")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Bioequivalence of paclitaxel protein-bound particles in patients with breast cancer: determining total and unbound paclitaxel in plasma by rapid equilibrium dialysis and liquid chromatography–tandem mass spectrometry
Authors Li J , Li W, Dai X, Zhong D, Ding Y, Chen X
Received 7 January 2019
Accepted for publication 23 March 2019
Published 20 May 2019 Volume 2019:13 Pages 1739—1749
DOI https://doi.org/10.2147/DDDT.S200679
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Cristiana Tanase
Junling Li,1 Wei Li,2 Xiaojian Dai,2 Dafang Zhong,2 Yaping Ding,1 Xiaoyan Chen2
1College of Sciences, Shanghai University, Shanghai, People’s Republic of China; 2Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, People’s Republic of China
Background and objective: Paclitaxel protein-bound particles for injectable suspension (nab-paclitaxel) showed many advantages in safety, effectiveness, and convenience. Different from conventional formulations, the bioequivalence evaluation of nab-paclitaxel formulations requires to determine the total amount of paclitaxel in plasma and the unbound paclitaxel to reflect their in vivo disposition. This study aimed to develop an analytical method to quantify the total and unbound paclitaxel in plasma and evaluate the bioequivalence of two formulations of nab-paclitaxel in patients with breast cancer.
Materials and methods: An open-label, randomized, two-period crossover study was completed among 24 Chinese patients with breast cancer. The patients were randomized to receive either the test formulation on cycle 1 day 1 and after 21 days in cycle 2 day 1 by the reference formulation (Abraxane®), or vice versa. Rapid equilibrium dialysis was adopted to separate the unbound paclitaxel in human plasma. Total and unbound paclitaxel concentrations were measured by the validated liquid chromatography–tandem mass spectrometry methods over the range of 5.00–15,000 and 0.200–200 ng/mL, respectively. The bioequivalence of the test formulation to the reference formulation was assessed using the Food and Drug Administration and European Medicines Agency guidelines.
Results: All the 90% confidence intervals (CIs) of the geometric mean ratios fell within the predetermined acceptance range. The 90% CIs for the area under the concentration–time curve (AUC) from 0 h to 72 h (AUC0–t), AUC from time zero to infinity (AUC0–∞), and peak plasma concentrations (Cmax) for total paclitaxel were 92.03%–98.05%, 91.98%–99.37%, and 91.37%–99.36%, respectively. The 90% CIs of AUC0–t, AUC0–∞, and Cmax for unbound paclitaxel were 86.77%–97.88%, 86.81%–97.88%, and 87.70%–98.86%, respectively.
Conclusion: Bioequivalence between the two nab-paclitaxel formulations was confirmed for total and unbound paclitaxel at the studied dose regimen.
Keywords: nab-paclitaxel, bioequivalence, rapid equilibrium dialysis, unbound fraction, pharmacokinetics
Introduction
Paclitaxel is a diterpenoid product extracted from the bark of the Western yew tree, Taxus brevifolia or semi-synthesized.1 The unique mechanism of paclitaxel of stabilizing tubulin polymer and promoting microtubule assembly effectively inhibits mitosis, motility, and intracellular transport within cancerous cells and results in antineoplastic activity against a wide variety of malignancies.2–4 Paclitaxel is administered intravenously instead of via oral administration because of low bioavailability (<6%) caused by P-glycoproteins-related efflux and first-pass metabolism.5
Paclitaxel is highly lipophilic (hydrophobic). Development of injection formulations suitable for clinical use has always been a focus and difficulty in the research. The first clinically available formulation used polyethoxylated castor oil (CrEL) as the cosolvent. However, CrEL had biological and pharmacological properties that caused severe anaphylactoid hypersensitivity reactions and peripheral neuropathy.6 To reduce CrEL-induced toxicities, formulations, such as albumin-bound nanoparticles (nab-paclitaxel), liposomes,7 micelles, polymeric nanoparticles,8 taxane analogs, and nanomedicine without drug carrier,9 were developed subsequently. Among these formulations, nab-paclitaxel is the only one that gained the approval of the Food and Drug Administration (FDA).10 All other formulations did not contain albumin that brought the potential for viral transmission and microbial growth, so they were more of solubilizing agents than stable drug carriers. Thus, the other formulations are most likely to result in similar (rather than superior) efficacy compared with nab-paclitaxel. Apart from the reduced potential risk of allergic reactions related to the solvent, nab-paclitaxel did not require pretreatment to prevent allergic reactions before medication and significantly reduced the infusion time to 30 min.11–13
Unlike traditional formulations, nab-paclitaxel primarily delivered the active ingredient to the tumor cells directly and bypassed the unbound form. The drug delivery mechanism of nab-paclitaxel involved glycoprotein 60-mediated endothelial cell transcytosis of paclitaxel-bound albumin and accumulation in the area of tumor by albumin binding to secreted protein, acidic and rich in cysteine.14–16 Therefore, the unbound paclitaxel in plasma was significantly less important in efficacy but is still being important for off-target exposure–toxicity relationships. Given the ultrafast distribution and decomposition of paclitaxel–carrier complexes, unbound paclitaxel in plasma significantly affected the extravascular tissue distribution of paclitaxel.17 Clearly, the plasma concentration of unbound paclitaxel was essential to predict the changes in pharmacokinetics or pharmacodynamics for nab-paclitaxel.18 These factors determined that the bioequivalence evaluation of nab-paclitaxel differed from conventional formulations. Thus, assessing the plasma exposure of total paclitaxel and also the unbound paclitaxel levels is necessary.
Numerous methods including equilibrium dialysis (ED), ultrafiltration, ultracentrifugation, and in vivo microdialysis are available for separating unbound drug in plasma samples.19–22 Each technique has its own advantages and shortcomings, and ED is the most commonly and generally considered as the “gold standard”.23 Rapid equilibrium dialysis (RED) device was designed based on ED, with a Teflon base plate and disposable dialysis cells. Compared with the standard ED method, the surface area to volume ratio of each dialysis cell was increased, providing the possibility of reducing equilibration time and increasing throughput.23
This study aimed to separate unbound paclitaxel in human plasma by RED and determine the total and unbound paclitaxel by liquid chromatography–tandem mass spectrometry (LC–MS/MS). Using the developed and validated methods, the bioequivalence study of nab-paclitaxel formulations was carried out in compliance with international rules and regulations adopted by the FDA and European Medicines Agency guidelines.24–26 The area under the concentration–time curve (AUC) from 0 h to 72 h (AUC0–t) and AUC from time zero to infinity (AUC0–∞) and peak plasma concentrations (Cmax) for total and unbound paclitaxel were defined as the main parameters to assess possible bioequivalence.
Materials and methods
The study was approved by the Ethics Committee of the First Hospital of Jilin University (Changchun, China) and conducted in compliance with Good Clinical Practice guidelines of the International Conference on Harmonization and the Declaration of Helsinki (2008). Written informed consent was obtained from all patients before participation.
Study drugs
Paclitaxel protein-bound particles for injectable suspension (equivalent to 100 mg paclitaxel; batch number: A1I1611004AL) manufactured by Qilu Pharmaceutical (Hainan) Co., Ltd. was selected as the test formulation, and Abraxane® (100 mg, batch number: 6,110,247) manufactured by Fresenius Kabi, USA, LLC. was selected as the reference formulation.
Subjects
Twenty-five patients with breast cancer were enrolled according to the inclusion/exclusion criteria.27 These patients were diagnosed by imaging, cytological or histological examination, or who were considered eligible for the injection of nab-paclitaxel antitumor therapy.
Study design
The study aimed to evaluate the bioequivalence of nab-paclitaxel formulations in Chinese patients with breast cancer. Subjects who passed the screening were completely randomized according the number table obtained by the Statistical Analysis System (SAS) 9.1 software. The test or reference formulation was administered in period 1, and the alternate formulation was administered in period 2. The volume of the prepared paclitaxel suspension (5 mg/mL) was calculated at 260 mg/m2 dose based on the body surface area (BSA) of the subject. The subjects were given a fasting period of at least 10 h prior to dosing. Approximately 30 min after eating a standard breakfast in the morning, the subjects were given a uniform infusion via an intravenous infusion pump, and the infusion was completed within 30±3 min.
Lunch and dinner were provided after 4 and 10 h of administration, respectively. The designated food was ingested at the designated time during hospitalization, and no other diet was taken. Caffeinated and alcoholic beverages and xanthine-containing food or beverages (including chocolate, tea, and coffee) were prohibited from the screening date to the end of the trial. During the administration period and within 2 h after administration, the subjects should remain in a lying position and must avoid prolonged bed rest or strenuous exercise.
Safety evaluation
Safety evaluation indicators included adverse events (AEs), serious adverse events, combined medication, changes in clinical laboratory tests (biochemistry, hematology, and urinalysis), clinical symptoms, vital signs, physical examinations, and 12-lead electrocardiograms. All patients who participated in the study were included in the safety analysis. The vital signs of the subjects were monitored at 1 h before administration and at 10 min, 30 min, 1 h, 2 h, 12 h, 24 h, 48 h, and 72 h after administration. The monitoring times can be increased according to the subjects’ conditions. AEs were recorded throughout the study period and at the follow-up visit. The subjects underwent follow-up visits on the 7±1 and 21±1 days of each trail period in the fasting state. Physical examinations, vital signs, laboratory examination, and electrocardiogram were conducted at the scheduled follow-up visit.
Sample collection
Blood samples were collected by inserting a catheter into the forearm vein prior to administration. Blood collection and drug administration should not be in the same arm. In each study period, approximately 5 mL blood was collected at 0, 0.25, 0.5, 0.75, 1, 1.5, 2, 4, 6, 8, 12, 24, 36, 48, and 72 h past dose to determine the plasma concentrations for total and unbound paclitaxel. The blood samples were centrifuged at 3000 rpm for 10 min. Plasma was separated and frozen at approximately −80 °C for further analysis.
Chromatography/mass spectrometry conditions
The LC–MS/MS system consisted of an LC-20A (Shimadzu Corporation, Japan) coupled to a Triple Quad 5500 mass spectrometer (AB Sciex, USA) equipped with electrospray ionization source. To measure the plasma samples and plasma chamber samples in the RED plate, chromatographic analysis was performed on Agilent Zorbax Eclipse Plus C18 (3.5 μm, 100×4.6 mm) column coupled with a Phenomenex Security Guard C18 (4.0×3.0 mm) column at a column temperature of 40 °C. The mobile phase consisted of acetonitrile: 0.2% formic water (70: 30, v/v) for a total run time of 3.0 min at a 0.70 mL/min flow rate. The mass spectrometer was operated in the positive ion mode, with electrospray ionization and multiple reaction monitoring by using the transitions of m/z 854.5→286.1 for the paclitaxel and m/z 859.5→291.1 for the d5-paclitaxel (internal standard). The ion spray voltage was set at 5500 V, and the source temperature was 300 °C. The gases 1 and 2, curtain gas, and collision gas were set at 50, 50, 30 and 8 psi respectively. Declustering potential and collision energy were, respectively, 50 and 24 V for paclitaxel and d5-paclitaxel with a dwell time of 80 ms per transition.
To determine the buffer chamber samples, chromatographic separation was employed using a Waters BEH C18 (1.7 µm 50×2.1 mm) analytical column at 40 °C. The mobile phases were (A) 0.2% formic acid in water and (B) acetonitrile, which delivered at a 0.55 mL/min flow rate. The gradient elution was 15% B at 0–0.2 min, 15–70% B at 0.2–0.5 min, 70–90% B at 0.5–1.0 min, 90% B at 1.0–1.6 min, 90–15% B at 1.6–1.8 min, and 1.8–2.3 min kept constant at 15% B.
The above two methods were characterized by adequate sensitivity, specificity, linearity, accuracy and precision, recovery, and stability. The validation data, as presented in Tables 1 and 2, were taken from our validation reports. The calibration curves for the determination of plasma samples showed good linearity over the range of 5.00–15,000 ng/mL. In method validation, a 10-fold dilution of plasma samples containing paclitaxel above the upper limit of quantification was considered acceptable. The calibration curves for the determination of buffer chamber samples showed good linearity over the range of 0.200–200 ng/mL with a low limit quantification (LLOQ) of 0.200 ng/mL.
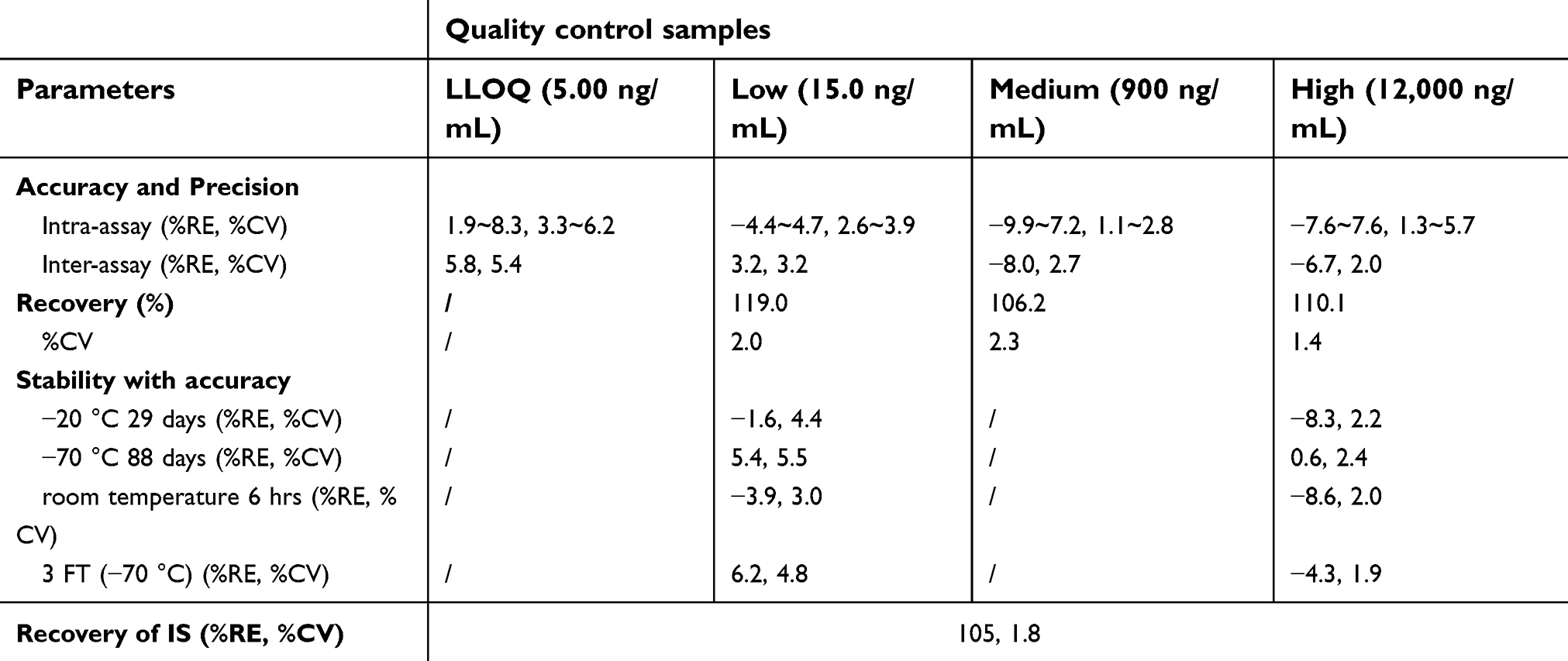 | Table 1 Validation data of the analytical method used to determine total paclitaxel in human plasma |
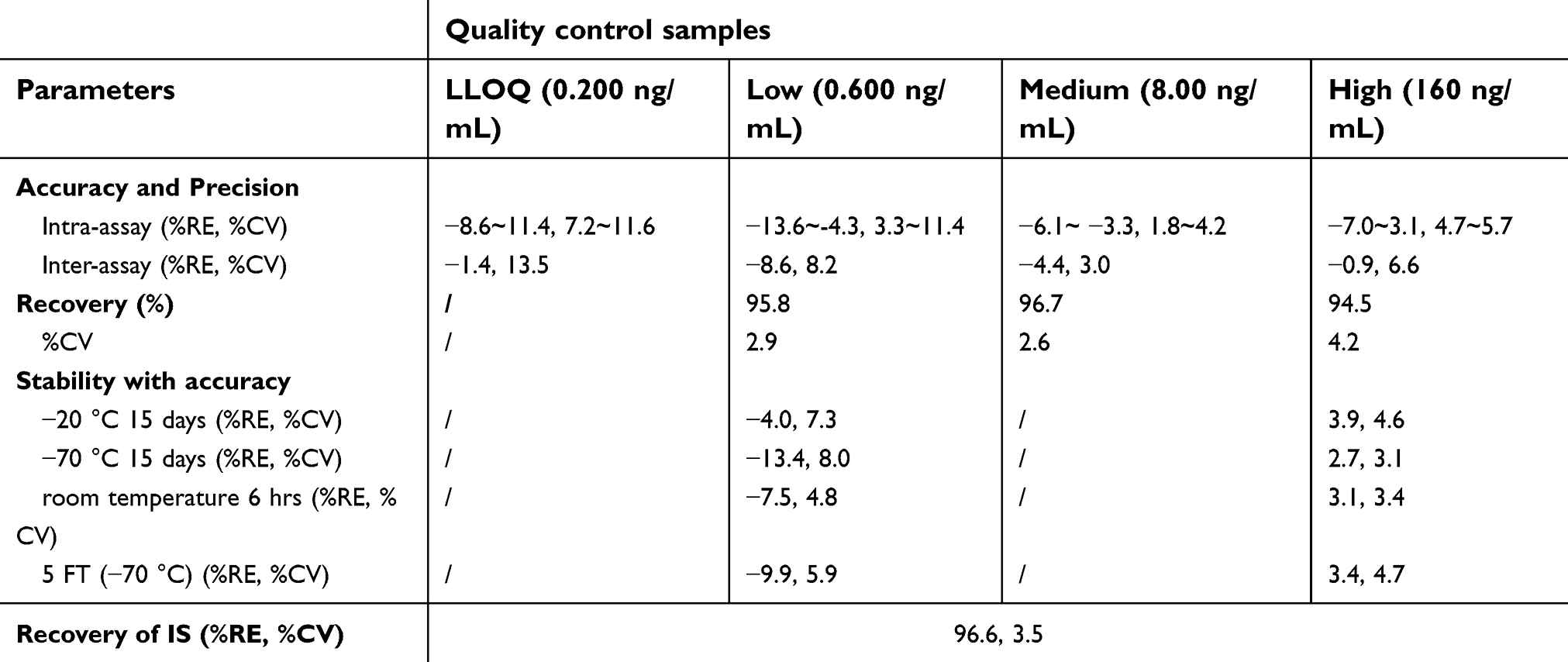 | Table 2 Validation data of the analytical method used to determine unbound paclitaxel in human plasma |
Sample pretreatment
To determine the total plasma concentration of paclitaxel, 50 μL plasma sample was pretreated in 96-deep well by adding 200 μL of acetonitrile and 25 μL internal standard solution (d5-paclitaxel, 10.0 μg/mL). The mixture was vortexed for 15 min and subsequently centrifuged at 3700 rpm for 15 min. A 100 μL aliquot of the supernatant was added to 50 μL of the mobile phase (acetonitrile/0.2% formic water=70:30, v/v) and was vortex-mixed. Aliquot (2 μL) of the resulting solution was injected into the LC–MS/MS system for analysis.
To separate unbound paclitaxel, the plasma samples were processed by RED by using inserts (Thermo-Fisher Scientific, Waltham, MA, USA) placed in a Teflon-coated base plate. Plasma samples (200 μL) were loaded into the plasma chambers, whereas the buffer chambers were filled with 350 μL phosphate-buffered saline (PBS, pH 7.4). The plate was sealed and incubated on an orbital shaker (250 rpm) at 37 °C for 4 h in a humidified atmosphere of 5% CO2 incubator. All volume of post dialysis sample was removed from the plasma chambers and the buffer chambers in separate microcentrifuge tubes. Equal volumes of acetonitrile were added to the buffer tubes and vortexed to obtain the mixed buffer chamber samples.
The mixed buffer chamber sample was pretreated by taking 100 μL of the sample in test tube, adding 50 μL d5-paclitaxel solution (20.0 ng/mL) and 100 μL acetonitrile. Then, the mixture was vortexed and centrifuged. A 15 μL aliquot of the supernatant was collected for LC–MS/MS system. Plasma chamber samples were pretreated in the same way as plasma samples.
Pharmacokinetic analyses
Unbound fraction (fu) in plasma was calculated from the ratio of the buffer chamber concentration (Creceiver) to the plasma chamber concentration (Cdonor). The unbound paclitaxel concentration in plasma (Cunbound) was calculated according to the following formula:
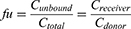
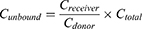
According to the plasma concentration of unbound paclitaxel and total paclitaxel (Ctotal), the pharmacokinetic parameters of the two formulations, including AUC0–t, AUC0–∞, Cmax, time to reach Cmax (Tmax), terminal elimination half-life (t1/2), and their mean value and standard deviation (SD), were evaluated by the noncompartmental model by using WinNonlin Software (Phoenix WinNonlin® version 6.4, Pharsight Corporation, NC, USA). Cmax and Tmax were obtained directly from the measured values. AUC0–t was calculated by trapezoid method, AUC0–∞=AUC0–t+Ctn/λz. Ctn is the plasma drug concentration at the last point measured. λz is the terminal elimination rate constant determined by least-squares regression analysis during the terminal loglinear phase of the concentration time curve. The t1/2 time (hours) was calculated as 0.693/λz.
SAS version 9.1 was used for data statistics. AUC0–t, AUC0–∞, and Cmax were tested for significance by multivariate analysis of variance (ANOVA) (significance level was 5%) with formulation effect, period effect, sequence effect, and subject effect as factors. The geometric mean ratio of the major pharmacokinetic parameters (AUC0–t, AUC0–∞, and C max) of the test formulation versus the reference formulation and their 90% confidence interval (CI) was calculated. Tmax was statistically evaluated using a nonparametric method.
Results
Background characteristics of the volunteers
In this clinical study, the subjects were screened strictly according to the inclusion/exclusion criteria of the test protocol. Forty-eight subjects were screened, and 25 subjects were actually enrolled. The age range of 25 subjects was 37–63 years old, and the average of BSA (SD) was 1.63 (0.146) kg/m2. Except for one subject who voluntarily withdrew from the study after the end of cycle 1 follow-up, the remaining 24 cases completed all the two cycles, of which 1 was male and 23 were females.
Development of the RED procedure
In this study, five factors, namely, the stability in the incubation, equilibrium time, recovery, freeze-thaw stability, and protein leakage, affecting the determination of unbound paclitaxel in human plasma were evaluated.
To ensure fu was not influenced by drug lability in PBS and plasma, paclitaxel at the concentrations of 15.0 and 12,000 ng/mL in plasma or 0.600 and 160 ng/mL in PBS was incubated at 37 °C for 4 and 6 h by using six replicates. After the incubation of plasma samples, the maximum concentration deviation from T0 was 10.2% (<15%) for 4 h, whereas it was 18.1% (>15%) for 6 h. The result indicated that plasma samples were stable within 4 h at 37 °C incubation conditions. For PBS samples in the CO2 incubator (37 °C) placed after 4 and 6 h, the concentration deviation from T0 is 8.8% or less.
Plasma samples spiked with nab-paclitaxel concentrations at 500 and 5000 ng/mL were incubated after 2, 4 and 8 h to determine the optimal equilibration time. The results showed that after 2, 4 and 8 h, fu at the low concentration (500 ng/mL) were 1.4%, 3.6% and 3.9%, respectively. fu at the high concentration (5000 ng/mL) were 1.2%, 3.6% and 4.1%, respectively. Considering the stability, the most suitable RED time was set at 4 h.
The recovery of the proposed method was also investigated. The recovery rate was calculated using formula 3. Paclitaxel concentrations of plasma samples were set at 50.0, 500 and 5000 ng/mL. Some of these concentrations were immediately treated as C0 samples. A 200 μL aliquot of plasma samples was added to the plasma chamber and 350 μL of PBS to the buffer chamber. After 4 h incubation, samples from plasma chamber and buffer chamber were measured by LC–MS/MS method. The recovery rate and recovery precision were 87.0% and 0.80%, respectively, indicating that the recovery of 200 μL plasma/350 μL PBS was high and reproducible during the RED.
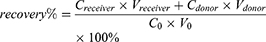
After five freeze-thaw cycles of nab-paclitaxel in plasma, the relative deviation between the average measured fu and the fresh plasma fu were 0.7% for 500 ng/mL and −1.0% for 5000 ng/mL, respectively. This result indicated that five freeze-thaw cycles did not affect the plasma fu of nab-paclitaxel.
The impact of protein leakage through the dialysis membrane was another possible source of error. A 200 μL aliquot of blank plasma samples was dialysed against PBS. Then, a bicinchoninic acid protein quantification kit (Meilun Biotechnology Co., Ltd., Dalian, China) was used to determine the protein content in the plasma chambers and PBS chamber samples. The measurement of protein concentration was performed at 562 nm. Protein leakage was only 0.113%, thereby the impact of protein leakage remains negligible.
Pharmacokinetics of paclitaxel
The mean plasma concentration–time curves of total and unbound paclitaxel after intravenous injection of 260 mg/m2 dose of the test or reference formulation of nab-paclitaxel are shown in Figure 1. The main pharmacokinetic parameters of total paclitaxel and unbound paclitaxel in plasma, AUC0–t, AUC0–∞, Cmax, Tmax, and t1/2 are shown in Table 3.
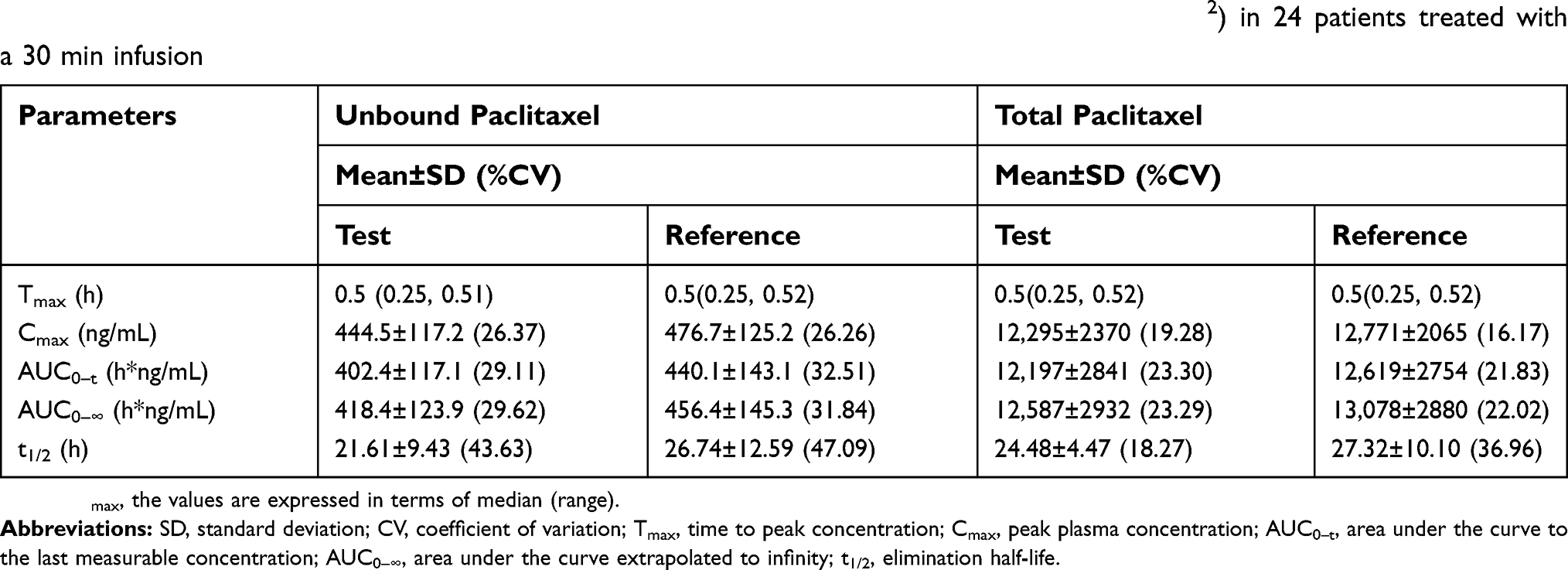 | Table 3 Pharmacokinetic parameters of total and unbound paclitaxel for nab-paclitaxel (260 mg/m2) in 24 patients treated with a 30 min infusion |
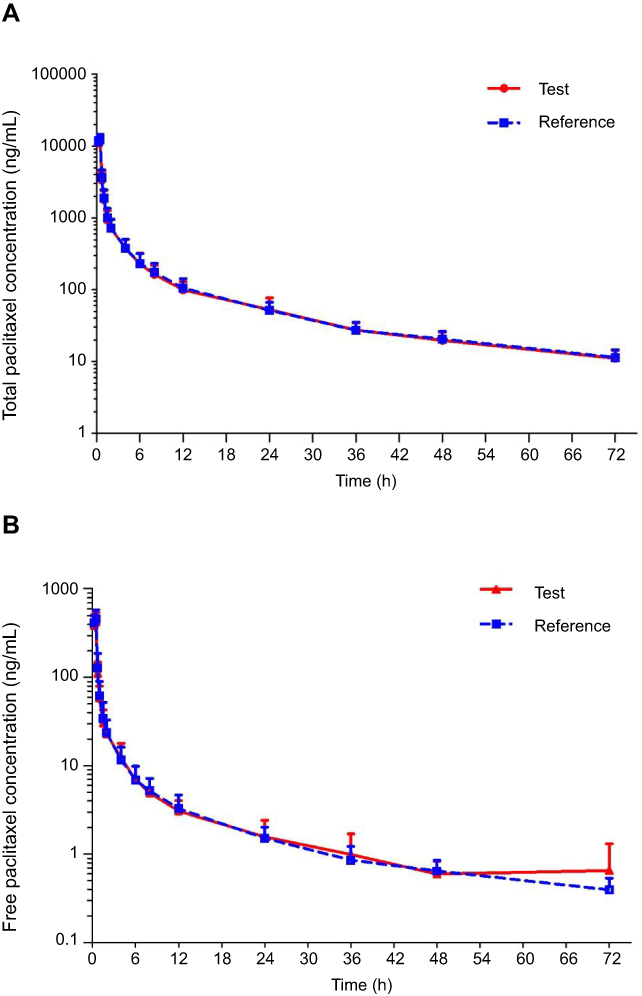 | Figure 1 Plasma concentration–time curves of total paclitaxel (A) and unbound paclitaxel (B) after 30 min infusion of the test formulation or the reference formulation (Abraxane®) at a dose of 260 mg/m2. Data are presented as Log10 mean ± SD. |
Bioequivalence assessments
Based on bioequivalence guidelines, the criteria for total and unbound paclitaxel bioequivalence is the 90% CI for the ratio of test/reference geometric means in the range of 80% to 125%.23–25 The geometric mean ratios (90% CI) for total paclitaxel were 95.60% (91.98%–99.37%) for AUC0–72 h, 95.28% (91.37%–99.36%) for AUC0–∞, and 94.99% (92.03%–98.055%) for Cmax. The geometric mean ratios (90% CI) for unbound paclitaxel were 92.16% (86.77%–97.88%) for AUC0–72 h, 92.18% (86.81%–97.88%) for AUC0–∞, and 93.12% (87.70%–98.86%) for Cmax. The differences between the test and reference formulation in the time to achieve Cmax values and elimination half-life values were not statistically significant. The 90% CIs of the test/reference AUC ratio and Cmax ratio of paclitaxel were within the acceptance range for bioequivalence (Tables 4 and 5).
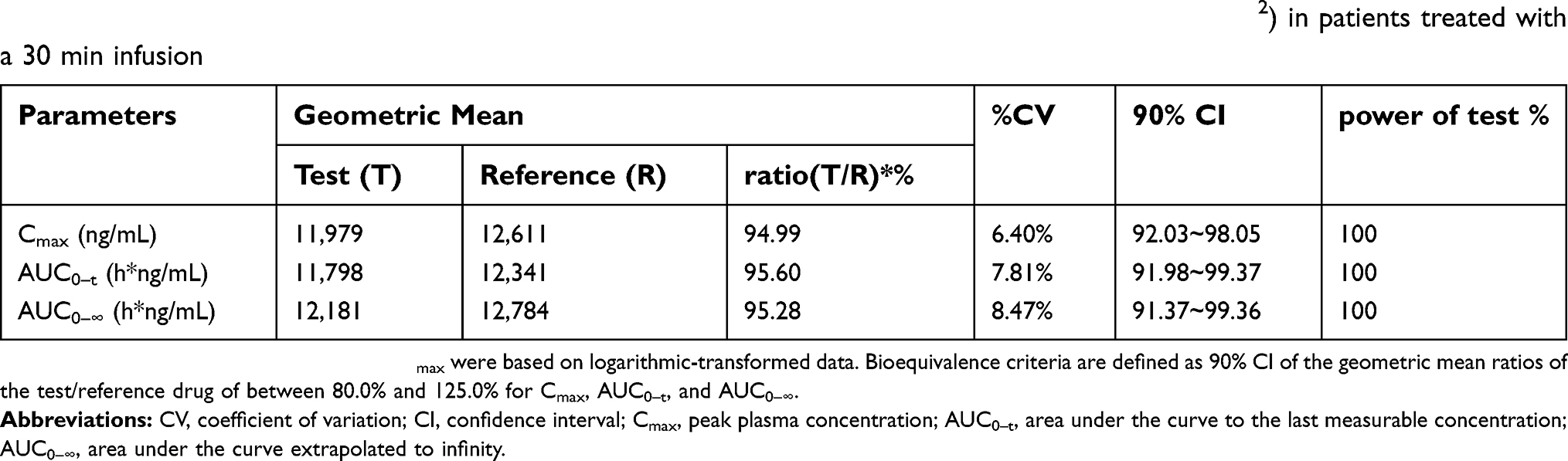 | Table 4 Geometric mean ratio and statistical comparison of total paclitaxel for nab-paclitaxel (260 mg/m2) in patients treated with a 30 min infusion |
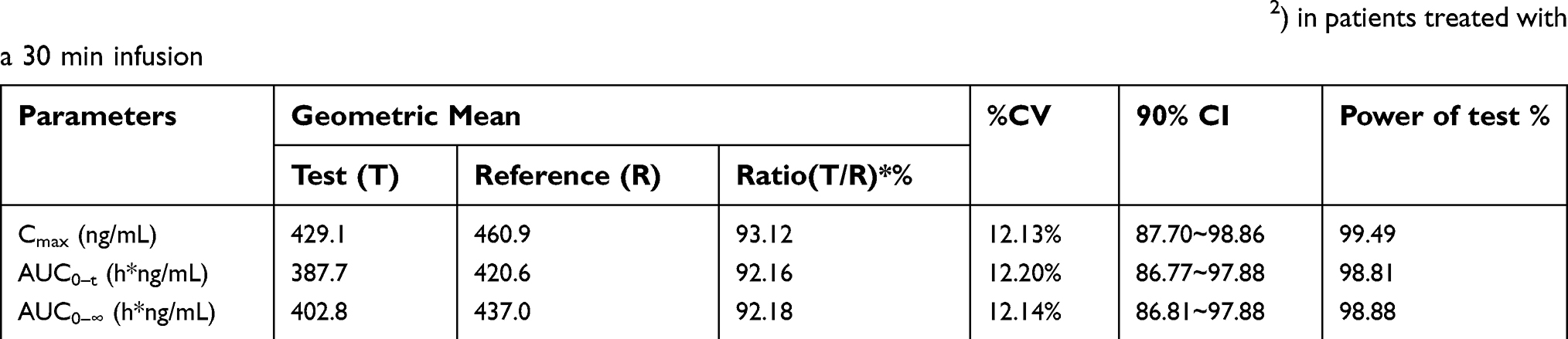 | Table 5 Geometric mean ratio and statistical comparison of unbound paclitaxel for nab-paclitaxel (260 mg/m2) in patients treated with a 30 min infusion |
Safety
This study included 25 participants in the safety evaluation, including one participant who voluntarily withdrew from the test after the first period of treating with the test drug. Therefore, 25 cases were used in the test formulation group and 24 cases in the reference formulation group. The 551AEs were reported by 100% (25/25) of the volunteers. Level III or high incidences of AEs were 34 cases (14/25). One serious adverse event occurred, which was definitely not related to the study drug. No dose adjustment was performed due to adverse events, and no adverse events led to withdrawal from the study. No significant difference was observed in the incidence of AEs between the two administration sequences. The difference in the incidence of AEs between the test formulation and the reference formulation was also not statistically significant.
Discussion
It has been reported that paclitaxel had a high binding affinity for human plasma proteins.28,29 As a result, a highly sensitive analytical method was required to determine unbound paclitaxel in plasma. Several LC-MS/MS methods have been reported to measure paclitaxel,30–37 where adduct ions [M+Na]+ or [M+Li]+ were used as precursor ion. To further enhance the sensitivity, the mobile phase was optimized including organic phase (methanol, acetonitrile) and additives (ammonium acetate, formic acid, lithium carbonate) in various concentration. By employing 0.2% formic acid and acetonitrile as the mobile phase with the precursor ion [M+H]+, the method had a LLOQ of 0.200 ng/mL for unbound paclitaxel. It was proved to be superior in sensitivity compared with the reported methods.36,37
This study referred to the Draft Guidance on Paclitaxel issued by the FDA.24 The results showed that the geometric mean ratios (T/R) and 90% CI of Cmax, AUC0–t, and AUC0–∞ for total paclitaxel and unbound paclitaxel of the two formulations were all within 80.00%–125.00% after the recommended dose of 260 mg/m2 was administered within 30 min. These results were in line with the bioequivalence evaluation criteria.
The washout period was 21 days, equivalent to 24 elimination half-lives. In addition, the paclitaxel concentrations in plasma samples collected before the second cycle of drug administration were all below the LLOQ of 5.00 ng/mL. This result can exclude the AUC period effect caused by insufficient washout period.
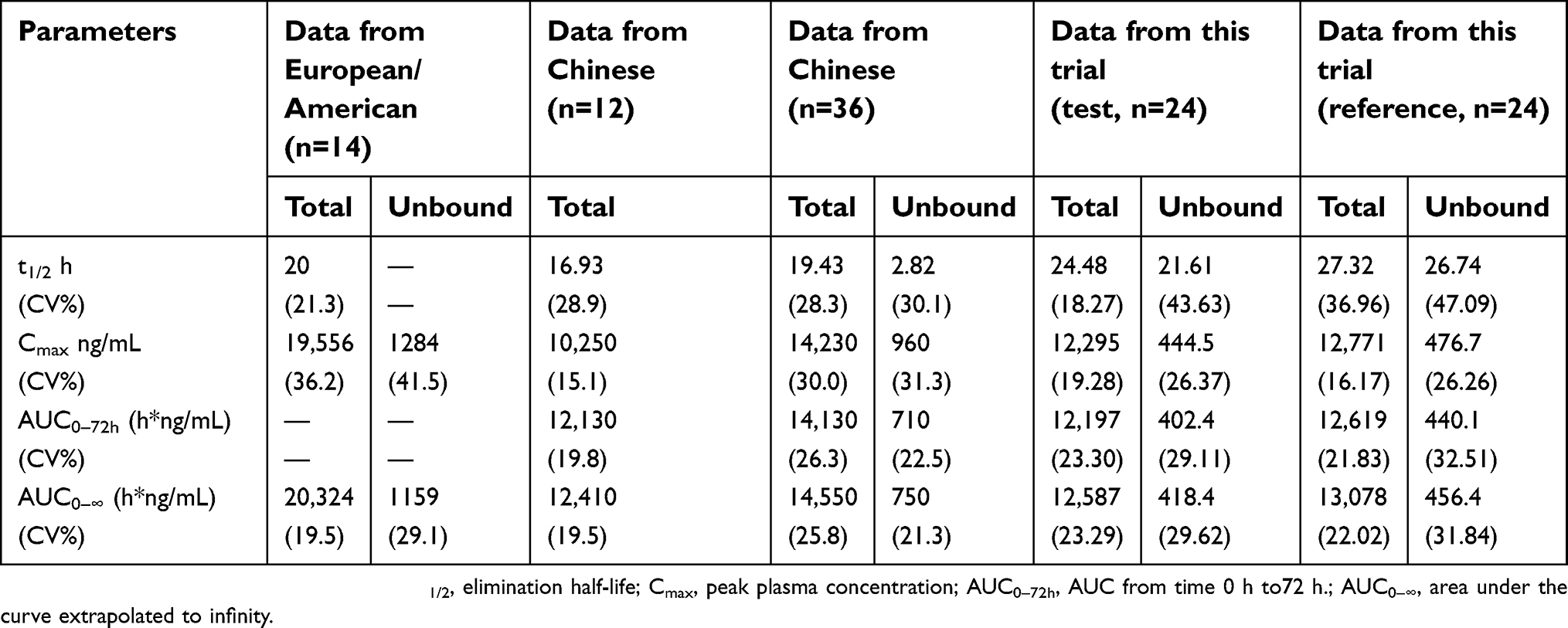
The main pharmacokinetic parameters of total paclitaxel and unbound paclitaxel, as shown in Table 6, were compared with those from the literature.19,38,39 It is worth noting that the reported t1/2 of unbound paclitaxel was 2.82 h,39 while the value in the present study was 21.61 h or 26.74 h. The t1/2 is the time that it takes for the plasma concentration to fall by half from its maximum levels. It is calculated by log-linear regression of plasma concentrations observed during the terminal phase of elimination. Therefore, the plasma concentrations of the elimination phase affect the calculated t1/2. In the published study, the plasma concentration of unbound paclitaxel was determined about to 7.5 h after dosing.39 In the present study, comprehensive method development and optimization led to a superior sensitivity and allowed the determination of unbound paclitaxel up to 72 h after an administration. Thus, the difference of the plasma concentrations in the elimination phase caused the difference of t1/2 observed in the two studies. Cmax and AUC values of total paclitaxel obtained in the present study were comparable with those from Chinese-reported literature38,39 but was lower than 38% to the reported data from Europeans and Americans.19 The clinical recommended dose of nab-paclitaxel is 260 mg/m2 based on the BSA of the subject. BSA of the Europeans and Americans is usually calculated using the Mosteller’s formula,40
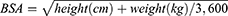
 | Table 6 Summary of total paclitaxel and unbound paclitaxel pharmacokinetic parameters of nab-paclitaxel |
BSA of Chinese adults is normally calculated by the Stevenson’s formula as follows:
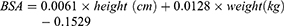
For a person with a height of 170 cm and a weight of 70 kg, the calculated BSA from formulas 4 and 5 were 1.82 and 1.78 m2, respectively. Therefore, the differences in Cmax and AUC values between the Chinese and Europeans/Americans were caused by different absolute doses of nab-paclitaxel (calculated based on the BSA formula). Sampling error and the effect of racial and genetic differences may also be contributing factors.
Few pharmacokinetic parameters of unbound paclitaxel for nab-paclitaxel were reported in the literature. The data from the Americans and Europeans showed that fu of nab-paclitaxel is 6.3±2.1%,19 but fu obtained in this study was approximately 3.3±0.7%. The reported literature and this study used different methods to separate unbound drugs, that is, one method used ultrafiltration, and another method employed RED. Although their working principles were based on membrane separation, the powers to drive unbound drugs through the membrane were different. In ultrafiltration, a pressure gradient rapidly forces the water-soluble component of plasma containing unbound drug through a semipermeable membrane.41 RED forms a diffusion pressure through the drug concentration gradient across the semipermeable membrane so that the unbound drug reaches the equilibrium. Compared with the ultrafiltration method, the RED method does not require an external force and can simulate in vivo environment and is a commonly used method for measuring unbound drugs. Ultrafiltration is affected greatly by test conditions, such as centrifugation time, centrifugation temperature, and centrifugation speed.41 The effect of different conditions of ultrafiltration on fu of paclitaxel was investigated. As Table 7 shows, we found that even in the same conditions, the results of the ultrafiltration varied widely. In addition, the molecular weight cut-off (MWCO) of the membranes was different between the two methods. MWCO of the ultrafiltration membrane was 30 kDa, whereas RED device with the dialysis membrane was only 8 kDa. As a result, the main reason why fu obtained in this study is different from that in the literature may be the different separation methods.
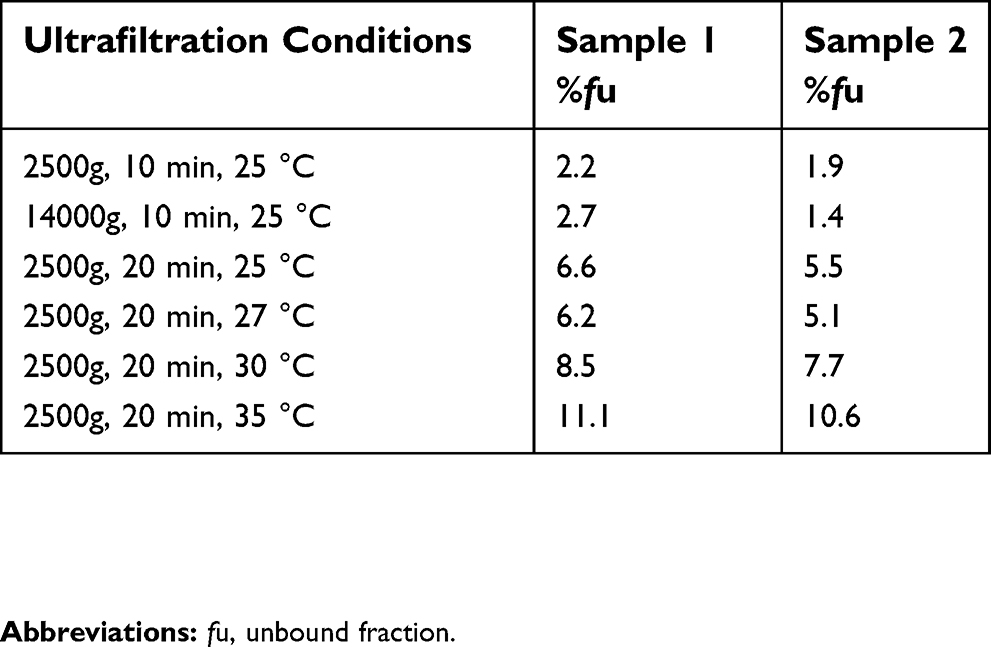 | Table 7 %fu values of nab-paclitaxel determined by ultrafiltration method under different conditions |
Conclusion
Overall, this study developed and validated a robust and reliable RED method to separate unbound paclitaxel, followed by LC–MS/MS method to quantify total and unbound paclitaxel in plasma. This method was successfully applied to bioequivalence studies of nab-paclitaxel formulations. Notably, nab-paclitaxel test demonstrated bioequivalence with the reference formulation Abraxane®.
Acknowledgments
We are grateful to Qilu Pharmaceutical (Hainan) Co., Ltd. for providing the test and reference formulation used in this study. We would also like to thank The First Hospital of Jilin University (Changchun, China) for performing the clinical trials.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Vidensek N, Lim P, Campbell A, Carlson C. Taxol content in bark, wood, root, leaf, twig, and seedling from several taxus species. J Nat Prod. 1990;53(6):1609−1610. doi:10.1021/np50072a039
2. Adams JD, Flora KP, Goldspiel BR, et al. Taxol: a history of pharmaceutical development and current pharmaceutical concerns. J Natl Cancer Inst Monogr. 1993;(15):141–147.
3. Riondel J, Jacrot M, Picot F, Beriel H, Mouriquand C, Potier P. Therapeutic response to taxol of six human tumors xenografted into nude mice. Cancer Chemother Pharmacol. 1986;17(2):137–142. doi:10.1007/BF00306742
4. Rowinsky EK, Cazenave LA, Donehower RC. Taxol: a novel investigational antimicrotubule agent. J Natl Cancer Inst. 1990;82(15):1247–1259.
5. Sparreboom A, Van AJ, Mayer U, et al. Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc Natl Acad Sci U S A. 1997;94(5):2031–2035. doi:10.1073/pnas.94.5.2031
6. Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37(13):1590–1598. doi:10.1016/S0959-8049(01)00171-X
7. Kong L, Askes SHC, Bonnet S, Kros A, Campbell F. Temporal control of membrane fusion through photolabile PEGylation of liposome membranes. Angew Chem Int Ed. 2016;55(4):1396–1400. doi:10.1002/anie.201509673
8. Sharma N, Madan P, Lin S. Effect of process and formulation variables on the preparation of parenteral paclitaxel-loaded biodegradable polymeric nanoparticles: a co-surfactant study. Asian J Pharm Sci. 2016;11(3):404–416. doi:10.1016/j.ajps.2015.09.004
9. Han X, Chen J, Jiang M, et al. Paclitaxel–paclitaxel prodrug nanoassembly as a versatile nanoplatform for combinational cancer therapy. ACS Appl Mater Interfaces. 2016;8(49):33506–33513. doi:10.1021/acsami.6b13057
10. Sofias AM, Dunne M, Storm G, Allen C. The battle of “nano” paclitaxel. Adv Drug Delivery Rev. 2017;122:20–30. doi:10.1016/j.addr.2017.02.003
11. Gradishar WJ, Tjulandin S, Davidson N, et al. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil–based paclitaxel in women with breast cancer. J Clin Oncol. 2005;23(31):7794–7803. doi:10.1200/JCO.2005.04.937
12. Cheng HR, Guan H, Chen J. Efficacy and safety of abraxane and paclitaxel liposome in treatment of recurrent ovarian cancer. J Pract Obstet Gynecol. 2015;31(3):229–232.
13. Gradishar WJ, Krasnojon D, Cheporov S, et al. Significantly longer progression-free survival with nab-paclitaxel compared with docetaxel as first-line therapy for metastatic breast cancer. J Clin Oncol. 2009;27(22):3611–3619. doi:10.1200/JCO.2008.18.5397
14. Desai N, Trieu V, Damascelli B, et al. SPARC expression correlates with tumor response to albumin-bound paclitaxel in head and neck cancer patients. Transl Oncol. 2009;2(2):59–64.
15. Gradishar WJ. Albumin-bound paclitaxel: a next-generation taxane. Expert Opin Pharmacother. 2006;7(8):1041–1053. doi:10.1517/14656566.7.8.1041
16. Desai N. Nab technology: a drug delivery platform utilizing endothelial gp60 receptor-based transport and tumor-derived SPARC for targeting. Drug Deliv Rep. 2007;37–41.
17. Desai N. Nanoparticle Albumin-Bound Anticancer Agents. In: Crommelin D., de Vlieger J. (eds) Non-Biological Complex Drugs. Non-Biological Complex Drugs. AAPS Advances in the Pharmaceutical Sciences Series, Springer, Cham; 2015;20:335–354
18. Stern ST, Martinez MN, Stevens DM. When is it important to measure unbound drug in evaluating nanomedicine pharmacokinetics? Drug Metabo Dispos. 2016;44(12):1934–1939. doi:10.1124/dmd.116.073148
19. Gardner ER, Dahut WL, Scripture CD, et al. Randomized crossover pharmacokinetic study of solvent-based paclitaxel and nab-paclitaxel. Clin Cancer Res. 2008;14(13):4200–4205. doi:10.1158/1078-0432.CCR-07-4592
20. Pacifici GM, Viani A. Methods of determining plasma and tissue binding of drugs. Pharmacokinetic consequences. Clin Pharmacokinet. 1992;23(6):449–468. doi:10.2165/00003088-199223060-00005
21. Guo J, Zhang Y, Huo Y, Chang Y-X, Liu E-W, Hao J. Monitoring unbound warfarin in drug combination therapy by pharmacokinetics and fluorospectrometry. Chin Herbal Med. 2019;11(1):92–97. doi:10.1016/j.chmed.2018.10.002
22. Tsai TH. Chapter 6.5 Assaying protein-unbound drugs using microdialysis techniques. Tech Behav Neural Sci. 2006;16:573–587.
23. Waters NJ, Jones R, Williams G, Sohal B. Validation of a rapid equilibrium dialysis approach for the measurement of plasma protein binding. J Pharm Sci. 2010;97(10):4586–4595. doi:10.1002/jps.21317
24.
25.
26.
27. Li QM, Zhang H, Zhu XX, et al. Tolerance, variability and pharmacokinetics of albumin-bound paclitaxel in chinese breast cancer patients. Front Pharmacol. 2018;9:1372. doi:10.3389/fphar.2018.01372
28. Kumar GN, Walle UK, Bhalla KN, et al. Binding of taxol to human plasma, albumin and alpha 1-acid glycoprotein. Res Commun Chem Pathol Pharmacol. 1993;80(3):337–344.
29. Brouwer E, Verweij J, De Bruijn P, et al. Measurement of fraction unbound paclitaxel in human plasma[J]. Drug Metab Dispos. 2000;28(10):1141–1145.
30. Sottani C, Minoia C, D’Incalci M, Paganini M, Zucchetti M. High‐performance liquid chromatography tandem mass spectrometry procedure with automated solid phase extraction sample preparation for the quantitative determination of paclitaxel (Taxol®) in human plasma. Rapid Commun Mass Spectrom. 1998;12(5):251–255. doi:10.1002/(ISSN)1097-0231
31. Parise RA, Ramanathan RK, Zamboni WC, Egorin MJ. Sensitive liquid chromatography-mass spectrometry assay for quantitation of docetaxel and paclitaxel in human plasma. J Chromatogr B. 2003;783(1):231–236. doi:10.1016/S1570-0232(02)00659-1
32. Mortier KA, Renard V, Verstraete AG, Van Gussem A, Van Belle S, Lambert WE. Development and validation of a liquid chromatography−tandem mass spectrometry assay for the quantification of docetaxel and paclitaxel in human plasma and oral fluid. Anal Chem. 2005;77(14):4677–4683. doi:10.1021/ac0500941
33. Zhang SQ, Chen GH. Determination of paclitaxel in human plasma by UPLC–MS–MS. J Chromatogr Sci. 2008;46(3):220–224. doi:10.1093/chromsci/46.3.220
34. Fan YX, Chen XY, Ma ZY, et al. Determination of paclitaxel and hydroxylated metabolites in rat plasma with lithium adduct ion by LC–MS/MS. J Chin Mass Spectr Soc. 2013;34(3):137–144.
35. Wu HL, Ma ZY, Zhang SQ, et al. Effects of different formulations on plasma free concentration of paclitaxel in rats. Chin J Pharm. 2016;47(11):1430–1435.
36. Gardner ER, Dahut W, Figg WD. Quantitative determination of total and unbound paclitaxel in human plasma following Abraxane treatment. J Chromatogr B. 2008;862:213–218. doi:10.1016/j.jchromb.2007.12.013
37. Bulitta JB, Zhao P, Arnold RD, et al. Mechanistic population pharmacokinetics of total and unbound paclitaxel for a new nanodroplet formulation versus Taxol in cancer patients. Cancer Chemother Pharmacol. 2009;63(6):1049–1063. doi:10.1007/s00280-008-0827-2
38. Li S, Liao H, Zhan J, et al. Comparison the pharmacokinetics of nanoparticle albumin bound paclitaxel and solvent based paclitaxel in breast cancer patients. Chin J Clin Pharmacol. 2010;26(8):606–610.
39. Bian L, Geng CZ, Ouyang QC, et al. Study of bioequiavailability of paclitaxel for injection (Albumin Bound) and abraxane and the efficacy of extension treatments in patients with metastatic breast cancer. Natl Med J China. 2018;98(16):1236–1241.
40. Mosteller RD. Simplified calculation of body surface area. N Engl J Med. 1987;317:1098. doi:10.1056/NEJM198707163170301
41. Mortier KA, Lambert WE. Determination of unbound docetaxel and paclitaxel in plasma by ultrafiltration and liquid chromatography-tandem mass spectrometry. J Chromatog A. 2006;1108(2):195–201. doi:10.1016/j.chroma.2005.12.103
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.