")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Bioequivalence of generic alendronate sodium tablets (70 mg) to Fosamax® tablets (70 mg) in fasting, healthy volunteers: a randomized, open-label, three-way, reference-replicated crossover study
Authors Zhang Y , Chen X, Tang Y, Lu Y, Guo L, Zhong D
Received 29 March 2017
Accepted for publication 22 May 2017
Published 11 July 2017 Volume 2017:11 Pages 2109—2119
DOI https://doi.org/10.2147/DDDT.S138286
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Yifan Zhang,1 Xiaoyan Chen,1 Yunbiao Tang,2 Youming Lu,1 Lixia Guo,1 Dafang Zhong1
1State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 2Department of Pharmacy, The General Hospital of Shenyang Military Region, Shenyang, People’s Republic of China
Purpose: The aim of this study was to evaluate the bioequivalence of a generic product 70 mg alendronate sodium tablets with the reference product Fosamax® 70 mg tablet.
Materials and methods: A single-center, open-label, randomized, three-period, three-sequence, reference-replicated crossover study was performed in 36 healthy Chinese male volunteers under fasting conditions. In each study period, the volunteers received a single oral dose of the generic or reference product (70 mg). Blood samples were collected at pre-dose and up to 8 h after administration. The bioequivalence of the generic product to the reference product was assessed using the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) reference-scaled average bioequivalence (RSABE) methods.
Results: The average maximum concentrations (Cmax) of alendronic acid were 64.78±43.76, 56.62±31.95, and 60.15±37.12 ng/mL after the single dose of the generic product and the first and second doses of the reference product, respectively. The areas under the plasma concentration–time curves from time 0 to the last timepoint (AUC0–t) were 150.36±82.90, 148.15±85.97, and 167.11±110.87 h·ng/mL, respectively. Reference scaling was used because the within-subject standard deviations of the reference product (sWR) for Cmax and AUC0–t were all higher than the cutoff value of 0.294. The 95% upper confidence bounds were -0.16 and -0.17 for Cmax and AUC0–t, respectively, and the point estimates for the generic/reference product ratio were 1.08 and 1.00, which satisfied the RSABE acceptance criteria of the FDA. The 90% CIs for Cmax and AUC0–t were 90.35%–129.04% and 85.31%–117.15%, respectively, which were within the limits of the EMA for the bioequivalence of 69.84%–143.19% and 80.00%–125.00%.
Conclusion: The generic product was bioequivalent to the reference product in terms of the rate and extent of alendronate absorption after a single 70 mg oral dose under fasting conditions.
Keywords: alendronate sodium, pharmacokinetics, highly variable drug, reference-scaled average bioequivalence
Introduction
Alendronate sodium, the monosodium salt of alendronic acid, is an important member of the bisphosphonate class of drugs, which inhibits bone resorption by binding to bone surfaces and slowing down the formation of hydroxyapatite crystals. This drug is used to treat osteoporosis, Paget’s disease, and other bone-related diseases.1–4 The bioavailability of alendronate after oral administration is <1%, which decreases in the presence of food or divalent ions, such as calcium, but increases with increasing gastric pH. Alendronate is rapidly distributed from plasma because >95% of the drug is cleared within 6 h after intravenous infusion. Alendronate is not metabolized, but it appears to be excreted renally through filtration and via an active channel that differs from the classical acid and base channels. Despite the rapid plasma clearance of alendronate, clinical studies estimated an elimination half-life of 10 years, suggesting that the drug exhibits prolonged sequestration in bone.5–7
Wante Pharmaceutical (Hainan) Co., Ltd. (Haiko, China) has developed a new generic product of alendronate sodium. An unpublished pilot bioequivalence study indicated that alendronate exhibits extremely high within-subject variability in terms of the area under the concentration–time curve (AUC) and maximum concentration (Cmax), with a within-subject coefficient of variation (CV) of up to 50%. Therefore, alendronate can be considered a highly variable drug. Highly variable drugs are defined as drugs in which the within-subject CV in one or more measures of bioequivalence (eg, Cmax or AUC) is ≥30%.8,9 In a survey of generic products reviewed by the US Food and Drug Administration (FDA) between 2003 and 2005 for marketing approval, approximately 20% of generic drugs are highly variable.10 A survey of the Canadian Contract Research Organization database showed that 18% of 580 studies fell into the highly variable category, where the failure rate was 54%.11 Therefore, determining the bioequivalence of highly variable generic drugs remains challenging for the pharmaceutical industry and regulatory authorities. In recent decades, several methods have been proposed to overcome this problem. These methods include multiple-dose studies, replicate study designs, individual bioequivalence studies, direct expansion of bioequivalence limits to other prefixed values, and scaled bioequivalence studies.11 Scientists and regulatory authorities introduced reference-scaled average bioequivalence (RSABE) method, which involves widening of bioequivalence limits based on reference variability.12–14 In 2010, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) issued substantially revised guidelines on bioequivalence;15 the guidelines include RSABE method for determining the bioequivalence of highly variable drugs. Similarly, the FDA published a draft guidance, which includes RSABE method, in 2011.16 The methods recommended by the EMA and FDA are all based on scaled average bioequivalence or a closely related variant. A partial replicated (three-way crossover: TRR, RTR, or RRT [T is the test product, and R is the reference product]) or fully replicated (four-way crossover: TRTR or RTRT) study should be performed to estimate the within-subject CV for the reference product (CVWR) and confirm that the drug is highly variable for a given parameter. According to these guidelines, we performed a randomized-sequence, single-dose, open-label, reference-replicated, three-period, crossover study in healthy male Chinese volunteers under fasting conditions. The RSABE methods recommended by the FDA and EMA were used to assess the bioequivalence of the generic product to the reference product.
Materials and methods
Study drugs
We used generic alendronate sodium tablets (equivalent to 70 mg alendronic acid; batch number: 20130504; expiry date: May 2015) manufactured by Wante Pharmaceutical (Hainan) Co., Ltd. as the test product and Fosamax® (equivalent to 70 mg alendronic acid; batch number: 130200; expiry date: February 2015) manufactured by Hangzhou MSD Pharmaceutical Co., Ltd. (Hangzhou, China) as the reference product.
Volunteers
Healthy Chinese male nonsmokers, aged 18–40 years, with a body mass index (BMI) of 18–24 kg/m2 were recruited as volunteers. The health of the participants was assessed in terms of medical history, physical examination, 12-lead electrocardiography, and routine clinical laboratory tests (hematology, biochemistry, urinalysis, human immunodeficiency virus, hepatitis C antibodies, and hepatitis B surface antigen). Volunteers were not enrolled if they had taken any investigational drugs within 4 weeks or any prescription or nonprescription drugs within 2 weeks before the first dose of the study drug. Other exclusion criteria included known or suspected allergies to the study drugs or related compounds, a positive result in any drug screening, current smoking, significant blood loss (200 mL) within 2 months before the first dose of the study drugs, or consumption of grapefruit or grapefruit juice within 7 days before the first dose of the study drugs. All volunteers provided written informed consent prior to enrollment.
Based on the results of a previous pilot study and considering the within-subject CV of approximately 50% for AUC0–t and Cmax and the predicted geometric mean ratios (GMRs) of 0.95–1.05, the enrollment of 31 patients would provide power of ≥90% to show bioequivalence. Therefore, considering possible study withdrawals, we planned to enroll 36 volunteers.
Study design
This study involved a single-center, randomized-sequence, single-dose, open-label, reference-replicated, three-period, crossover design, with a washout period of 7 days between each dosing period. A follow-up visit was scheduled up to 7 days after the third dose of the study drugs. The study was conducted at the General Hospital of Shenyang Military Region and the protocol was approved by the hospital’s independent ethics committee.
In each dosing period, after an overnight fast of ≥10 h, each volunteer received the test product or the reference product according to the randomization scheme. Both products were administered as a single oral dose of one tablet containing 70 mg of alendronate sodium with 200 mL of water in the morning. The volunteers continued to fast for ≥4 h post dose, and water was forbidden for >2 h post dose. Standard meals, of similar compositions in each study period, were provided to the volunteers at 4 and 10 h after drug administration. Beverages, alcohol, extreme physical activity, and smoking were not allowed between enrollment and the final follow-up visit.
Safety evaluation
All volunteers who participated in the study were included in the safety analysis. Safety was evaluated in terms of physical examinations, vital signs (blood pressure, pulse, respiratory rate, and body temperature), 12-lead electrocardiography, laboratory tests (clinical chemistry, hematology, urinalysis, and serology), and adverse events (AEs). Vital signs were assessed before administration and at 2, 4, 6, and 8 h post dose. Clinical chemistry, hematology, and urinalysis were conducted in the screening period and at a follow-up visit scheduled up to 7 days after the third dose. AEs were recorded throughout the study period and at the follow-up visit.
Sample collection
In each study period, blood samples (4 mL) were collected from a forearm vein and were placed in heparinized tubes. Samples were obtained before administration and at 10, 20, 30, and 45 min and 1.0, 1.33, 1.67, 2.0, 2.5, 3.0, 4.0, 5.0, 6.0, and 8 h after administration. The samples were centrifuged at 1,500× g for 10 min. Plasma was separated and frozen at approximately −20°C until further analysis.
Measurement of plasma alendronate concentrations
Plasma alendronate (measured as alendronic acid) concentrations were determined using a validated liquid chromatography–tandem mass spectrometry (LC–MS/MS) assay (Supplementary materials). The procedure was based on derivatization with trimethylsilyldiazomethane during solid-phase extraction on a weak anion-exchange solid-phase cartridge, which integrated sample purification and derivatization into one step. The alendronic acid derivative was eluted with methanol. Chromatographic separation was performed on a Capcell PAK-C18 column for a total run time of 7.0 min. A Qtrap 5500 triple quadrupole mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) was used for mass analysis and detection. Quantification was performed by multiple reaction monitoring of the transitions m/z 348.2→289.0 for the alendronic acid derivative and m/z 354.2→295.0 for the d6-alendronic acid (internal standard) derivative.
The calibration curve for alendronic acid was linear within the range of 0.200–30.0 ng/mL with a low limit quantification of 0.200 ng/mL. The intra- and inter-day precision values were <7.3% and 8.9%, respectively, and the accuracy ranged from 97.8% to 106.7% of the nominal value.
In method validation, dilution test was performed to assess the reliability of the method at concentration levels outside the calibration range. Based on the results, a fivefold dilution of human plasma samples containing alendronic acid above the upper limit of quantification (ULOQ) was considered acceptable. In an analysis of unknown samples with concentrations higher than ULOQ, they were reassayed after appropriate dilution.
Pharmacokinetic analyses
Pharmacokinetic parameters were calculated using standard non-compartmental methods. The Cmax and the time to reach Cmax (tmax) were derived from the plasma concentration–time curves. Elimination rate constant (λz) was calculated by linear regression of the terminal linear portion of the ln(concentration–time) curve, and the apparent elimination half-life (t1/2) was determined as 0.693/λz. The AUC from time 0 to the last timepoint (AUC0–t) was calculated using linear trapezoidal method. The AUC from 0 to infinity (AUC0–∞) was calculated as AUC0–t + Ct/λz, where Ct is the last measurable concentration. Phoenix WinNonlin software version 6.3 (Pharsight Corporation, St Louis, MO, USA) was used for all pharmacokinetic analyses.
Statistical analysis
The protocol was prespecified using the method recommended by the FDA for assessing bioequivalence. EMA method was performed in a post hoc manner to detect any differences in bioequivalence between the two methods.
RSABE method recommended by the FDA
The FDA posted guidance for industry with step-by-step instructions for statistical analysis of bioequivalence study data by using RSABE method.16 The first step involves the determination of sWR, which is the within-subject standard deviation (SD) of the reference product estimated from the study, for AUC and Cmax. If sWR <0.294 for either measure, the two one-sided test (TOST) procedure should be used to determine the absolute bioequivalence for that measure. If sWR ≥0.294, RSABE method should be used for that measure. If RSABE method needs to be applied to a bioequivalence parameter, the next step involves calculating the 95% upper confidence bound for 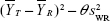 , where
, where 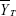 and
and 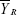 are the mean values of the ln-transformed AUC and/or Cmax for the test and reference products, respectively. In θ = (ln (1.25)2/σW0)2, the bioequivalence limit σW0 is a predetermined constant set at 0.25 by the FDA. The method used to calculate the upper confidence bound was based on “Howe’s Approximation I.”17 The test and reference products are concluded to be bioequivalent if, 1) the 95% upper confidence bound for is ≤0, and 2) the GMR for test/reference is within the range of 0.80–1.25.
are the mean values of the ln-transformed AUC and/or Cmax for the test and reference products, respectively. In θ = (ln (1.25)2/σW0)2, the bioequivalence limit σW0 is a predetermined constant set at 0.25 by the FDA. The method used to calculate the upper confidence bound was based on “Howe’s Approximation I.”17 The test and reference products are concluded to be bioequivalent if, 1) the 95% upper confidence bound for is ≤0, and 2) the GMR for test/reference is within the range of 0.80–1.25.
Wilcoxon’s test was performed on the mean tmax for both treatments. All statistical tests were performed at the α-level of 0.05.
RSABE method recommended by the EMA
The ln-transformed AUC0–t, AUC0–∞, and Cmax were first analyzed using general linear models according to Method A recommended by the EMA.18 The statistical model included sequence, period, treatment, and subject-within sequence as fixed factors. The sequence effect was tested using the subject-within sequence effect as error term. The treatment and period effects were tested against the residual mean square error. CVWR was calculated by using analysis of variance (ANOVA) on the reference data only, with sequence, subject-within sequence, and study period as fixed effects. The point estimates and the 90% CIs for the test/reference GMR were calculated for AUC0–t, AUC0–∞, and Cmax, and presented as least-squares means.
According to the regulatory requirements of the EMA, the hypothesis of bioequivalence between a generic product and a reference product is acceptable if the 90% CIs of the test/reference ratio of least-squares means for ln-transformed AUC0–t are within the acceptance range of 80.00%–125.00%. For Cmax, a scaled average bioequivalence method can be used. This method was based on CVWR. For CVWR ≤30%, the 90% CIs of the test/reference ratio of least-squares means of ln-transformed Cmax should be within the acceptance range of 80.00%–125.00%. However, if the CVWR >30% for Cmax, the acceptance range should be scaled according to the within-subject variability of the reference product, to a maximum of 69.84%–143.19%.
All statistical procedures recommended by the FDA and EMA were performed using applications developed in WinNonlin 6.3 and validated in SAS 9.2.1 (SAS Institute Inc., Cary, NC, USA).
Results
Background characteristics of the volunteers
A total of 36 Chinese males were enrolled and completed this study. The mean ± SD (range) age, height, weight, and BMI of the participants were 25.0±3.7 years (19–33 years), 1.73±0.06 m (1.64–1.92 m), 66.8±7.8 kg (58–83 kg), and 22.1±2.1 kg/m2 (19.1–25.0 kg/m2), respectively.
Pharmacokinetics of alendronate
The mean plasma concentration–time curves of alendronic acid after a single dose of the test or reference products containing 70 mg of alendronate sodium are shown in Figure 1. The pharmacokinetic parameters are summarized in Table 1.
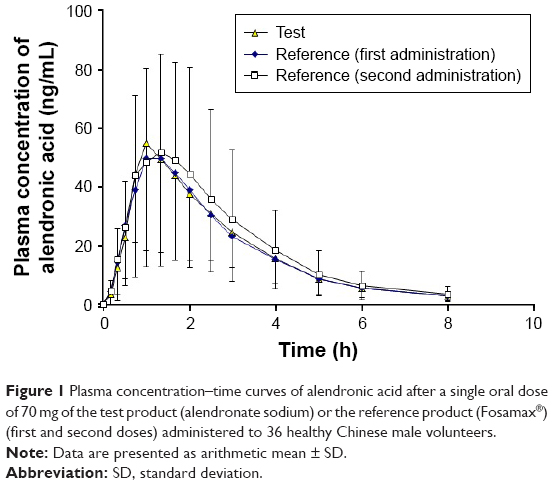 | Figure 1 Plasma concentration–time curves of alendronic acid after a single oral dose of 70 mg of the test product (alendronate sodium) or the reference product (Fosamax®) (first and second doses) administered to 36 healthy Chinese male volunteers. |
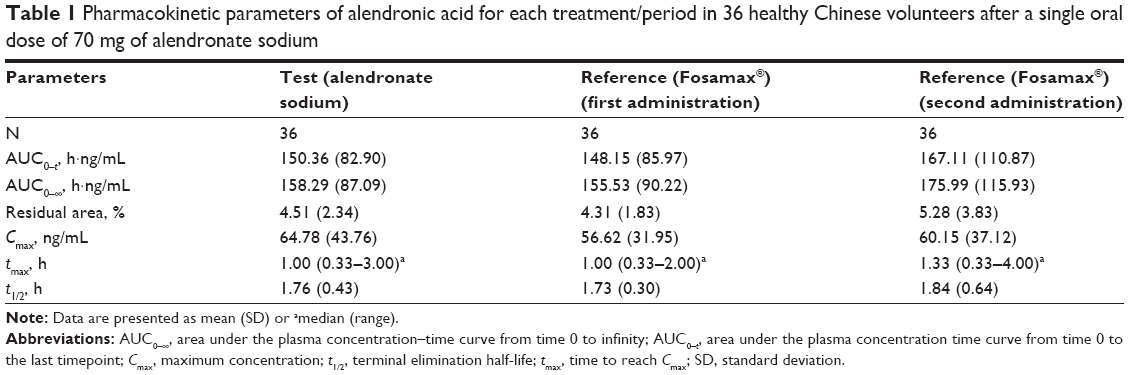 | Table 1 Pharmacokinetic parameters of alendronic acid for each treatment/period in 36 healthy Chinese volunteers after a single oral dose of 70 mg of alendronate sodium |
The Cmax values of alendronic acid after the single dose of the test product and the first and second doses of the reference product were 64.78, 56.62, and 60.15 ng/mL, respectively, and they occurred approximately 1 h after administration. The AUC0–t values were 150.36, 148.15, and 167.11 h·ng/mL, respectively. Alendronic acid was rapidly eliminated with t1/2 values of 1.76, 1.73, and 1.84 h, respectively.
Bioequivalence assessments
The bioequivalence values determined using the method recommended by the FDA are summarized in Table 2. The first step of the analysis showed that the sWR values were 0.546, 0.527, and 0.571 for AUC0–t, AUC0–∞, and Cmax, respectively, which exceeded the regulatory limit of 0.294. Thus, RSABE method was conducted for all three parameters. The 95% upper confidence bound was <0 for all three parameters. The GMR values for the point estimates of AUC0–t, AUC0–∞, and Cmax were 1.00, 1.00, and 1.08, respectively, which are within the range of 0.80–1.25. Therefore, the test product satisfied the RSABE acceptance criteria and can be considered bioequivalent to the reference product Fosamax.
 | Table 2 Statistical analysis of the results obtained using the FDA’s RSABE method |
The results of the analysis using EMA method are summarized in Table 3. The sWR values for AUC0–t, AUC0–∞, and Cmax were 0.569, 0.548, and 0.607, respectively, and the CVWR values were 61.83%, 59.14%, and 66.71%, respectively. The acceptance limit for Cmax based on the within-subject variability of the reference product was set at 69.84%–143.19% because CVWR >50%. The 90% CIs for the test/reference GMRs were 85.31%–117.15% for AUC0–t, 85.29%–116.15% for AUC0–∞, and 90.35%–129.04% for Cmax, which are within the accepted bioequivalence limits.
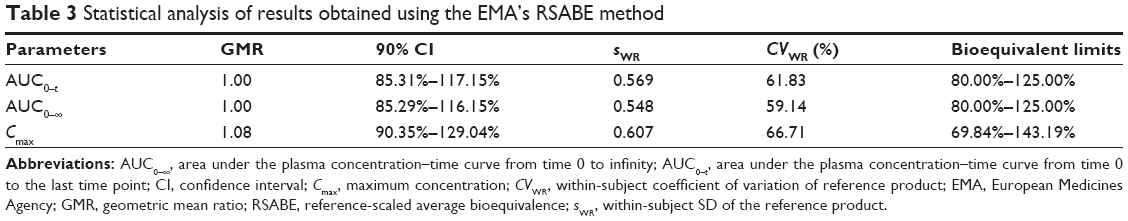 | Table 3 Statistical analysis of results obtained using the EMA’s RSABE method |
Wilcoxon’s test showed that tmax was not significantly different between the two products (P=0.72).
Safety
A total of 36 AEs were reported in 19 of 36 subjects who received a minimum of one dose of the study drugs; 15 AEs were reported by 22.2% (8/36) of the volunteers after the administration of the test product, and 21 AEs were reported by 38.9% (14/36) of the volunteers after the administration of the reference product. The most common AEs were fatigue (10/36), joint pain (9/36), and fever (9/36). After the assessment of the causality of the 36 AEs, most AEs were judged as “probable/likely” to be related to the study products. A total of 14 AEs were related to the test product, and 20 AEs were related to the reference product. The severity of all AEs was classified as mild, and none of the volunteers withdrew from the study because of an AE. No serious AEs were observed.
Discussion
Urine alendronate concentrations were used to assess the bioequivalence of alendronate formulations because of the low concentrations of alendronate in plasma and the limitations of assay methods. In a classical, typical bioequivalence study, 122 subjects were enrolled because of the high within-subject CV for urine parameters, 37.42% for Ae 0–t, and 40.05% for Rmax.19 Rhim et al20 were the first to assess the bioequivalence of alendronate by determining its plasma concentrations in a two-period crossover study involving 23 Korean subjects; the results demonstrated bioequivalence even though the study power was <60%. Wright et al21 performed a classic bioequivalence study of the combination of alendronate and vitamin D3 tablet in 68 healthy Taiwanese volunteers. The combination tablet was bioequivalent to the coadministration of separate tablets based on the AUC0–∞ for alendronate but not on Cmax. The Cmax exceeded the bioequivalent limit of 80%–125%, because the high variability of alendronate was not considered in sample size calculation; therefore, the sample size was not sufficiently large to confirm bioequivalence. We conducted a pilot study that provided valuable information, including differences in pharmacokinetics between the test and reference products, and within-subject CV for AUC and Cmax; these data were used to estimate the sample size in our follow-up study. Assuming a difference of 5%, we estimated that ≥98 subjects would be necessary for 80% power to ensure that the 90% CI of the ratios are within the bioequivalence limit of 80%–125% by using the classical two-way crossover design. However, by using a partial replicated design (ie, a three-way crossover study) and RSABE method of the FDA, a sample size of 31 subjects would achieve 90% power. The current study demonstrates that the new method not only reduces the sample size required but also increases the study power. Considering the increasing cost of bioequivalence studies, the new method, which uses fewer subjects, is an economic choice for the sponsor.
The methods proposed by both the FDA and EMA involved mixed scaling, that is, the reference-scaled procedure should be used for bioequivalence measures with sWR ≥0.294 (or CVWR >30%), and the TOST procedure must be used for pharmacokinetic parameters with sWR <0.294 (or CVWR ≤30%). In the current study, we preferred the FDA method to assess the bioequivalence between the two formulations. We also used the EMA method to compare the two methods. The differences between the FDA and EMA methods were reported in previous papers.13,14,22 For highly variable drugs, the FDA method is more lenient than the EMA method and requires fewer subjects. The FDA method permits the scaling of both Cmax and AUC and does not restrict the scaling limits, but the test/reference GMRs must be within the range of 0.80–1.25. By contrast, the EMA method only permits reference scaling for Cmax and imposes an extreme limit of CVWR, in which the scaled limits should be applied. For CVWR values >50%, the extreme values of 143.19% and 69.84% are imposed on the upper and lower limits, respectively. Another major discrepancy is the different scaling factors. The scaling factor k approximately equals 0.893 in the FDA method but equals 0.760 in the EMA method.22 Moreover, we found that the sWR values differed between the FDA and EMA methods. For example, the sWR value for Cmax was 0.571 with the FDA method compared with 0.607 with the EMA method. This variation is due to the differences in the calculations used in both methods. The EMA questions and answers’ document contains a section “Clarification on the recommended statistical method for the analysis of a bioequivalence study,” which describes the statistical models for analyzing data from replicated bioequivalence trials and the differences between the EMA and the FDA methods.18 Thus, although the FDA and EMA methods are based on similar principles, they yield slightly different results in bioequivalence studies. Considering these differences, researchers should carefully select appropriate scaling methods when designing the protocol prior to conducting the study. In addition, the guidelines of the relevant application region or the recommendations of the appropriate regulatory authority should be considered.
Conclusion
The current study demonstrated the bioequivalence of generic alendronate sodium tablets to the reference product Fosamax® by using RSABE method proposed by the FDA and EMA in a partial replicated crossover study. The new reference scaling methods are more effective and economic than the classical method for assessing bioequivalence.
Disclosure
The authors report no conflicts of interest in this work.
References
Hosking D, Chilvers CE, Christiansen C, et al. Prevention of bone loss with alendronate in postmenopausal women under 60 years of age. N Engl J Med. 1998;338(8):485–492. | ||
Russell RG, Rogers MJ. Bisphosphonates: from the laboratory to the clinic and back again. Bone. 1999;25(1):97–106. | ||
Finkelstein JS, Wyland JJ, Lee H, Neer RM. Effects of teriparatide, alendronate, or both in women with postmenopausal osteoporosis. J Clin Endocrinol Metab. 2010;95(4):1838–1845. | ||
Fleisch H. Bisphosphonates in osteoporosis. Eur Spine J. 2003;12(suppl 2):S142–S146. | ||
Ananchenko G, Novakovic J, Tikhomirova A. Alendronate sodium. Profiles Drug Subst Excip Relat Methodol. 2013;38:1–33. | ||
Porras AG, Holland SD, Gertz BJ. Pharmacokinetics of alendronate. Clin Pharmacokinet. 1999;36(5):315–328. | ||
Cocquyt V, Kline WF, Gertz BJ, et al. Pharmacokinetics of intravenous alendronate. J Clin Pharmacol. 1999;39(4):385–393. | ||
Blume HH, Midha KK. Bio-International 92, conference on bioavailability, bioequivalence, and pharmacokinetic studies. J Pharm Sci. 1993;82(11):1186–1189. | ||
Shah VP, Yacobi A, Barr WH, et al. Evaluation of orally administered highly variable drugs and drug formulations. Pharm Res. 1996;13(11):1590–1594. | ||
Davit BM, Conner DP, Fabian-Fritsch B, et al. Highly variable drugs: observations from bioequivalence data submitted to the FDA for new generic drug applications. AAPS J. 2008;10(1):148–156. | ||
Tothfalusi L, Endrenyi L, Arieta AG. Evaluation of bioequivalence for highly variable drugs with scaled average bioequivalence. Clin Pharmacokinet. 2009;48(11):725–743. | ||
Zhang X, Zheng N, Lionberger RA, Yu LX. Innovative approaches for demonstration of bioequivalence: the US FDA perspective. Ther Deliv. 2013;4(6):725–740. | ||
Davit BM, Chen ML, Conner DP, et al. Implementation of a reference-scaled average bioequivalence approach for highly variable generic drug products by the US Food and Drug Administration. AAPS J. 2012;14(4):915–924. | ||
García-Arieta A, Gordon J. Bioequivalence requirements in the European Union: critical discussion. AAPS J. 2012;14(4):738–748. | ||
Committee for Medicinal Products for Human Use (CHMP). Guideline on the Investigation of Bioequivalence. London: European Medicines Agency (EMA); 2010. | ||
Office of Generic Drugs, Food and Drug Administration (FDA). Draft Guidance for Industry on Bioequivalence Recommendations for Progesterone Capsules. 2011. Available from: http://www.fda.gov/downloads/Drugs/GuidanceCompliance RegulatoryInformation/Guidances/UCM209294.pdf. Accessed May 29, 2017. | ||
Howe WG. Approximate confidence limits on the mean of X + Y where X and Y are two tabled independent random variables. J Am Stat Assoc. 1974;69(347):789–794. | ||
EMEA, CHMP Efficacy Working Party Therapeutic Subgroup on Pharmacokinetics (EWP-PK). EMA Questions & Answers: Positions on Specific Questions Addressed to the Pharmacokinetics Working Party, EMA/618604/2008 Rev. 3, January 26, 2011. | ||
Thudi NR, Gagnon S, Hussain S, et al. Two-way crossover bioequivalence study of alendronate sodium tablets in healthy, non-smoking male volunteers under fasted conditions. Arzneimittelforschung. 2009;59(10):521–525. | ||
Rhim SY, Park JH, Park YS, et al. Bioavailability and bioequivalence of two oral formulations of alendronate sodium 70 mg: an open-label, randomized, two-period crossover comparison in healthy Korean adult male volunteers. Clin Ther. 2009;31(5):1037–1045. | ||
Wright DH, Mols R, Brown KR, et al. Bioequivalence of alendronate and vitamin d3 in a combination tablet versus corresponding-dose individual tablets in healthy Taiwanese volunteers, determined using a novel plasma alendronate assay. Curr Ther Res Clin Exp. 2015;77:116–121. | ||
Karalis V, Symillides M, Macheras P. Bioequivalence of highly variable drugs: a comparison of the newly proposed regulatory approaches by FDA and EMA. Pharm Res. 2012;29(4):1066–1077. |
Supplementary materials
Plasma alendronate (measured as alendronic acid) concentrations were determined using a validated liquid chromatography–tandem mass spectrometry (LC–MS/MS) assay modified based on the method of Chen et al.1
Instrumentation and LC–MS/MS analytical conditions
The high-performance liquid chromatography system consisted of an LC-30AD pump, a DGU-20A5 degasser, an SIL-30AC autosampler, and a CTO-30A thermostatted column compartment (Shimadzu, Kyoto, Japan). Alendronic acid and the internal standard (IS) d6-alendronic acid derivatives were separated on a Capcell PAK-C18 column (100×4.6 mm, 5 μm particle size; Shimadzu) protected by a Security Guard C18 column (4×3.0 mm, 5 μm; Phenomenex, Torrance, CA, USA). The mobile phase was composed of, 1) acetonitrile with 0.1% of formic acid, and 2) 10 mM ammonium acetate with 0.1% formic acid. The initial mobile phase was composed of 15% A at a flow rate of 0.5 mL/min. After 4.5 min, the composition was changed to 90% A and the flow rate was changed to 1.0 mL/min in 0.3 min. These conditions were maintained for 1 min, after which the column was quickly equilibrated with the initial mobile phase at 1.0 mL/min. The total run time was 7.0 min.
Mass spectrometric detection was performed on a Qtrap5500 triple quadrupole instrument (Thermo Fisher Scientific, Waltham, MA, USA) in multiple-reaction monitoring mode. An electrospray ionization (ESI) interface was used in positive ionization mode. Data were processed using Analyst 1.5.2 software (Thermo Fisher Scientific). The instrument was operated with an ion spray voltage of +4.0 kV, source temperature of 400°C, ion source gas 1 of 50 psi, ion source gas 2 of 50 psi, curtain gas of 30 psi, and collision gas of medium value. Declustering potential (DP) and collision energy (CE) were, respectively, set at 60 V and 30 eV for alendronic acid and the d6-alendronic acid derivatives. The multiple-reaction monitoring fragmentation transitions were set at m/z 348.2→m/z 289.0 for the alendronic acid derivative, and m/z 354.2→m/z 295.0 for the d6-alendronic acid derivative, with a dwell time of 80 ms per transition.
Sample preparation
The derivatization reagent was prepared in dark conditions just before the derivatization procedure. A 3.0 mL aliquot of trimethylsilyldiazomethane was added to 9.75 mL of methanol:water (3:0.25; v/v) under nitrogen, mixed immediately, and stored under nitrogen in dark conditions.
Frozen plasma samples from subjects were thawed to room temperature before processing. A 500 μL aliquot of plasma was spiked with the IS, 100 μL of 50 ng/mL d6-alendronic acid, and then diluted with 1.4 mL of water. The pH was adjusted to 4 using 50 μL of 1M HCl. The mixture was vortexed for 1 min, and then loaded on Waters Oasis WAX 6 cc/150 mg cartridges which had been conditioned with 2.0 mL of methanol and equilibrated with 2.0 mL of 20 mM HCl followed by 2.0 mL of water. The cartridges were washed with 2.0 mL of 20 mM HCl and 2.0 mL of methanol. Then, the cartridges were eluted in sequence by 2.0 mL of the derivatization reagent and 1 mL of methanol. The elutions were collected in silylated capped glass tubes and reacted for 30 min and dried at 40°C under a stream of nitrogen in a TurboVap evaporator (Zymark Corp., Hopkinton, MA, USA). The residues were reconstituted in 300 μL of the mobile phase, and 20 μL of the sample was injected into the LC–MS/MS system for analysis.
Method validation
LC–MS/MS method for the determination of alendronic acid in human plasma was developed and validated according to the requirements of the European Medicines Agency (EMA) guidance on bioanalytical method validation.2
Selectivity
The typical chromatograms of blank plasma samples, blank samples spiked with alendronic acid at 0.200 ng/mL, and plasma samples from a volunteer at 20 min after the administration of 70 mg of alendronate sodium are shown in Figure S1. No interfering peaks from endogenous compounds were observed at the retention time of the derivatives of the analyte and the IS.
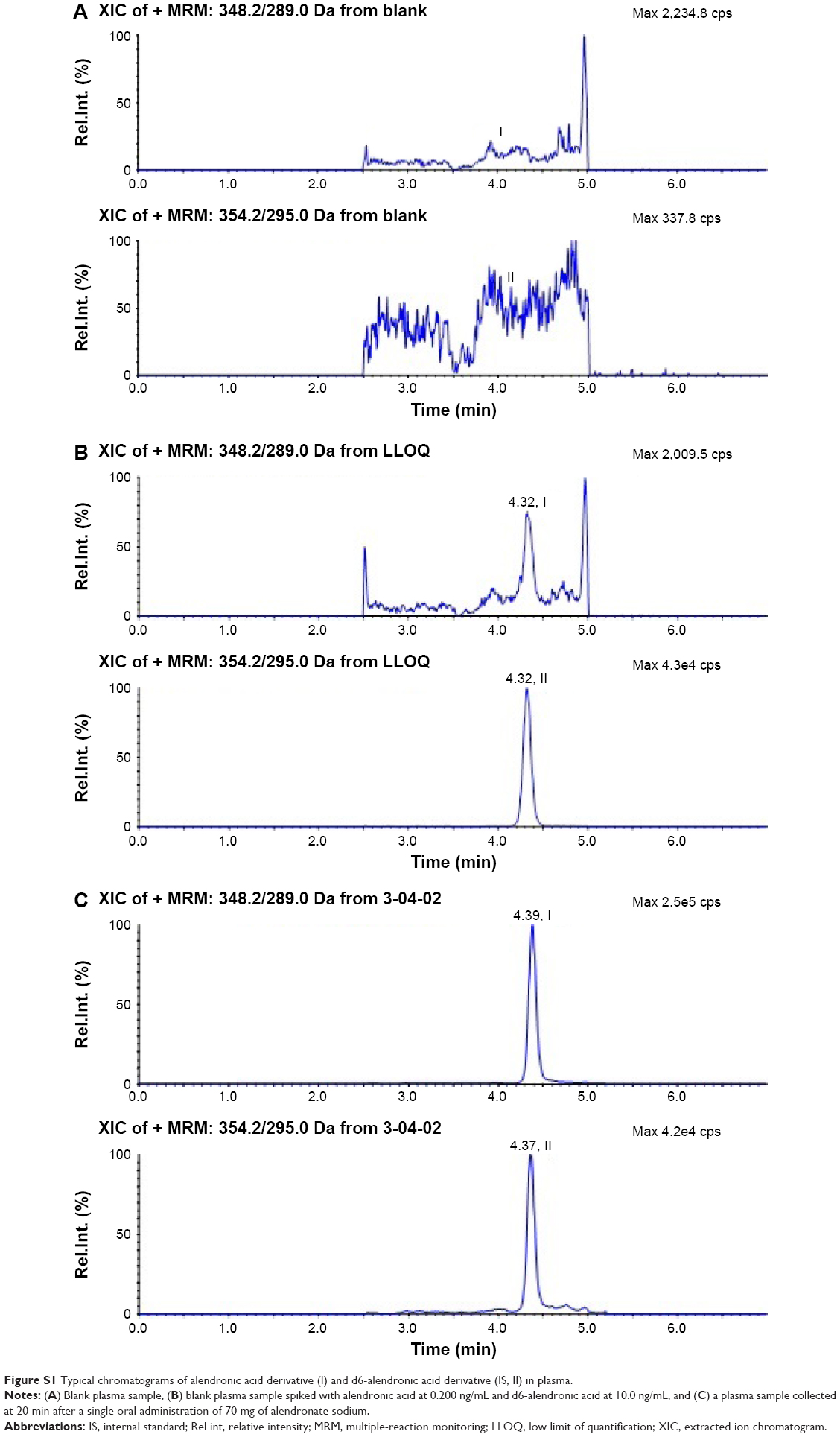 | Figure S1 Typical chromatograms of alendronic acid derivative (I) and d6-alendronic acid derivative (IS, II) in plasma. |
Linearity of calibration curves and low limit of quantification (LLOQ)
The calibration curve was linear over the range of alendronic acid concentrations of 0.200–3.00 ng/mL with a coefficient of correlation (r2) >0.99. The typical equation of the calibration curve was y =0.133x −5.01×10−3 (r=0.9988), where y represents the peak area ratio of analyte derivative to the IS derivative, and x represents the plasma concentration of alendronate. The LLOQ of this assay was 0.200 ng/mL, with a relative standard deviation (RSD) <9.0% and a relative error (RE) within ±4.0%.
Precision and accuracy
Intra- and inter-day precision and accuracy are summarized in Table S1. The intra- and inter-day precision were <7.3% and 8.9%, respectively, and the accuracy ranged from 97.8% to 106.7% of the nominal concentration. Therefore, the intra- and inter-assay precision and the accuracy for the determination of alendronic acid were acceptable.
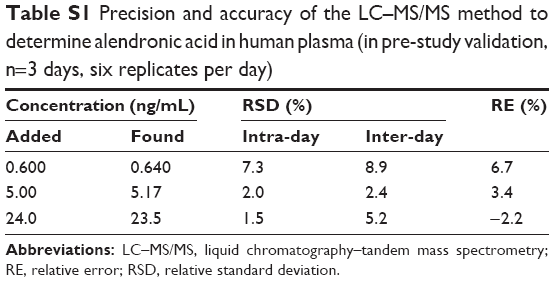 | Table S1 Precision and accuracy of the LC–MS/MS method to determine alendronic acid in human plasma (in pre-study validation, n=3 days, six replicates per day) |
A dilution test was also performed to evaluate the precision and accuracy of the method for diluted samples. The quality control samples containing 100 ng/mL alendronic acid were diluted fivefold with blank plasma in six replicates. All of the back-calculated values were within ±15% of the nominal concentration. The precision and accuracy values were 1.2% and 99.7%, respectively, which were acceptable for a fivefold dilution of human plasma samples with alendronic acid concentrations above the upper limit of quantification.
Recovery and matrix effect
The mean recoveries of alendronic acid determined at concentrations of 0.600, 5.00, and 24.0 ng/mL were 87.6%, 91.6%, and 99.4%, respectively. The extents of recovery from analyte and IS were consistent, precise, and reproducible.
The absolute matrix effects at concentrations of 0.600 and 24.0 ng/mL were 109% and 96.4%, respectively, and the relative matrix effects were less than 7.2%. These results indicated that ion suppression or enhancement from the plasma matrix was negligible under the current conditions.
Stability
The stability of alendronic acid in plasma was evaluated for short-term, long-term, post-preparation, and freeze–thaw conditions. Alendronic acid was stable in plasma stored at room temperature for 6 h (RSD ≤4.4%; RE in the range of 1.1%–9.2%), in plasma stored at −20°C for 54 days (RSD ≤3.2%; RE in the range of 1.0%–8.9%), and for three freeze–thaw treatments (RSD ≤12.0%; RE in the range of −8.8% to 9.9%). The alendronic acid derivative was stable in plasma extracts stored at room temperature for 48 h (RSD ≤8.4%; RE in the range of −10.5% to 4.2%).
References
Chen M, Liu K, Zhong D, Chen X. Trimethylsilyldiazomethane derivatization coupled with solid-phase extraction for the determination of alendronate in human plasma by LC-MS/MS. Anal Bioanal Chem. 2012;402(2):791–798. | ||
European Medicines Agency (EMA), Committee for Medicinal Products for Human Use (CHMP). Guideline on Bioanalytical Method Validation. EMEA/CHMP/EWP/192217/2009, 21 July 2011. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.