")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Bioequivalence of HTX-019 (aprepitant IV) and fosaprepitant in healthy subjects: a Phase I, open-label, randomized, two-way crossover evaluation
Authors Ottoboni T, Keller MR, Cravets M , Clendeninn N , Quart B
Received 2 November 2017
Accepted for publication 5 January 2018
Published 1 March 2018 Volume 2018:12 Pages 429—435
DOI https://doi.org/10.2147/DDDT.S155875
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tuo Deng
Video abstract presented by Thomas Ottoboni.
Views: 1069
Tom Ottoboni,1 Mary Rose Keller,2 Matt Cravets,3 Neil Clendeninn,4 Barry Quart5
1Pharmaceutical and Translational Sciences, Heron Therapeutics, Inc., San Diego, CA, USA; 2Clinical Operations, Heron Therapeutics, Inc., San Diego, CA, USA; 3Biometrics, Heron Therapeutics, Inc., San Diego, CA, USA; 4Clinical, Heron Therapeutics, Inc., San Diego, CA, USA; 5Heron Therapeutics, Inc., San Diego, CA, USA
Introduction: Fosaprepitant, an intravenous (IV) aprepitant prodrug for chemotherapy-induced nausea and vomiting prophylaxis, is associated with systemic and infusion-site reactions attributed in part to its surfactant, polysorbate 80. HTX-019 is an IV aprepitant formulation free of polysorbate 80 and other synthetic surfactants.
Materials and methods: This open-label, single-dose, randomized, two-way crossover bioequivalence study compared pharmacokinetics and safety of HTX-019 and fosaprepitant. Healthy subjects received single-dose HTX-019 (130 mg) or fosaprepitant (150 mg) IV over 30 min, with ≥7-day washout between doses. Blood samples were evaluated for pharmacokinetics and bioequivalence; safety evaluation included treatment-emergent adverse events (TEAEs) and serious adverse events. Ninety-seven of one hundred enrolled subjects completed the study.
Results: Baseline characteristics were comparable between treatment sequences. For HTX-019, mean (percent coefficient of variation) area under the curve (AUC) from time 0 to time of last measurable plasma concentration (AUC0-t), AUC from time 0 to infinity (AUC0-inf), and plasma concentration at 12 h (C12 h) for HTX-019 were 43,729 h*ng/mL (32.7), 45,460 h*ng/mL (36.8), and 988.4 ng/mL (27.5), respectively; corresponding fosaprepitant values were 44,130 h*ng/mL (32.0), 46,163 h*ng/mL (36.6), and 1,022 ng/mL (28.5). Also, 90% CIs (94.186–101.354) were within bioequivalence bounds (80%–125%). Within 1 h following infusion start, one (1%) HTX-019 recipient reported one TEAE, while 20 (20%) fosaprepitant recipients reported 32 TEAEs. Dyspnea occurred in three fosaprepitant recipients (at <1 min in two subjects and at 18 min in one subject, considered study drug related) and one HTX-019 recipient (at 120 h, associated with a respiratory tract infection and considered not related to the study drug). No severe TEAEs, serious adverse events, or deaths occurred; all TEAEs resolved.
Conclusion: HTX-019 was bioequivalent to fosaprepitant and may provide a safer alternative to fosaprepitant for chemotherapy-induced nausea and vomiting prophylaxis.
Keywords: antiemetics, polysorbate 80, safety, surfactant
Introduction
Chemotherapy-induced nausea and vomiting (CINV) is one of the most dreaded effects of chemotherapy.1 Poorly controlled CINV can have negative consequences, necessitating chemotherapy dose delays or dose reductions, requiring additional medical care or hospitalization, and affecting patient quality of life.2–4 The severity of chemotherapy emetogenicity is a major factor determining the type of antiemetic therapy that is prescribed, and is categorized by the rate of emesis in the absence of prophylaxis, with moderately emetogenic chemotherapy (MEC) and highly emetogenic chemotherapy (HEC) causing emesis in 30%–90% and >90% of patients, respectively.5,6 For CINV prophylaxis with HEC and some MEC regimens, antiemetic guidelines recommend a three-drug regimen of a 5-hydroxytryptamine type 3 receptor antagonist (RA), dexamethasone, and a neurokinin 1 (NK-1) RA,6–9 with the addition of olanzapine to this regimen being the recommended regimen by the American Society of Clinical Oncology and being an option in the National Comprehensive Cancer Network guidelines for HEC regimens.6,7,9 Despite available antiemetics, suboptimal CINV control following MEC or HEC remains a significant challenge.4
Aprepitant is a selective high-affinity NK-1 RA with little or no affinity for 5-hydroxytryptamine type 3, dopamine, or corticosteroid receptors, which are targets of existing CINV therapies. Because of its low water solubility, aprepitant is available only in an oral formulation.10 In 2008, an intravenous (IV) formulation was approved in the USA for CINV prevention in combination with other antiemetics using the more water-soluble prodrug fosaprepitant.11 Aprepitant and its IV prodrug, fosaprepitant, are the most commonly used NK-1 RAs. In patients receiving HEC, NK-1 RAs may significantly reduce nausea and vomiting in both acute phase (0–24 h after chemotherapy) and delayed phase (24–120 h after chemotherapy).6–9,12,13 Fosaprepitant includes the surfactant polysorbate 80 to fully solubilize the drug; and although polysorbate 80 is used in several drug formulations (eg, docetaxel [Taxotere; Sanofi-Aventis U.S. LLC, Bridewater, NJ, USA]), it has been associated with systemic hypersensitivity reactions and infusion-site adverse events (ISAEs), such as pain, erythema, swelling, induration, and thrombophlebitis, and may impact the bioavailability of certain drugs.13–22 Several publications have demonstrated that the use of IV fosaprepitant may come with significant risk of ISAEs, with patients at particularly high risk being those receiving anthracyclines and those receiving chemotherapy through a peripheral line.16,17,20 Consequently, in 2017, the labeling of fosaprepitant was changed to include a warning regarding serious systemic hypersensitivity reactions such as anaphylaxis and anaphylactic shock, either during or soon after infusion of the drug.11 An IV formulation of aprepitant that is free of polysorbate 80 and other synthetic surfactants may provide patients with a safer and effective NK-1 RA option with a lower risk of systemic hypersensitivity reactions and ISAEs.
HTX-019 is a polysorbate 80–free IV aprepitant formulation in development for the prevention of CINV. This open-label, single-dose, randomized, two-way crossover bioequivalence study evaluates the pharmacokinetics (PK) and safety of HTX-019 and fosaprepitant.
Materials and methods
Study design and objectives
This study was conducted in accordance with the International Conference on Harmonisation Guideline for Good Clinical Practice and the Declaration of Helsinki. The study was approved by the institutional review board for the single study site, Spaulding Clinical Research, LLC, West Bend, WI, USA. Written informed consent was obtained from each subject.
This was a Phase I, single-center, randomized, open-label, single-dose, two-period, two-sequence crossover bioavailability study (Figure 1). The primary objective was to determine bioequivalence, based on area under the curve (AUC) from time 0 to time of last measurable plasma concentration (AUC0-t), AUC from time 0 extrapolated to infinity (AUC0-inf), and plasma concentration at 12 h (C12 h), of IV infusions of single doses of aprepitant emulsion (HTX-019) 130 mg compared with commercially available fosaprepitant (Emend IV; Merck Sharp & Dohme Corp., Whitehouse Station, NJ, USA) 150 mg in healthy adult subjects. Secondary objectives were to assess safety and tolerability of HTX-019 130 mg and fosaprepitant infusions. A 130 mg HTX-019 dose was selected, as it was determined to be equivalent to the 150 mg fosaprepitant dose in terms of aprepitant delivery. Healthy men and women aged 18–55 years with body weight ≥50 kg and body mass index 18–35 kg/m2 were enrolled. Subjects who were pregnant, were breastfeeding, or had any condition possibly affecting drug absorption were excluded. Subjects were excluded if they had a history or presence of a clinically significant abnormal 12-lead electrocardiogram (ECG) or an ECG with QT interval corrected (QTc) by Bazett’s formula >470 ms (females) or >450 ms (males), or a PR interval >240 ms, QRS complex >110 ms, or history of QT prolongation at screening.
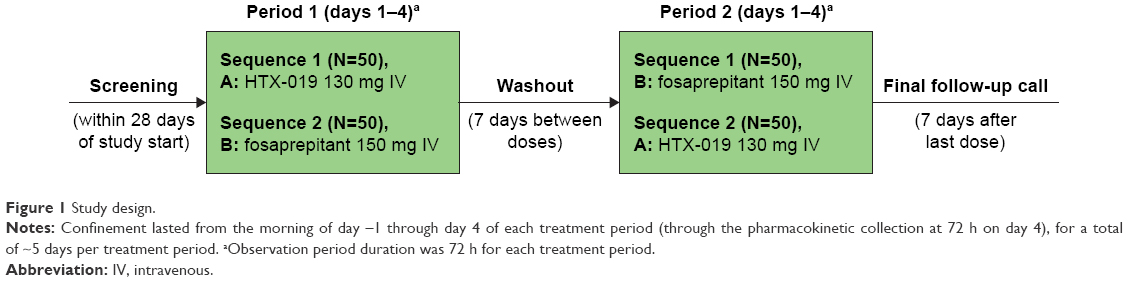 | Figure 1 Study design. |
Treatment regimens and administration
Subjects received HTX-019 130 mg or fosaprepitant 150 mg, administered IV over 30 min on day 1 of periods 1 (AB) and 2 (BA) in a crossover fashion (Figure 1). Each subject was randomly assigned to one of the two treatment sequences (AB [HTX-019 then fosaprepitant] or BA [fosaprepitant then HTX-019]) for crossover dosing. Subjects were confined to the clinic from the morning of day 1 through 72 h after dosing, for each treatment period, with a washout period of at least 7 days between doses. A 7-day washout period was considered ample time to prevent the possibility of carryover effect of medications between treatment periods. In humans, the apparent terminal elimination half-life (t1/2) of aprepitant ranges approximately from 9 to 13 h. Therefore, the 7-day washout period was more than five half-lives of aprepitant.
Assessments
Blood samples for measurement of aprepitant PK analysis were collected before dosing and at 0.5, 0.75, 1, 1.5, 2, 4, 6, 8, 10, 11, 12, 18, 24, 36, 48, 60, and 72 h after infusion start in each treatment period. Additional samples for fosaprepitant were collected before dosing and at 0.5, 0.75, 1, and 1.5 h after infusion start. Plasma concentrations were determined using validated liquid chromatography/mass spectrometry. The following PK parameters were assessed: AUC0-t, AUC0-inf, maximum plasma concentration (Cmax), C12 h, last measurable plasma concentration (Clast), time of Cmax (Tmax), t1/2, and apparent terminal elimination rate (λz).
Safety assessments included treatment-emergent adverse events (TEAEs; overall, by severity, and by relation to study treatment). Physical examination, 12-lead ECG, and clinical laboratory assessments were performed and vital signs taken.
Statistical analysis
A sample size of 90 evaluable subjects provided 90% statistical power to show that the test-to-reference ratio CI for the log-transformed PK parameters was within the bioequivalence criteria if the ratio was within 13% of the reference and the coefficient of variation (CV) was 23%. Bioequivalence evaluation used a mixed-effects model based on aprepitant AUC0-t, AUC0-inf, and C12 h for the PK population. Cmax was not included as a bioequivalence criterion because of the expected lower Cmax due to the ongoing conversion of fosaprepitant to aprepitant at the end of infusion. Ratios and 90% CIs of geometric least squares means for AUCs and C12 h were determined. Bioequivalence was declared if the 90% CI was within 80%–125%.
Continuous variables (eg, n, mean, geometric mean, median, SD, standard error of the mean, percent CV [CV%]) and categorical variables (frequency and percentage) were assessed using descriptive statistics. Mean and individual plasma concentration–time profiles were plotted on linear and semi-logarithmic scales, and PK parameters were calculated using noncompartmental methods for all subjects and treatments with sufficient concentration–time data.
Safety analyses included TEAEs, ISAEs, serious AEs, ECGs, clinical laboratory testing, vital signs, and 12-lead ECG. Related TEAEs included all those considered by the investigator to be possibly, probably, or definitively related to the study drug. Events for which the investigator did not record relationship to the study drug were considered related to the study drug. TEAEs occurring within 30 min and 1 h after infusion start were also recorded. A post hoc analysis of TEAE rate per subject-day was performed using a negative binomial regression model.
Results
One hundred healthy subjects were enrolled and randomly assigned to treatment sequence (Table 1). The safety population included all 100 subjects who were enrolled and received at least one dose of the study drug. The 97 (97%) subjects who completed the study comprised the PK population. Three subjects discontinued the study: two discontinuations were due to AEs (dyspnea, related to fosaprepitant) and one was due to protocol deviation (positive drug screen result for opiates). For the overall study population, patients had a mean age of 35 years and 64% were male. Demographics and baseline characteristics were similar between treatment sequences.
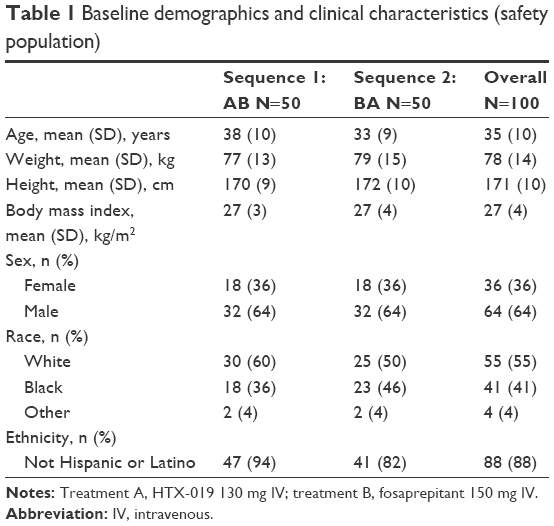 | Table 1 Baseline demographics and clinical characteristics (safety population) |
The primary endpoint of the study was mean AUC0-t, AUC0-inf, and C12 h values for aprepitant after IV administration of HTX-019 and fosaprepitant, which were evaluated for bioequivalence (Table 2). For HTX-019 and fosaprepitant, the mean (CV%) values for AUC0-t were 43,729 (32.7%) and 44,130 (32.0%) h*ng/mL, and the mean (CV%) values for AUC0-inf were 45,460 (36.8%) and 46,163 (36.6%) h*ng/mL, respectively. The mean C12 h (CV%) values after IV administration of HTX-019 and fosaprepitant were similar (988.4 [27.5%] and 1,022 [28.5%] ng/mL, respectively).
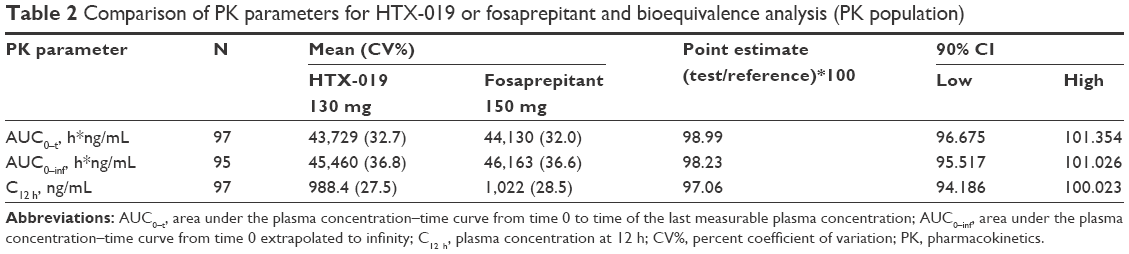 | Table 2 Comparison of PK parameters for HTX-019 or fosaprepitant and bioequivalence analysis (PK population) |
The 90% CIs for AUC0-t, AUC0-inf, and C12 h were all within bioequivalence bounds (80%–125%), consistent with comparable aprepitant exposure (Table 2). The ratio of (AUC0-inf − AUC0-t/AUC0-t) ×100 was 5.8% for HTX-019 and 6.2% for fosaprepitant, indicating that extrapolated AUC values were <6%, and plasma samples were collected for a time period sufficient for an accurate assessment of bioequivalence and terminal t1/2. Terminal phase t1/2 (CV%) values for aprepitant were 13.5 (50.9%) and 13.5 (44.5%) h after IV administration of HTX-019 and fosaprepitant, respectively.
Pharmacokinetics
PK data were analyzed for all subjects who completed the study (N=97).
Rapid conversion of the prodrug fosaprepitant to aprepitant was confirmed, with no fosaprepitant detected in the plasma at 0.75 h after administration (Figure 2). The plasma concentrations of aprepitant over time after administration of HTX-019 and fosaprepitant are shown in Figure 3 and were almost superimposable at 0.75 h after administration. For HTX-019, the mean (CV%) Cmax for aprepitant was 6,265 (26.2%) ng/mL at a median Tmax of 0.5 h. For fosaprepitant, the mean Cmax (CV%) for aprepitant was 4,298 (32.5%) ng/mL at a median Tmax of 0.5 h; at this time, the fosaprepitant Cmax was 4,446 (23.1%) ng/mL.
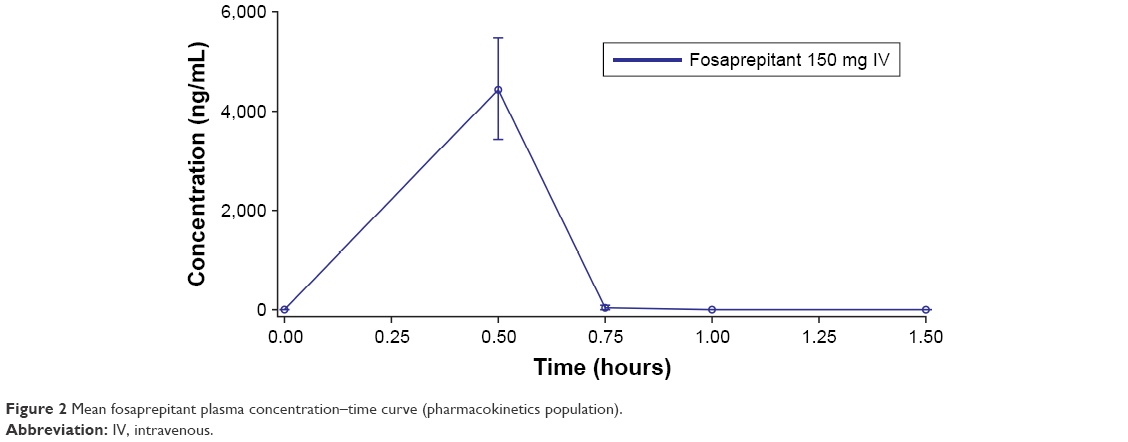 | Figure 2 Mean fosaprepitant plasma concentration–time curve (pharmacokinetics population). |
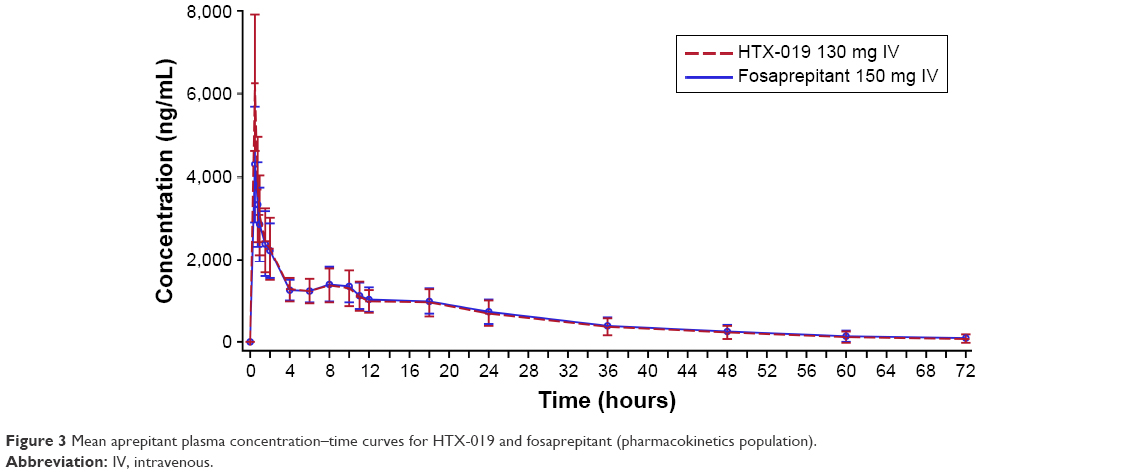 | Figure 3 Mean aprepitant plasma concentration–time curves for HTX-019 and fosaprepitant (pharmacokinetics population). |
Safety
All 100 subjects received at least one dose of the study drug and were included in the safety analyses. Overall, 41% of subjects reported at least one TEAE during the entire study period; 21% and 28% of subjects reported TEAEs in the HTX-019 and fosaprepitant arms, respectively. The most common TEAEs overall were headache and infusion-site pain (Table 3). Within 1 h following infusion start, 1% of subjects receiving HTX-019 reported one TEAE (infusion-site pain) and 20% of subjects receiving fosaprepitant reported 32 TEAEs (including eight subjects with ISAEs), as shown in Table 4.
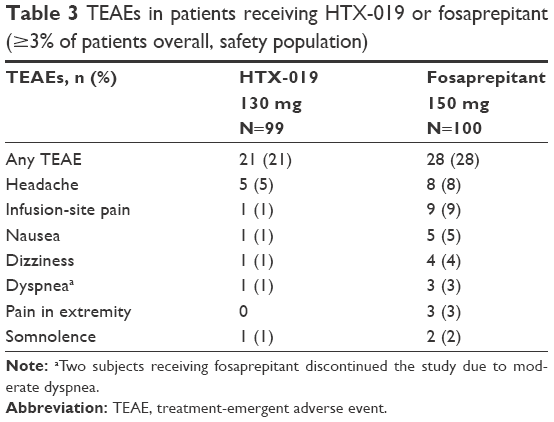 | Table 3 TEAEs in patients receiving HTX-019 or fosaprepitant (≥3% of patients overall, safety population) |
 | Table 4 TEAEs occurring within 1 h or 30 min of infusion start in patients receiving HTX-019 or fosaprepitant (safety population) |
TEAEs considered by the investigator to be related to study drug administration were reported in 15% and 28% of patients receiving HTX-019 and fosaprepitant, respectively. All TEAEs of headache, infusion-site pain, somnolence, nausea, and dizziness were considered related to the study drug. All TEAEs of pain in extremity were considered related to fosaprepitant; there were no TEAEs of pain reported with HTX-019. Importantly, three subjects reported dyspnea after receiving fosaprepitant (observed at <1 min in two subjects and at 18 min in one subject, all considered related to the study drug), as presented in Table 3. Two of the three subjects who experienced dyspnea with fosaprepitant discontinued the infusion and were taken off the study. One subject experienced dyspnea 5 days after receiving HTX-019 (likely due to an upper respiratory infection and considered not related to the study drug) and completed the study. The majority of subjects reported TEAEs that were considered mild in severity (39%).
There were no severe TEAEs, deaths, or serious AEs, and all TEAEs had resolved by the end of the study. There were no clinically meaningful changes in clinical laboratory, physical examination, vital signs, or ECG findings.
A negative binomial analysis of TEAEs found the estimated event rate per subject-day for HTX-019 was approximately half the rate observed for fosaprepitant (0.03 vs 0.06 TEAEs per subject-day; P=0.0274).
Discussion
In this study, a single dose of HTX-019 130 mg was bioequivalent to a single dose of fosaprepitant 150 mg in healthy subjects, in terms of aprepitant PK. Furthermore, the study design allowed evaluation of the NK-1 RA safety profile independently of AEs associated with concomitant chemotherapy. A single dose of HTX-019 130 mg IV appeared to be well tolerated, without the systemic hypersensitivity associated with fosaprepitant, and no new safety findings considered drug-related were identified. TEAEs were evaluated within the first hour of infusion start to assess safety associated with peak concentrations of aprepitant and commercially available fosaprepitant. Of note, considerably fewer subjects receiving HTX-019 (1%) vs fosaprepitant (20%) reported TEAEs within 1 h of infusion start, with most occurring within the first 30 min. Furthermore, there were fewer TEAEs with HTX-019 (one TEAE of infusion-site pain) than with fosaprepitant (32 TEAEs, including eight subjects with ISAEs). No subject receiving HTX-019 had to discontinue from the study due to a TEAE, whereas two subjects receiving fosaprepitant had to stop their infusions and discontinue because of dyspnea. These observations regarding TEAEs are consistent with previous reports of significant risk of ISAEs with IV fosaprepitant, especially in patients receiving anthracyclines and patients receiving chemotherapy through a peripheral line.16,17,20 These findings led to a change in the fosaprepitant prescribing information in 2017 to include a warning regarding serious systemic hypersensitivity reactions, such as anaphylaxis and anaphylactic shock, either during or shortly after infusion.11
A limitation of this study is that it was unblinded; however, the open-label design is typical of bioavailability studies. Furthermore, the crossover design minimizes intersubject variation and variability between the two routes of administration.
Several antiemetic guidelines have recommended inclusion of NK-1 RAs within the antiemetic regimen.6–9 A recent meta-analysis of 38 randomized clinical trials has shown improvement in the complete response rates in the acute, delayed, and overall phases of CINV, when NK-1 RAs such as fosaprepitant, aprepitant, netupitant, and casopitant were part of an antiemetic regimen.23 Recently, IV formulations of rolapitant (IV Varubi; Tesaro, Inc., Waltham, MA, USA) and aprepitant (Cinvanti; Heron Therapeutics, Inc., San Diego, CA, USA) were approved by the US Food and Drug Administration for prevention of CINV.24,25 Improvement in the complete response rates in the acute and delayed phases of CINV were shown by a meta-analysis that focused on the NK-1 RA rolapitant.26 The IV formulation of aprepitant was approved based on data demonstrating its bioequivalence to fosaprepitant, supporting the efficacy of IV aprepitant for the prevention of acute and delayed phases of CINV.25 Extensive clinical trial experience with aprepitant has established its efficacy and safety profile in the context of CINV prophylaxis.27 Consequently, aprepitant and its prodrug fosaprepitant remain the most commonly prescribed NK-1 RAs for CINV (Heron Therapeutics, Inc. data on file, 2016) However, a range of AEs has been observed in patients receiving fosaprepitant, frequently attributed to the synthetic surfactant, polysorbate 80, leading to the inclusion of a warning in its prescribing information regarding the risk of systemic hypersensitivity reactions.11 The incidence of these ISAEs is unclear because of reporting heterogeneity, although they have been reported in up to 42% of patients,19 and an increasing number of AEs with fosaprepitant were reported to the US Food and Drug Administration during 2017.9
HTX-019 is the first polysorbate 80– and synthetic surfactant–free NK-1 RA formulation that delivers aprepitant IV. Results presented here indicate that HTX-019 is bioequivalent to fosaprepitant in terms of plasma aprepitant and appears to be well tolerated. HTX-019 may provide a safer and effective alternative to fosaprepitant for patients with CINV.
Conclusion
HTX-019, a polysorbate 80– and synthetic surfactant–free aprepitant IV formulation, was shown to be generally well tolerated and bioequivalent to commercially available fosaprepitant (Emend IV). HTX-019 may provide a safer alternative to fosaprepitant for patients with CINV, without the risk of polysorbate 80 surfactant–associated systemic hypersensitivity and ISAEs.
Acknowledgments
This study was funded by Heron Therapeutics, Inc., Redwood City, CA, USA. Medical writing support was provided by Joanna K Sandilos Rega, PhD, of SciStrategy Communications and funded by Heron Therapeutics, Inc.
This was presented in part as an oral presentation at the 2017 MASCC/ISOO Annual Meeting, June 22–24, Washington, DC, and as a poster at the 2017 Hematology/Oncology Pharmacy Association (HOPA) Annual Conference, March 29–April 1, Anaheim, CA.
Disclosure
TO, MRK, MC, NC, and BQ are Heron Therapeutics, Inc., employees and report stock ownership. TO, NC, and BQ report leadership roles with Heron Therapeutics, Inc. TO reports travel, accommodation, and expenses reimbursed by Heron Therapeutics, Inc. The authors report no other conflicts of interest in this work.
References
Bloechl-Daum B, Deuson RR, Mavros P, Hansen M, Herrstedt J. Delayed nausea and vomiting continue to reduce patients’ quality of life after highly and moderately emetogenic chemotherapy despite antiemetic treatment. J Clin Oncol. 2006;24(27):4472–4478. | ||
Haiderali A, Menditto L, Good M, Teitelbaum A, Wegner J. Impact on daily functioning and indirect/direct costs associated with chemotherapy-induced nausea and vomiting (CINV) in a U.S. population. Support Care Cancer. 2011;19(6):843–851. | ||
Navari RM. Treatment of breakthrough and refractory chemotherapy-induced nausea and vomiting. Biomed Res Int. 2015;2015:595894. | ||
Van Laar ES, Desai JM, Jatoi A. Professional educational needs for chemotherapy-induced nausea and vomiting (CINV): multinational survey results from 2388 health care providers. Support Care Cancer. 2015;23(1):151–157. | ||
Hesketh PJ, Kris MG, Grunberg SM, et al. Proposal for classifying the acute emetogenicity of cancer chemotherapy. J Clin Oncol. 1997;15(1):103–109. | ||
NCCN Clinical Practice Guidelines in Oncology: Antiemesis (version 2); 2017. Available from: www.nccn.org/professionals/physician_gls/f_guidelines.asp. Accessed February 6, 2018. | ||
Herrstedt J, Roila F, Warr D, et al. 2016 Updated MASCC/ESMO consensus recommendations: prevention of nausea and vomiting following high emetic risk chemotherapy. Support Care Cancer. 2017;25(1):277–288. | ||
Roila F, Warr D, Hesketh PJ, et al. 2016 updated MASCC/ESMO consensus recommendations: prevention of nausea and vomiting following moderately emetogenic chemotherapy. Support Care Cancer. 2017;25(1):289–294. | ||
Hesketh PJ, Kris MG, Basch E, et al. Antiemetics: American Society of Clinical Oncology Clinical Practice Guideline Update. J Clin Oncol. 2017;35(28):3240–3261. | ||
Emend (aprepitant) capsules, for oral use [prescribing information]. Whitehouse Station, NJ: Merck & Co; 2003. | ||
Emend (fosaprepitant dimeglumine) for injection, for intravenous use [prescribing information]; Whitehouse Station, NJ: Merck & Co; 2017. | ||
Hesketh PJ, Grunberg SM, Gralla RJ, et al. The oral neurokinin-1 antagonist aprepitant for the prevention of chemotherapy-induced nausea and vomiting: a multinational, randomized, double-blind, placebo-controlled trial in patients receiving high-dose cisplatin–the Aprepitant Protocol 052 Study Group. J Clin Oncol. 2003;21(22):4112–4119. | ||
Saito H, Yoshizawa H, Yoshimori K, et al. Efficacy and safety of single-dose fosaprepitant in the prevention of chemotherapy-induced nausea and vomiting in patients receiving high-dose cisplatin: a multicentre, randomised, double-blind, placebo-controlled phase III trial. Ann Oncol. 2013;24(4):1067–1073. | ||
Grunberg S, Chua D, Maru A, et al. Single-dose fosaprepitant for the prevention of chemotherapy-induced nausea and vomiting associated with cisplatin therapy: randomized, double-blind study protocol–EASE. J Clin Oncol. 2011;29(11):1495–1501. | ||
Weinstein C, Jordan K, Green SA, et al. Single-dose fosaprepitant for the prevention of chemotherapy-induced nausea and vomiting associated with moderately emetogenic chemotherapy: results of a randomized, double-blind phase III trial. Ann Oncol. 2016;27(1):172–178. | ||
Hegerova LT, Leal AD, Grendahl DC, et al. An analysis of fosaprepitant-induced venous toxicity in patients receiving highly emetogenic chemotherapy. Support Care Cancer. 2015;23(1):55–59. | ||
Leal AD, Kadakia KC, Looker S, et al. Fosaprepitant-induced phlebitis: a focus on patients receiving doxorubicin/cyclophosphamide therapy. Support Care Cancer. 2014;22(5):1313–1317. | ||
Tsuda T, Kyomori C, Mizukami T, et al. Infusion site adverse events in breast cancer patients receiving highly emetic chemotherapy with prophylactic anti-emetic treatment with aprepitant and fosaprepitant: a retrospective comparison. Mol Clin Oncol. 2016;4(4):603–606. | ||
Fujii T, Nishimura N, Urayama KY, et al. Differential impact of fosaprepitant on infusion site adverse events between cisplatin- and anthracycline-based chemotherapy regimens. Anticancer Res. 2015;35(1):379–383. | ||
Sato Y, Kondo M, Inagaki A, et al. Highly frequent and enhanced injection site reaction induced by peripheral venous injection of fosaprepitant in anthracycline-treated patients. J Cancer. 2014;5(5):390–397. | ||
Cummings J, Forrest GJ, Cunningham D, Gilchrist NL, Soukop M. Influence of polysorbate 80 (Tween 80) and etoposide (VP-16-213) on the pharmacokinetics and urinary excretion of Adriamycin and its metabolites in cancer patients. Cancer Chemother Pharmacol. 1986;17(1):80–84. | ||
Azmin MN, Stuart JF, Calman KC, Florence AT. Effects of polysorbate 80 on the absorption and distribution of oral methotrexate (MTX) in mice. Cancer Chemother Pharmacol. 1982;9(3):161–164. | ||
Yuan DM, Li Q, Zhang Q, et al. Efficacy and safety of neurokinin-1 receptor antagonists for prevention of chemotherapy-induced nausea and vomiting: systematic review and meta-analysis of randomized controlled trials. Asian Pac J Cancer Prev. 2016;17(4):1661–1675. | ||
Varubi (rolapitant) injectable emulsion, for intravenous use [prescribing information]; Waltham, MA: Tesaro, Inc; 2017. | ||
Cinvanti (aprepitant) injectable emulsion, for intravenous use [prescribing information]; San Diego, CA: Heron Therapeutics; 2017. | ||
Ahmed H, Hammad AM, Abushouk AI, et al. Meta-analysis of safety and efficacy of rolapitant, NK-1 receptor antagonist for prevention of chemotherapy induced nausea and vomiting. Curr Probl Cancer. 2017. pii:S0147-0272(17)30052-1. | ||
Aapro M, Carides A, Rapoport BL, Schmoll HJ, Zhang L, Warr D. Aprepitant and fosaprepitant: a 10-year review of efficacy and safety. Oncologist. 2015;20(4):450–458. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.