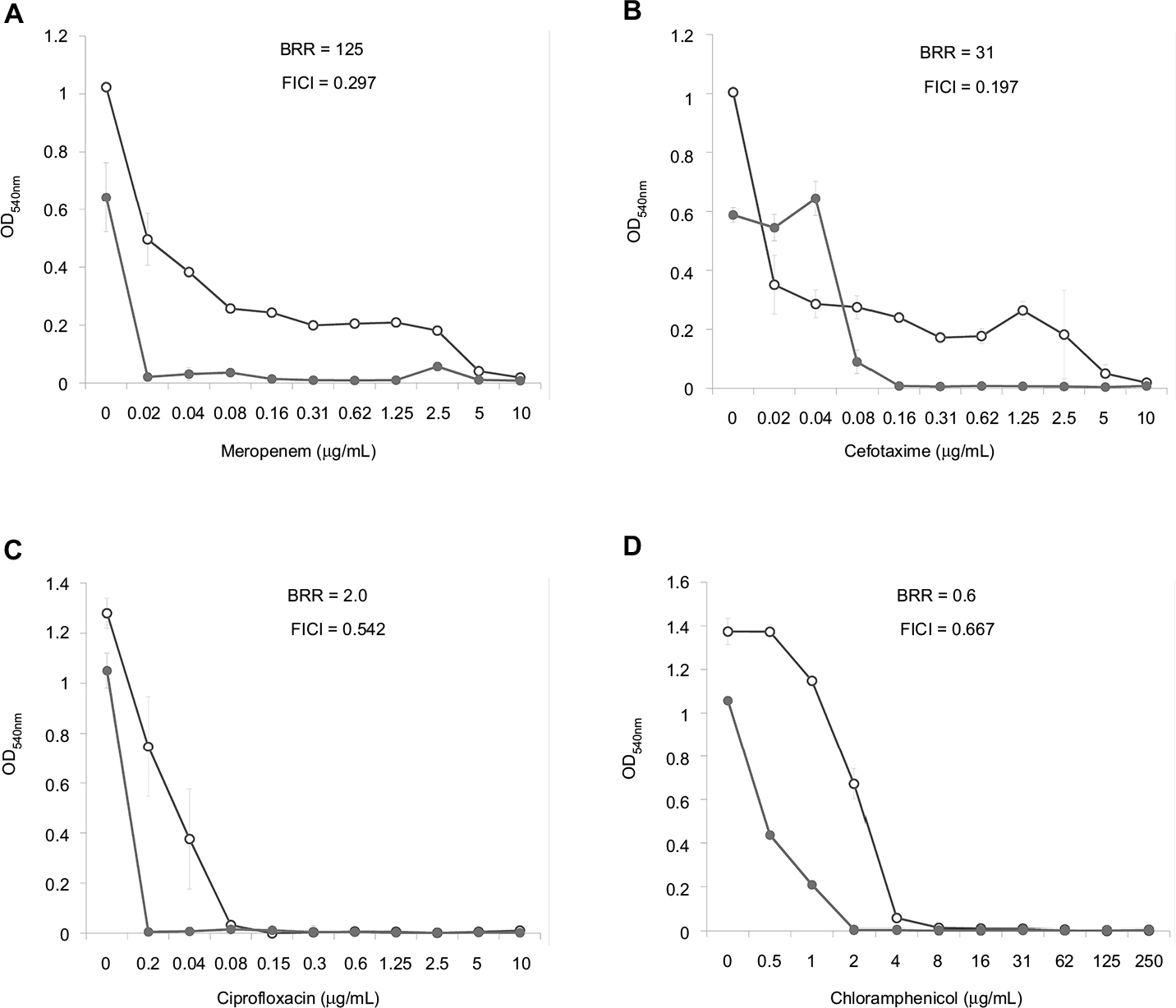 | Figure 2 Doze–effect curves of antibiotics alone and in combination with FLIP7 against Escherichia coli biofilm (TTC assay). Notes: (A) Meropenem versus meropenem + FLIP7 500 µg/mL. (B) Cefotaxime versus cefotaxime + FLIP7 500 µg/mL. (C) Ciprofloxacin versus ciprofloxacin + FLIP7 500 µg/mL. (D) Chloramphenicol versus chloramphenicol + FLIP7 250 µg/mL. Abbreviations: TTC, trimethyl tetrazolium chloride; OD, optical density; BRR, biofilm-related resistance; FICI, fractional inhibitory concentration index. |
FLIP7 and antibiotics interaction (crystal violet biofilm eradication assay)
The interaction of FLIP7 and antibiotics was also studied by staining the biofilms with crystal violet dye (biofilm eradication assay). In total, 862 combinations organized in accordance with standard checkerboard scheme were tested. The analysis of the FICI and fold reduction values showed results almost identical to the TTC method (Table 3). In S. aureus biofilm, FLIP7 demonstrated high levels of synergy and fold reduction with meropenem, amikacin and kanamycin and a lower level in combination with chloramphenicol. In E. coli, the interaction with meropenem and cefotaxime was synergistic, whereas in combination with ciprofloxacin the effect was only additive. Synergy with meropenem was also found in P. aeruginosa and A. baumannii, while the interaction was only additive in K. pneumoniae.
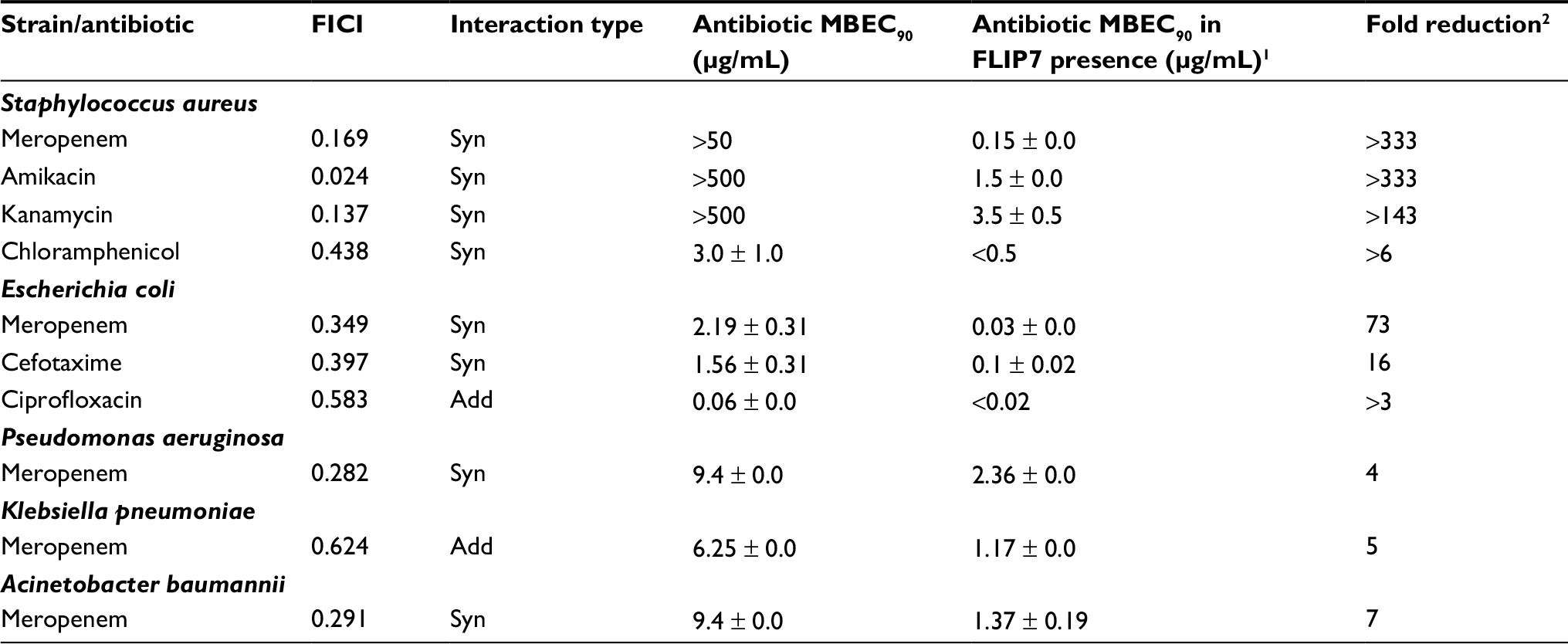 | Table 3 FLIP7 and antibiotics interaction in crystal violet biofilm eradication assay Notes: 1MBEC90 of antibiotic in combination with FLIP7 concentration causing maximum antibiotic fold reduction. 2Ratio of antibiotic MBEC90 in the absence and presence of FLIP7 concentration causing maximum antibiotic fold reduction. Data presented as mean ± SE. Abbreviations: FICI, fractional inhibitory concentration index; MBEC, minimum biofilm eradication concentration; Syn, synergy; Add, additivity. |
Analysis of the dose–effect curves showed that meropenem, amikacin and kanamycin are not capable of destroying the S. aureus biofilm material, although they almost completely eradicated the material in the presence of an effective FLIP7 concentration (Figure 3). In contrast to these antibiotics, chloramphenicol effectively destroyed the biofilm material at a concentration of 2 µg/mL and more. Under these conditions, combination with FLIP7 enhanced the effect of the antibiotic in the range of its low concentrations. In experiments with E. coli (Figure 4), similar results were obtained: FLIP7 drastically improved the limited ability of meropenem and cefotaxime to destroy biofilm material and strengthened the action of subthreshold concentrations of ciprofloxacin. Potentiation of the effect of meropenem was also well expressed in P. aeruginosa and A. baumannii biofilms and less considerable although reliable in K. pneumoniae.
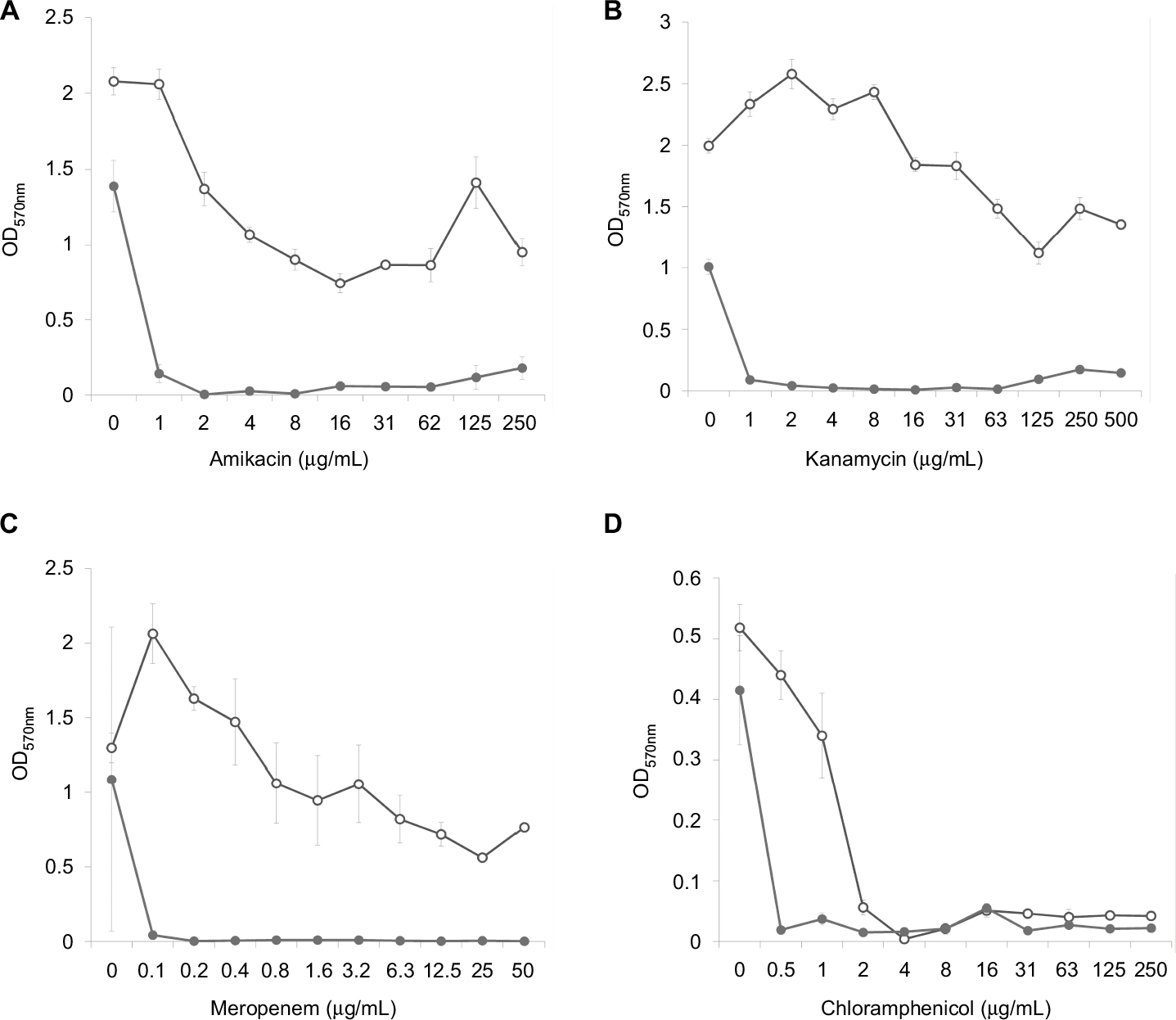 | Figure 3 Doze–effect curves of antibiotics alone and in combination with FLIP7 against Staphylococcus aureus biofilm (crystal violet assay). Notes: (A) Amikacin versus amikacin + FLIP7 500 µg/mL. (B) Kanamycin versus kanamycin + FLIP7 500 µg/mL. (C) Meropenem versus meropenem + FLIP7 250 µg/mL. (D) Chloramphenicol versus chloramphenicol + FLIP7 250 µg/mL. Open circles: antibiotic; closed circles: antibiotic + FLIP7. Abbreviation: OD, optical density. |
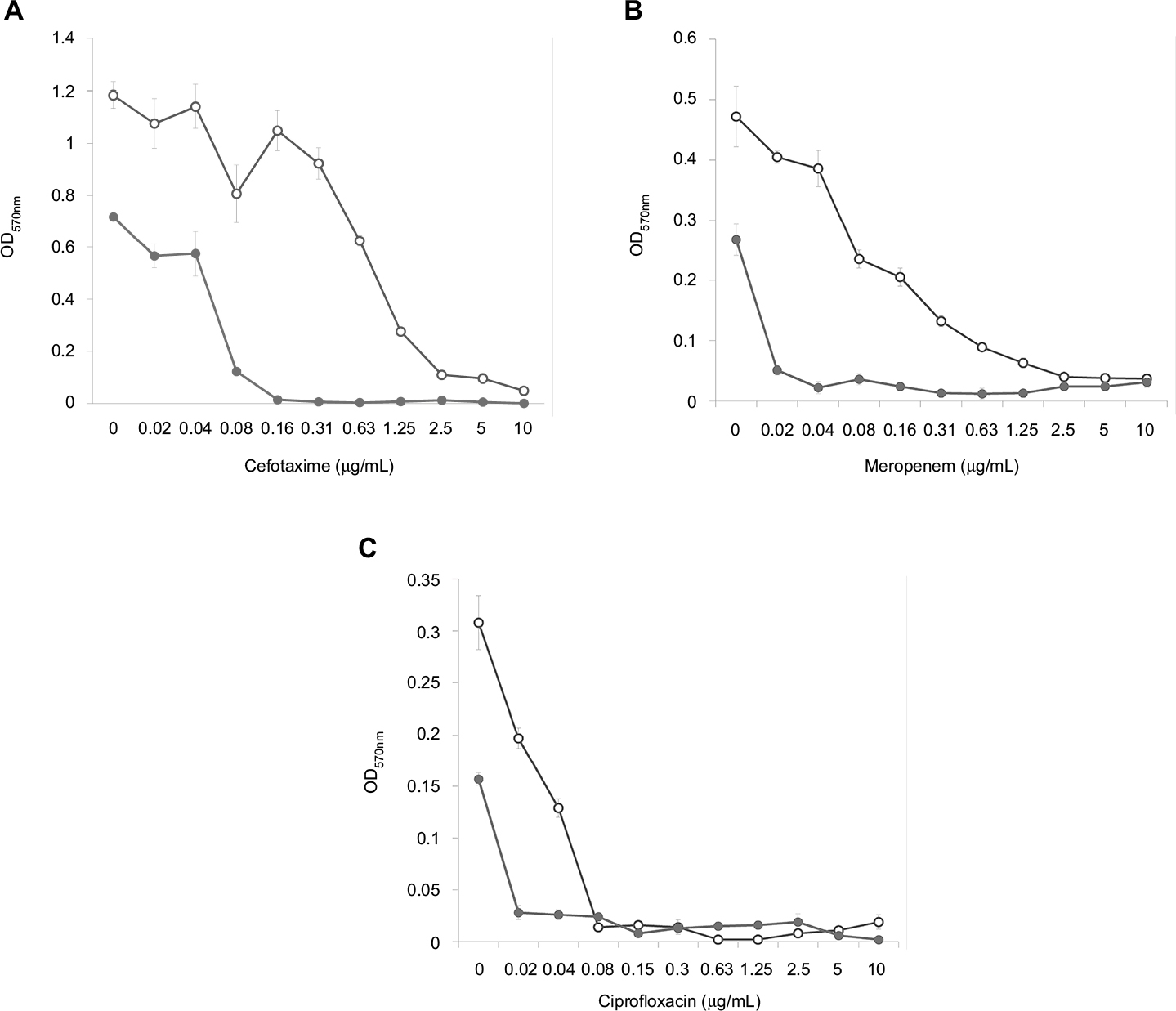 | Figure 4 Doze–effect curves of antibiotics alone and in combination with FLIP7 against Escherichia coli biofilm (crystal violet assay). Notes: (A) Cefotaxime versus cefotaxime + FLIP7 500 µg/mL. (B) Meropenem versus meropenem + FLIP7 500 µg/mL. (C) Ciprofloxacin versus ciprofloxacin + FLIP7 250 µg/mL. Open circles: antibiotic; closed circles: antibiotic + FLIP7. Abbreviation: OD, optical density. |
Biofilm microscopic visualization
In addition to obtaining the quantitative data of the TTC and crystal violet tests, changes in the biofilm state were visualized at the level of light microscopy. An S. aureus biofilm treated with meropenem alone (0.2 µg/mL) or the same concentration of meropenem and FLIP7 (125 μg/mL) was chosen as the model. High-synergy concentrations of the active substances were selected based on the results of the checkerboard experiments discussed above. The data of the TTC and crystal violet tests showed that this combination causes complete suppression of metabolic activity and destruction of the biofilm material, respectively. Based on this, it was expected that processing the biofilm with the combination would cause the killing of living cells and biofilm material destruction, which could be microscopically observed. The results of the microscopic analysis corresponded well to the expectations (Figure 5). In the control, bacteria formed a multilayered biofilm that was firmly attached to a glass surface and consisted of cells of regular round shape with no signs of damage. Treatment with meropenem alone led to a strong thinning of the population represented mainly by damaged cells of irregular shape, although apparently normal cells (presumable persisters) were also present in the biofilm. After treatment with the combination, only a small number of dead or obviously damaged “ghost” cells remained on the glass. No evidence of live intact cells was observed with this treatment option. Thus, microscopic examination provided independent confirmation of the results of spectrophotometric analyses.
 | Figure 5 Staphylococcus aureus biofilm microscopic visualization (Nomarski optics ×1000). Notes: Twenty-four-hour biofilms were formed on the coverslip and processed as follows: (A) control; (B) treated with meropenem 0.2 µg/mL; and (C) treated with meropenem 0.2 µg/mL + FLIP7 125 µg/mL. In control, bacteria formed a multilayered biofilm consisting of live cells of regular shape. Treatment with meropenem killed most of the bacteria but a portion of the cells (presumable persisters) survived (survivors are marked with arrows). After treatment with the combination, no signs of living cells were seen on the glass. |
Discussion
Along with the growing prevalence of antibiotic-resistant strains, BRR represents one of the two main obstacles in the fight against bacterial infections. There are different approaches to the treatment of biofilms in the research stage, but the problem remains unresolved so far.14,15,27
While studying insect immunity, we noted that insects respond to infection with synchronous synthesis of not one but a handful of AMPs. A particularly rich repertoire and a high concentration of AMPs were found in the hemolymph of bacteria-challenged maggots of the blue blowfly C. vicina. The C. vicina AMP complex comprises four AMP families: defensins, cecropins, diptericins and proline-rich peptides.23,24 AMPs similar in structure and functions were found in various combinations in many other insects.28,29 Defensins are peptides with a three-dimensional structure containing α-helix/β-sheet elements coordinated by three disulfide bridges and predominantly active against Gram-positive bacteria by means of bacterial cell wall disruption/permeabilization. Cecropins are linear amphipathic α-helical peptides particularly active against Gram-negative bacteria and known to have pore-forming and cell membrane-permeabilizing activity. Diptericins are members of a glycine-rich AMP family selectively toxic to some Gram-negative bacteria by means of cell wall disruption. Calliphora proline-rich peptides belong to the family of proline-/arginine-rich AMPs. In contrast to defensins, cecropins and diptericins, proline-/arginine-rich AMPs kill bacteria by damaging DNA and/or protein synthesis. Thus, FLIP7 comprises three structurally distinct families of cell wall-disrupting AMPs targeted predominantly to the membranes of Gram-negative (cecropins, diptericins) or Gram-positive (defensins) bacteria and one group affecting intracellular targets (proline-rich peptides). From a mechanistic point of view, the composition looks like a well-organized system of complementary elements the combined action of which protects the host organism from various potentially dangerous bacteria.
In the current work, we studied the interaction of FLIP7 and antibiotics using several in vitro models of mature biofilms and planktonic cultures. As expected, bacteria in the biofilm state showed increased recalcitrance to antibiotics compared to planktonic cells of the same strain. However, the level of BRR differed greatly, depending on the type of antibiotic and the species of microorganism. The S. aureus biofilm became almost completely insensitive to the beta-lactams meropenem and ampicillin, and aminoglycosides amikacin and kanamycin, which effectively killed planktonic cells. An intermediate BRR level was found for vancomycin, erythromycin and antiseptic benzalkonium chloride, while for other antibiotics (oxacillin, tetracycline and chloramphenicol) BRR was weakly expressed. Varying BRR was found for the E coli biofilm as well.
Checkerboard experiments with biofilm models demonstrated the positive interaction of FLIP7 with almost all of the antibiotics in all bacteria studied except polymyxin B in E. coli. In S. aureus, high synergy (FICI <0.25) was observed with five antibiotics of the 11 tested (Table 1). In publications concerning S. aureus biofilms, we were able to find the FICI values of 37 diverse two-component AMP–antibiotic combinations.30–32 FICIs <0.25 were registered only in two cases: cecropin A synthetic analog + ciprofloxacin30 and peptide 1018 + ceftazidime.32 Comparison with the literature data showed that a high synergy with antibiotics is much more common in FLIP7 than in individual AMPs referenced above (45% versus 5%, P = 0.005, Z-test)
The interaction in planktonic cell models was, in most cases, significantly weaker than in biofilms of the same strains and never reached a high synergy level. This observation is in agreement with the fact that a high synergy level was only found in high-BRR antibiotics. If the antibiotic did not encounter a BRR barrier (planktonic cells) or the barrier was weak (low-BRR antibiotics), the interaction strength was greatly reduced. From this point of view, FLIP7 can be considered a synergist helping antibiotics to overcome the BRR barrier.
The analysis of the dose–effect curves points to persister cells as a likely target of FLIP7 synergistic effect. It is known that the biphasic character of the curve indicates the presence in the population of persisters insensitive to the antibiotic.33 In the S. aureus biofilm, dose–effect curves of high-BRR/high-synergy antibiotics (meropenem, ampicillin, amikacin and kanamycin) had a typical biphasic shape (Figure 1). The combination of these antibiotics with FLIP7 resulted in complete elimination of survivors detectable by TTC method, while the antibiotic alone could not cope with this task. The dose–effect curves of antibiotics with a minimal BRR level (oxacillin, chloramphenicol) did not reveal persister cells in the biofilm population. A similar pattern was observed in the E. coli biofilm: persisters were suppressed by the combination of FLIP7 and antibiotic in all cases when such cells were found in the population (Figure 2).
However, the TTC test can only detect metabolically active cells and may overlook the persisters. Microscopic analysis of the S. aureus biofilm treated with a highly synergistic combination of FLIP7 and meropenem confirmed TTC test data (Figure 5). After the treatment, we were unable to detect living cells, although they were visible in the biofilm treated with meropenem alone. Demonstration of this fact confirms the possibility of complete elimination of bacterial cells in the biofilm by a combination of FLIP7 and antibiotic and thus preventing the recurrence of the disease.
The biofilm eradication assay revealed another possible mechanism of the interaction. In this test, the crystal violet binding is directly proportional to the total content of all biofilm components (extracellular matrix, dead and living cells). The binding of the dye was sharply reduced after the treatment of S. aureus and E. coli biofilms with the combination of FLIP7 and antibiotics in comparison with the antibiotic alone (Figures 3 and 4). Similar results were obtained in the study of meropenem action on P. aeruginosa and A. baumannii biofilms, even though the effect was less pronounced in K. pneumoniae. The total destruction of the biofilm evidently means the elimination of the extracellular matrix, which, along with persisters, plays a key role in protecting biofilms from antibiotic assault. Thus, destruction of the matrix is another possible mechanism for the interaction of FLIP7 and antibiotics.
In summary, it can be concluded that FLIP7 is a highly efficient natural antimicrobial system helping antibiotics to overcome strong biofilm barriers through persisters’ sensitization and biofilm material destruction. The system comprises four AMP families with distinct structures and mechanisms of action. Inevitably, the question arises whether it is technically and economically feasible to use such a complex system in medical practice. Even if it is theoretically possible to synthesize all the active components of the complex, the manufacturing cost of such a man-made composition is likely to be unacceptably high. Therefore, we opted for biotechnology for the parallel biosynthesis of natural AMPs in a C. vicina culture. The technology allows scalable cost-effective production of the AMPs, the composition of which is identical to natural FLIP7.34
Another problem comes with choosing an appropriate FLIP7 partner among conventional antibiotics. Judging from the results of the study, the standard checkerboard method is well suited for this purpose. At present, beta-lactams meropenem and ampicillin, aminoglycosides amikacin and kanamycin, glycopeptide vancomycin and cephalosporin cefotaxime look like the most suitable candidates in respect to the synergy level.
Currently, these and many other antibiotics are being used successfully to treat early stages of infections. At later stages accompanied by the emergence of biofilms, their use is much less effective even with the greatest possible increase in dosage. The synergistic effect of FLIP7 allows reducing the effective anti-biofilm concentration of antibiotic to a level well below the one clinically achievable (Table 4). From this point of view, the best partners of FLIP7 in the treatment of S. aureus and/or E. coli biofilms are meropenem, ampicillin, oxacillin, ciprofloxacin and cefotaxime the effective anti-biofilm concentrations of which can be reduced by 2–3 orders of magnitude compared with clinically achievable concentrations. The acceptable width of the pharmacological window is also characteristic of vancomycin, amikacin, gentamicin and tetracycline where the clinically achievable concentration exceeds that required for destruction of the biofilm by >20 times. Biofilms of A. baumannii, K. pneumoniae and P. aeruginosa can also be sensitized to the action of the antibiotic with the help of FLIP7; however, the data available now are limited to the example of meropenem.
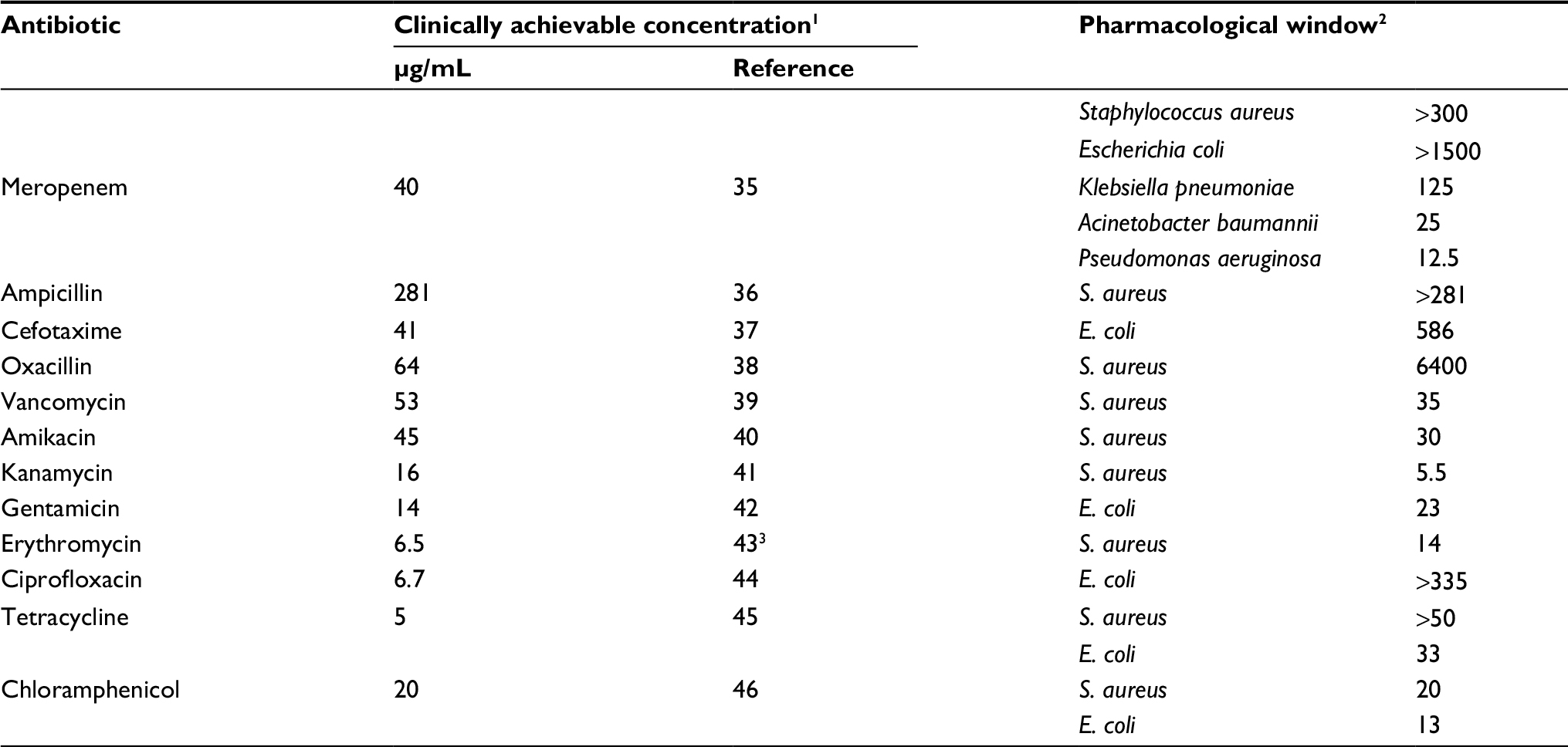 | Table 4 Pharmacological windows of antibiotics used in combination with FLIP7 Notes: 1Plasma Cmax at intravenous administration. 2The ratio of clinically achievable concentration and MBIC90 of the antibiotic in the presence of FLIP7. The values of MBIC90 are taken from Table 1. 3Plasma Cmax at oral administration. Abbreviation: MBIC, minimum biofilm-inhibiting concentration. |
In evaluating the clinical prospects of FLIP7, attention should be paid to the totality of its properties that are well suited to the tasks of treating biofilm infections: efficacy against planktonic and biofilm bacteria of various species; sensitization of persisters to the action of antibiotics; the ability to destroy biofilm material; high synergy with antibiotics of different classes. The ability to prevent acquired resistance development in Gram-negative bacteria23 and low toxicity for human cells24 supplement the list of advantages of FLIP7 as a platform for the future development of new antibacterial drugs.
Acknowledgments
This study was supported by a grant from the Russian Science Foundation (http://rscf.ru/, grant number 16-14-00048). The funder had no role in study design, data collection and analysis and decision to publish or preparation of the manuscript. The authors are grateful to Resource Centers “Development of molecular and cellular technologies”, “Chemical analysis and materials research” and “Culture collections of microorganisms” of St. Petersburg State University Research Park for technical assistance.
Disclosure
SC and NG are inventors of patent RU2447896 “Antimicrobial material” (patentee Allopharm company). SC is a shareholder of Allopharm company. DT and AY have no conflicts of interests to declare in this work.
References
Grants.nih.gov [homepage on the Internet]. Grants and fundings. Bethesda, MD: National Institutes of Health (NIH) [updated September 25, 2015; cited October 15, 2017]. Available from: https://grants.nih.gov/grants/guide/pa-files/PA-03-047.html. Accessed January 3, 2018. | ||
Pinto-Santini D, Salama NR. The biology of Helicobacter pylori infection, a major risk factor for gastric adenocarcinoma. Cancer Epidemiol Biomarkers Prev. 2005;14(8):1853–1858. | ||
Collins D, Hogan AM, Winter DC. Microbial and viral pathogens in colorectal cancer. Lancet Oncol. 2011;12(5):504–512. | ||
Johnson CH, Dejea CM, Edler D, et al. Metabolism links bacterial biofilms and colon carcinogenesis. Cell Metab. 2015;21(6):891–897. | ||
Fong IW. New perspectives of infections in cardiovascular disease. Curr Cardiol Rev. 2009;5(2):87–104. | ||
Lanter BB, Sauer K, Davies DG. Bacteria present in carotid arterial plaques are found as biofilm deposits which may contribute to enhanced risk of plaque rupture. mBio. 2014;5(3):e01206–e01214. | ||
Aslam S. Effect of antibacterials on biofilms. Am J Infect Control. 2008;36(10):S175.e9–S175.e11. | ||
Bryers JD. Medical biofilms. Biotechnol Bioeng. 2008;100(1):1–18. | ||
Leid JG. Bacterial biofilms resist key host defenses. Microbe. 2009;4(2):66–70. | ||
López D, Vlamakis H, Kolter R. Biofilms. Cold Spring Harb Perspect Biol. 2010;2(7):a000398. | ||
Høiby N, Bjarnsholt T, Givskov M, Molin S, Ciofu O. Antibiotic resistance of bacterial biofilms. Int J Antimicrob Agents. 2010;35(4):322–332. | ||
Dufour D, Leung V, Lévesque CM. Bacterial biofilm: structure, function, and antimicrobial resistance. Endod Topics. 2012;22(1):2–16. | ||
Römling U, Balsalobre C. Biofilm infections, their resilience to therapy and innovative treatment strategies. J Intern Med. 2012;272(6):541–561. | ||
Gupta P, Sarkar S, Das B, Bhattacharjee S, Tribedi P. Biofilm, pathogenesis and prevention – a journey to break the wall: a review. Arch Microbiol. 2016;198(1):1–15. | ||
Wu H, Moser C, Wang HZ, Høiby N, Song ZJ. Strategies for combating bacterial biofilm infections. Int J Oral Sci. 2015;7(1):1–7. | ||
Di Luca M, Maccari G, Maisetta G, Batoni G. BaAMPs: the database of biofilm-active antimicrobial peptides. Biofouling. 2015;31(2):193–199. | ||
Strempel N, Strehmel J, Overhage J. Potential application of antimicrobial peptides in the treatment of bacterial biofilm infections. Curr Pharm Des. 2015;21(1):67–84. | ||
Batoni G, Maisetta G, Esin S. Antimicrobial peptides and their interaction with biofilms of medically relevant bacteria. Biochim Biophys Acta. 2016;1858(5):1044–1060. | ||
Pletzer D, Coleman SR, Hancock RE. Anti-biofilm peptides as a new weapon in antimicrobial warfare. Curr Opin Microbiol. 2016;33:35–40. | ||
Rahnamaeian M, Cytryńska M, Zdybicka-Barabas A, et al. Insect antimicrobial peptides show potentiating functional interactions against Gram-negative bacteria. Proc Biol Sci. 2015;282(1806):20150293. | ||
Yu G, Baeder DY, Regoes RR, Rolff J. Combination effects of antimicrobial peptides. Antimicrob Agents Chemother. 2016;60(3):1717–1724. | ||
Dosler S, Karaaslan E, Alev Gerceker A. Antibacterial and anti-biofilm activities of melittin and colistin, alone and in combination with antibiotics against Gram-negative bacteria. J Chemother. 2016;28(2):95–103. | ||
Chernysh S, Gordya N, Suborova T. Insect antimicrobial peptide complexes prevent resistance development in bacteria. PLoS One. 2015;10(7):e0130788. | ||
Gordya N, Yakovlev A, Kruglikova A, et al. Natural antimicrobial peptide complexes in the fighting of antibiotic resistant biofilms: Calliphora vicina medicinal maggots. PLoS One. 2017;12(3):e0173559. | ||
Leber A. Synergism testing: broth microdilution checkerboard and broth macrodilution methods. Clinical Microbiology Procedures Handbook. 4th ed. Washington, DC: ASM Press; 2016:5.16.1–5.16.23. | ||
Lewis K. Persister cells and the paradox of chronic infections. Microbe. 2010;5(10):429–437. | ||
Ciofu O, Rojo-Molinero E, Macià MD, Oliver A. Antibiotic treatment of biofilm infections. APMIS. 2017;125(4):304–319. | ||
Bulet P, Stöcklin R. Insect antimicrobial peptides: structures, properties and gene regulation. Protein Pept Lett. 2005;12(1):3–11. | ||
Mylonakis E, Podsiadlowski L, Muhammed M, Vilcinskas A. Diversity, evolution and medical applications of insect antimicrobial peptides. Philos Trans R Soc Lond B Biol Sci. 2016;371(1695):20150290. | ||
Mataraci E, Dosler S. In vitro activities of antibiotics and antimicrobial cationic peptides alone and in combination against methicillin-resistant Staphylococcus aureus biofilms. Antimicrob Agents Chemother. 2012;56(12):6366–6371. | ||
Hwang IS, Hwang JS, Hwang JH, et al. Synergistic effect and antibiofilm activity between the antimicrobial peptide coprisin and conventional antibiotics against opportunistic bacteria. Curr Microbiol. 2013;66(1):56–60. | ||
Reffuveille F, de la Fuente-Núñez C, Mansour S, Hancock RE. A broad-spectrum antibiofilm peptide enhances antibiotic action against bacterial biofilms. Antimicrob Agents Chemother. 2014;58(9):5363–5371. | ||
Kim KP, Kim YG, Choi CH, et al. In situ monitoring of antibiotic susceptibility of bacterial biofilms in a microfluidic device. Lab Chip. 2010;10(23):3296–3299. | ||
Chernysh SI, Gordja NA. The immune system of maggots of the blow fly (Calliphora vicina) as a source of medicinal drugs. J Evol Biochem Physiol. 2011;47(6):524–533. | ||
Ikawa K, Nakashima A, Morikawa N, et al. Clinical pharmacokinetics of meropenem and biapenem in bile and dosing considerations for biliary tract infections based on site-specific pharmacodynamic target attainment. Antimicrob Agents Chemother. 2011;55(12):5609–5615. | ||
Pacifici GM. Clinical pharmacology of ampicillin in neonates and infants: effects and pharmacokinetics. Int J Pediatr. 2017;5(12):6383–6410. | ||
Patel KB, Nicolau DP, Nightingale CH, Quintiliani R. Pharmacokinetics of cefotaxime in healthy volunteers and patients. Diagn Microbial Infect Dis. 1995;22(1–2):49–55. | ||
Nesseler N, Verdier MC, Launey Y, et al. High-dose continuous oxacillin infusion results in achievement of pharmacokinetics targets in critically ill patients with deep sternal wound infections following cardiac surgery. Antimicrob Agents Chemother. 2014;58(9):5448–5455. | ||
James JK, Palmer SM, Levine DP, Rybak MJ. Comparison of conventional dosing versus continuous-infusion vancomycin therapy for patients with suspected or documented gram-positive infections. Antimicrob Agents Chemother. 1996;40(3):696–700. | ||
Burdet C, Pajot O, Couffignal C, et al. Population pharmacokinetics of single-dose amikacin in critically ill patients with suspected ventilator-associated pneumonia. Eur J Clin Pharmacol. 2015;71(1):75–83. | ||
Aung TY, Thwin MH, Zin T, Nwe Oo K, Aye NN, Sin S. Comparative pharmacokinetics of kanamycin between multi-drug resistant tuberculosis patients and healthy volunteers. Int J Med Pharm. 2014;2(2):45–59. | ||
Medellín-Garibay SE, Rueda-Naharro A, Peña-Cabia S, García B, Romano-Moreno S, Barcia E. Population pharmacokinetics of gentamicin and dosing optimization for infants. Antimicrob Agents Chemother. 2015;59(1):59482–59489. | ||
Josefsson K, Bergan T, Magni L. Dose-related pharmacokinetics after oral administration of a new formulation of erythromycin base. Br J Clin Pharmacol. 1982;13(5):685–691. | ||
Lipman J, Scribante J, Gous AG, Hon H, Tshukutsoane S. Pharmacokinetic profiles of high-dose intravenous ciprofloxacin in severe sepsis. Antimicrob Agents Chemother. 1998;42(9):2235–2239. | ||
Agwuh KN, MacGowan A. Pharmacokinetics and pharmacodynamics of the tetracyclines including glycylcyclines. J Antimicrob Chemother. 2006;58(2):256–265. | ||
Ambrose PJ. Clinical pharmacokinetics of chloramphenicol and chloramphenicol succinate. Clin Pharmacokinet. 1984;9(3):222–238. | ||
National Committee for Clinical Laboratory Standards. Performance standards for antimicrobial disk susceptibility tests. Approved standard M2-A8. Wayne, PA: NCCLS; 2003. | ||
Clinical Laboratory Standards Institute. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. Approved standard, 10th edition, M07-A10. Wayne, PA: CLSI; 2015. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.