")
Back to Journals » Drug Design, Development and Therapy » Volume 15
Bioequivalence and Pharmacokinetic Profiles of Generic and Branded Obeticholic Acid in Healthy Chinese Subjects Under Fasting and Fed Conditions
Authors Wang MN, Yu HT, Li YQ, Zeng Y, Yang S, Yang GP , Pei Q , Huang J
Received 27 October 2020
Accepted for publication 9 December 2020
Published 14 January 2021 Volume 2021:15 Pages 185—193
DOI https://doi.org/10.2147/DDDT.S289016
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Meng-Na Wang,1,* Hai-Tao Yu,2,* Ya-Qian Li,1 Yun Zeng,1 Shuang Yang,3 Guo-Ping Yang,1,3– 6 Qi Pei,6 Jie Huang1,3
1Center for Clinical Pharmacology, The Third Xiangya Hospital, Central South University, Changsha, Hunan 410013, People’s Republic of China; 2Research and Development Center, Nanjing Chia Tai Tianqing Pharmaceutical Co., Ltd., Nanjing 210038, People’s Republic of China; 3Research Center of Drug Clinical Evaluation of Central South University, Changsha, Hunan 410013, People’s Republic of China; 4XiangYa School of Pharmaceutical Sciences, Central South University, Changsha, Hunan 410013, People’s Republic of China; 5National-Local Joint Engineering Laboratory of Drug Clinical Evaluation Technology, Changsha, Hunan 410013, People’s Republic of China; 6Department of Pharmacy, The Third Xiangya Hospital, Central South University, Changsha, Hunan 410013, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jie Huang; Qi Pei
The Third Xiangya Hospital, Central South University, Yinpenling Street, Yuelu District, Changsha, Hunan 410013, People’s Republic of China
Tel +86 15116469024
Fax +86 731 8861 8326
Email [email protected]; [email protected]
Objectives: This study was conducted to evaluate the bioequivalence (BE) of a generic form of obeticholic acid (OCA) and OcalivaTM under fasting and fed conditions and to determine the effects of food on the pharmacokinetic (PK) profiles of OCA in healthy Chinese subjects.
Methods: A randomized, single-dose, three-sequence, three-period, partial replicated crossover study was conducted with a 21-day washout interval between periods under fasting (n=48) and fed (n=48) conditions. Blood samples for OCA and its metabolites Glyco-OCA and Tauro-OCA were collected up to 168 hours after administration in each period. PK parameters were calculated using the non-compartmental method. Geometric mean ratios for PK parameters of the test to reference drug under fasting and fed conditions and their 90% confidence intervals were estimated. Safety evaluations were carried out all through the study.
Results: A total of 91 subjects completed the study with 45 in a fasted state and 46 receiving a high-fat diet. There were no serious or unexpected drug-related adverse events occurring during the study. There was no significant difference in the main PK parameters of the two preparations, irrespective of the fasting or fed conditions. Under fasting and fed conditions, the SWR of lnCmax, lnAUC0-t and lnAUC0-∞ were 0.445, 0.370, 0.448, 0.340, 0.168, and 0.180, respectively. Thus, the average BE or the reference-scaled average BE was used to verify that the two preparations were bioequivalent under fasting and fed conditions. Compared with the fasting state, the AUC0-t of the test drug, the AUC0-t, and AUC0-∞ of the reference drug were higher in the fed state.
Conclusion: The test drug and the reference drug were BE and well tolerated in Chinese healthy subjects under both fasting and fed conditions. Food-intake may cause a significant difference in the main PK parameters of the two preparations.
Keywords: obeticholic acid, bioequivalence, pharmacokinetics, food effect
Introduction
Primary biliary cholangitis (PBC) is a progressive cholestatic liver disease that leads to the gradual destruction of intrahepatic bile ducts resulting in periportal inflammation, cholestasis, and fibrosis and may potentially lead to cirrhosis in a proportion of patients.1 The etiology of PBC remains unknown, and both environmental factors and genetic variants increase disease susceptibility.2–5 The number of patients with PBC in China has been increasing annually.6
Obeticholic acid (OCA, OcalivaTM), an oral potent selective agonist of farnesoid X receptor (FXR), is indicated for the treatment of PBC in ursodeoxycholic acid (UDCA)-refractory or UDCA intolerant patients.3,7 The FXR is a nuclear receptor abundantly expressed in tissues that engage in the enterohepatic circulation of bile acid.8 Unlike UDCA which operates at a post-translational level, FXR-signaling directly regulates genes involved in bile acid synthesis, secretion, transportation, absorption, and detoxification.9 The time to peak plasma concentrations (Tmax) of OCA has been reported to occur after 1.5 hours, and systemic exposures to OCA increased the dose proportionally.10 OCA metabolites glyco-obeticholic acid (Glyco-OCA) and tauro-obeticholic acid (Tauro-OCA) have similar pharmacological activities, which are secreted into bile and are absorbed in the small intestine leading to enterohepatic recirculation.11 OCA is mainly excreted in feces through biliary secretion.10
The United States Food and Drug Administration (FDA) has recommended the implementation of a reference-scaled average bioequivalence (RSABE) approach scaled to the high variability of reference drugs to be administered to patients more than once.12 The primary objective of the present partially replicated experimental study was to evaluate the bioequivalence (BE) of the generic and reference drug in healthy Chinese subjects under both fasting and fed conditions and to determine the effect of food intake on the pharmacokinetic (PK) profile, the secondary objective was to observe and evaluate the safety of the two formulations.
Methods
Ethics Statement of Human Rights
This study was reviewed and approved by the Institutional Review Board of the Third Xiangya Hospital of Central South University. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. All subjects provided written informed consent and the study was registered at the Chinese Clinical Trial Registry [http://www.chictr.org.cn] (number ChiCTR1900026043).
Subjects
This study enrolled healthy Chinese subjects aged >18 years, weighing > 55 kg for males and >45 kg for females, with a body mass index (BMI) ranging from 19.0 to 26.0 kg/m2, no pregnancy plan within 3 months after the end of the trial, both fasting and fed conditions were required. Exclusion criteria included history of circulatory disease, endocrine disease, or any other clinically significant diseases that could affect PK of the drug, history of alcoholism or drug dependence, history of sensitivity to pharmaceutical products, taking any other medication within 14 days before the study, participation in other trials in the previous 3 months, surgery within the last 1 month, blood donation in the previous 3 months, pregnancy, breast-feeding or participation in high-performance athletics.
Sample Size
This study adopts a three-cycle partly repeated design. When SWR is greater than or equal to 0.294 (that is, when the intra-individual CVw ≥30%), RSABE is used to evaluate BE. Assuming one-sided a=0.05, β=0.1, Intra-CV=50%, and the geometric mean ratio (GMR) of the test drug to the reference drug is 0.95–1.05, the sample sizes that meets the FDA and EMA equivalence standard are 31 and 39 cases. Therefore, this study intends to enroll 48 healthy subjects, both male and female.
Study Design
This study was performed as a single-dose, randomized, open-label, 3-sequence, 3-period partial replicated crossover trial design, including two parts: fasting and fed. A total of 96 healthy subjects were enrolled, 48 in the fasting study and 48 in the fed study. Eligible subjects were randomly assigned to three sequences (T-R-R, R-T-R, R-R-T, where T was the test drug, and R was the reference drug). Given that the effective elimination half-life (t1/2eff) was 24 hours, the actual elimination half-life (t1/2z) could not be calculated due to the significant enterohepatic circulation, a 21-day washout period was used between the periods.
The subjects in the fasting-condition study received a single oral dose (10-mg tablet) of the reference (Intercept Pharmaceuticals, Inc.) or test (Nanjing Zhengda Tianqing Pharmaceutical Co., Ltd) formulation of OCA with 240 mL of water after fasting overnight (≥10 hours) and accepting a thorough examination of the oral cavity and hand to guarantee administration compliance. The subjects were constrained from drinking for 2 hours before and after dosing. Light meals were served at 4 and 10 hours after each dose, which should be eaten within 30 minutes, with identical standardized diets provided across all dosing periods.
In total, 48 subjects participated in the fed-condition study. All the procedures were the same as those in the fasting condition, except for eating high-fat, high-calorie standard meals (approximately 150 kcal protein, 250 kcal carbohydrates, and 500–600 kcal fat, a total of about 800–1000 kcal, three periods should ensure that the standard meal formula was consistent) 30 minutes before administration, to be finished within 30 minutes.
Blood Sampling and Determination of Concentration
A peripheral blood sample (5 mL) from each subject was collected in tubes containing sodium heparin at time 0 (within 60 minutes before dosing) and at 15, 30, 45 minutes and 1.0, 1.5, 2.0, 2.5, 3.0, 4.0, 6.0, 8.0, 10.0, 12.0, 14.0, 16.0, 24.0, 36.0, 48.0, 72.0, 96.0, 120.0, and 168.0 hours after dosing in the study under both fasting and fed conditions. Plasma was obtained from the supernatants of the collected blood samples after centrifugation at 2150×g for 10 minutes and were stored at –60°C until use.
Briefly, the plasma concentration of OCA, Glyco-OCA and Tauro-OCA was measured by HPLC-MS/MS (Triple QuadTM 6500+; Sciex) after liquid-liquid extraction (cyclohexane:ethyl acetate=1:3, v/v), and separated on a Poroshell 120 EC-C18, 2.1×50 mm column (Agilent, USA) with mobile phase A (water with 0.1% formic acid) and a mobile phase B (acetonitrile with 0.1% formic acid) for OCA, and mobile phase A (Water with 0.05% formic acid) and mobile phase B (100% acetonitrile) for Glyco-OCA and Tauro-OCA, gradient elution. Glimepiride was used as the internal standard (IS). Multiple reaction monitoring was employed, and transmission masses for OCA, Glyco-OCA, Tauro-OCA, and the IS were 465.3 to 419.3, 476.3 to 74.1, 526.3 to 123.9, and 489.3 to 224.8, respectively. The linear ranges of OCA, Glyco-OCA and Tauro-OCA were 0.15–100 ng·mL−1, 1.00–120 ng·mL−1, and 0.15–18.0 ng·mL−1, respectively.
PK and BE Evaluation
A non-compartmental approach (Phoenix® WinNonlin 7.0) was used to calculate the PK parameters for OCA, and its metabolites Glyco-OCA and Tauro-OCA from the concentration-time profiles. The following PK parameters were evaluated: the peak plasma concentration (Cmax), time to reach the peak plasma concentration (Tmax), area under the plasma concentration-time (AUC) from dosing to the last measurable concentration (AUClast), AUC from dosing to infinity (AUC0-∞), t1/2Z, apparent clearance, λz.
The PK profiles of Glyco-OCA and Tauro-OCA were also calculated, but no BE analysis was performed. The within-subject standard deviation (SWR) of the log-transformed values of Cmax, AUC0-t and AUC0-∞, were calculated. The BE analysis method selection was based on the SWR: the average bioequivalence (ABE) method was adopted when SWR <0.294, while the RSABE method was adopted when SWR ≥0.294. The BE acceptance limits were scaled to the variability of the reference formulation as directed by the RSABE approach.
BE was determined by ABE method when the 90% confidence interval (CI) of the test preparation/reference preparation GMR of Cmax, AUC0-t, and AUC0-∞ of OCA was within 80.00%-125.00%.
The RSABE can be applied to a BE measure, when the test and reference formulations are considered bioequivalent, calculated as:
95% upper confidence limit for 
Where  and
and  are the means of the log-transformed PK endpoints for the test formulation and that for the reference formulation, θ is
are the means of the log-transformed PK endpoints for the test formulation and that for the reference formulation, θ is  , σw0 is the regulatory limit (FDA set σw0 at 0.25). Point estimates of test/reference GMR and 90% CIs of the GMR of test to reference were also calculated. BE conducted by RSABE method need to meet both the BE limits and the GMR constraints (0.80–1.25).
, σw0 is the regulatory limit (FDA set σw0 at 0.25). Point estimates of test/reference GMR and 90% CIs of the GMR of test to reference were also calculated. BE conducted by RSABE method need to meet both the BE limits and the GMR constraints (0.80–1.25).
Safety Evaluation
The safety and tolerability of the two formulations were assessed based on the occurrence of adverse events (AEs), vital signs, clinical laboratory tests, electrocardiogram, and physical examination throughout the study period. The severity of AEs was classified as mild, moderate, and severe. The causality of AEs was evaluated as five categories: unrelated, unlikely, possibly, probably, or definitely related. Among them, AEs determined by the trial doctor to be possibly, probably, or definitely related to the treatment were recorded as adverse reactions (ADRs).
Statistical Analysis
Statistical description methods were used to identify the demographic characteristics, the occurrence of AEs and PK parameters. Based on the above two methods of assessing BE, if the 90% CI of the GMR (test drug/reference drug) of Cmax, AUC0-t and AUC0-∞ was within 80.00%-125.00% under fasting or fed conditions, we should consider the two preparations to be bioequivalent. In addition, we also used analysis of variance (ANOVA) to compare the PK parameters (Cmax, AUC0-t, AUC0-∞, and t1/2z) under fasting and fed conditions to explore the effects of food on the PK profiles of the test/reference drug. The Mann–Whitney U-test was used to perform statistical analysis on Tmax.
Results
Subjects
A total of 96 subjects were enrolled and randomized to one of two groups: a fasting group (n=48) and a fed group (n=48). The gender, average age, height, weight and BMI of the subjects were similar for the two parts of the study (Table 1). Of the 96 subjects, 5 subjects discontinued the study (fasting group: 3 subjects; fed group: 2 subjects). In the fasting group, one subject discontinued the study before the period 1, two subjects discontinued because of personal reasons before period 3. In the fed group, one subject withdrew because of AE and the other one discontinued because of personal reasons before period 2.
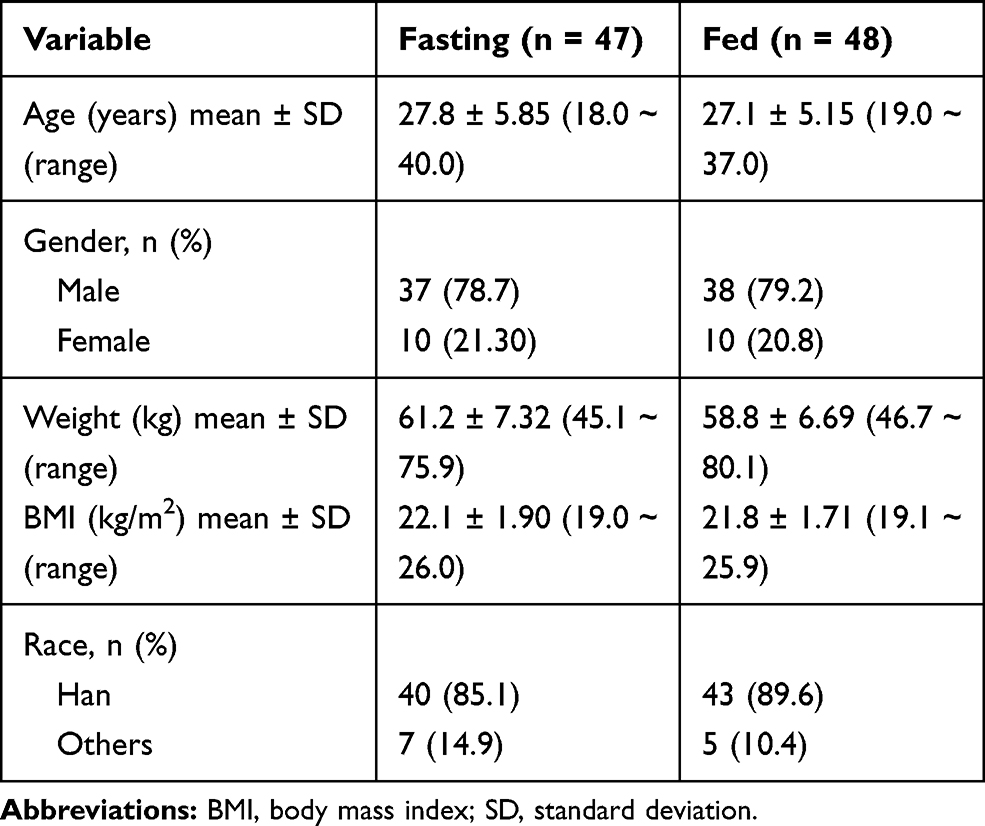 |
Table 1 Demographic Data of the Subjects Under Fasting and Fed Conditions |
Safety Evaluations
In the fasting group, 29 AEs were reported from 19 (41.3%) of 46 subjects included in the safety evaluation in the test drug part; of these, 14 AEs in 10 subjects were defined as ADRs. One subject experienced a severe AE (diarrhea), one subject experienced moderate AE (abdominalgia), and all other AEs were of mild severity. Overall, 21 AEs were reported from 16 (34.0%) of 47 subjects in the R1 part (ie, first time taking the reference drug); among them, 10 AEs in eight subjects were defined as ADRs. Except for one severe AE (presyncope), the others were mild AEs. A total of 38 AEs were identified in 24 (52.2%) of 46 subjects in the R2 part (ie, the second time taking the reference drug); 16 AEs in 15 subjects were ADRs. All AEs were of mild severity. In the fasting-condition study, elevated triglycerides, acne, diastolic hypotension and low high-density lipoprotein cholesterol were the most common AEs.
In the fed group, 16 AEs were observed in 13 (28.3%) of 46 subjects in the test drug part; among them, 11 AEs in eight subjects were judged as ADRs. All AEs were mild. Four AEs were found in 4 (8.3%) of 48 subjects in R1 part; of these, two AEs in two subjects were defined as ADRs. Except for one case of moderate AEs (urinary tract obstruction), the rest were mild. Overall, 29 AEs were observed in 20 (43.5%) of 46 subjects in R2 part, of which, 13 AEs in 12 subjects were defined as ADRs. All AEs were moderate (elevated bilirubin, urinary tract obstruction, and eczematous) and mild in severity. Common AEs in this fed-condition study were low high-density lipoprotein cholesterol, increased radial pulse and elevated bilirubin.
All ADRs were mild or moderate in severity, see Table 2 for details.
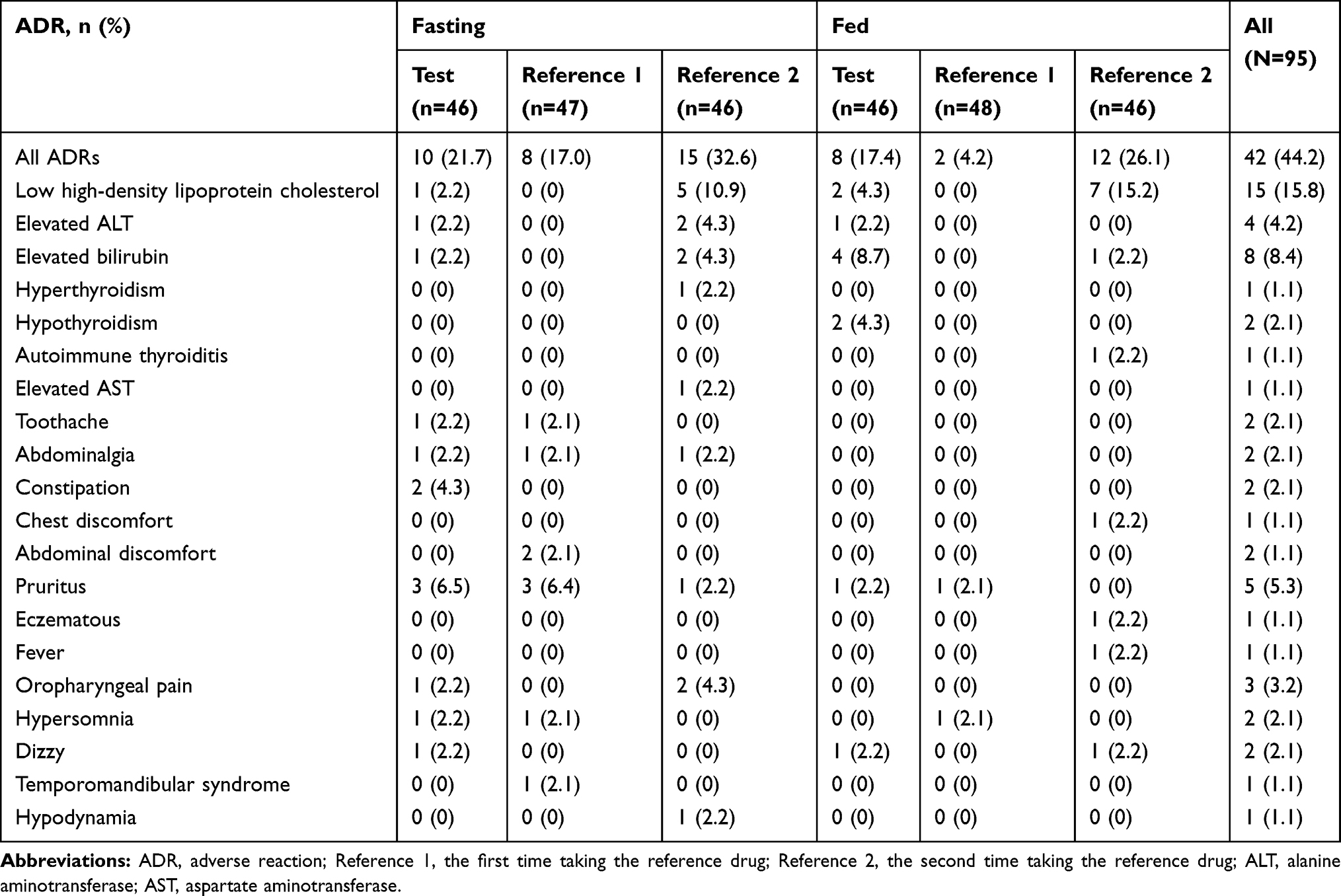 |
Table 2 Adverse Reaction of Subjects After Study Drug |
BE Studies and Effect of Food on PK
The mean concentration-time curves of the test drug was similar to the reference drug in both the fasting-condition study and the fed-condition study (Figures 1 and 2). According to the actual blood collecting time, the main PK parameters of OCA, Glyco-OCA and Tauro-OCA were calculated by non-atrioventricular model in the two groups (Table 3). There was no significant difference in the PK profile of the test drug and the reference drug, irrespective of the fasting group or the fed group.
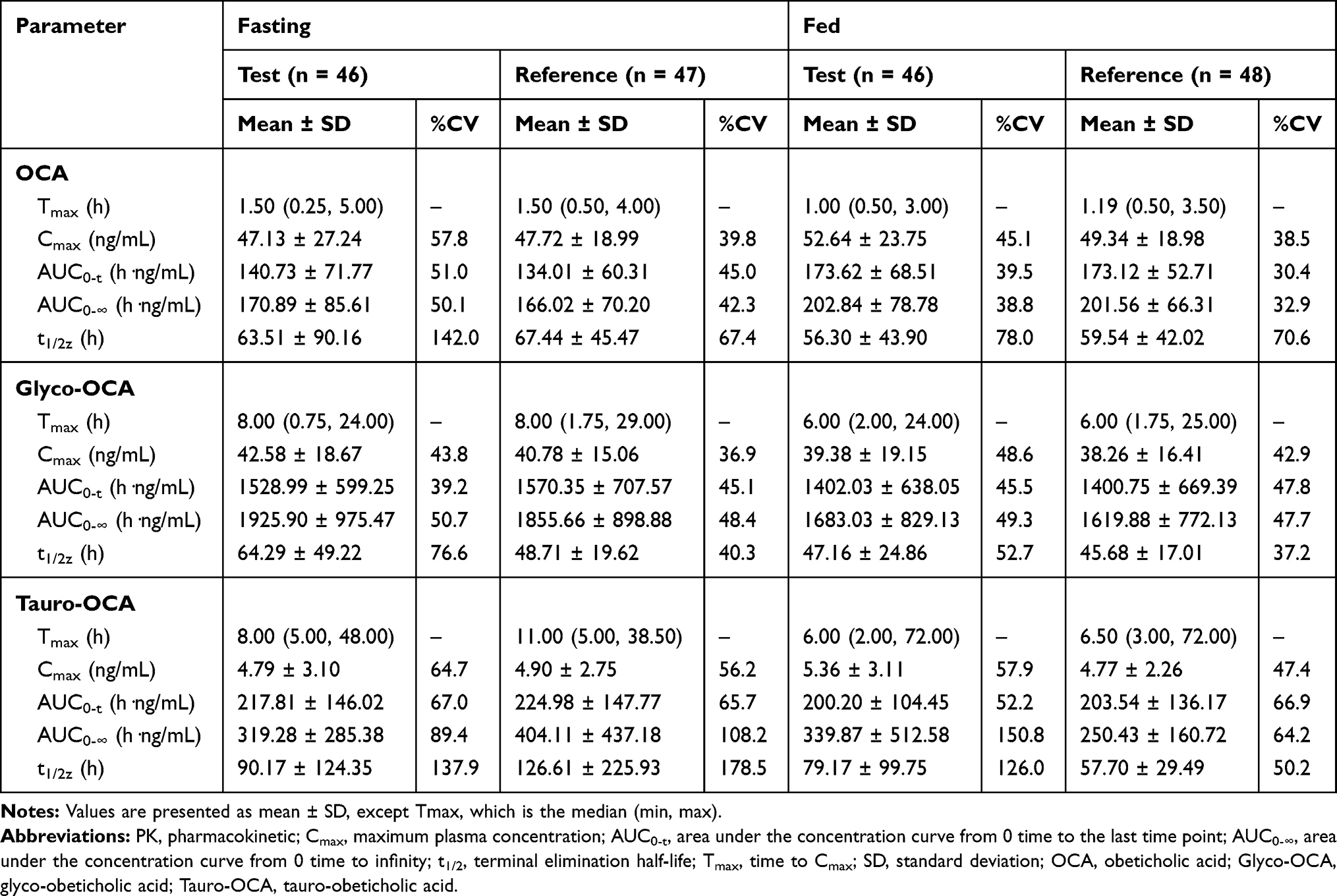 |
Table 3 PK Parameters of the Test and Reference Formulations Under Fasting And Fed Conditions |
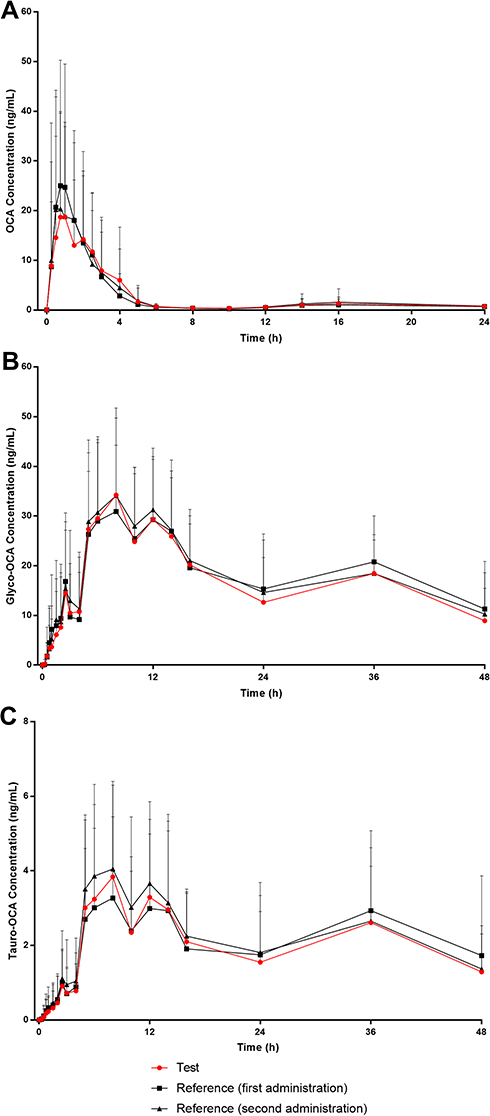 |
Figure 1 Mean plasma concentration-time curves of OCA, Glyco-OCA and Tauro-OCA after a single 10 mg dose of the test/reference drug in healthy subjects under fasting conditions. (A) OCA. (B) Glyco-OCA. (C) Tauro-OCA. Abbreviations: OCA, obeticholic acid; Glyco-OCA, glyco-obeticholic acid; Tauro-OCA, tauro-obeticholic acid. |
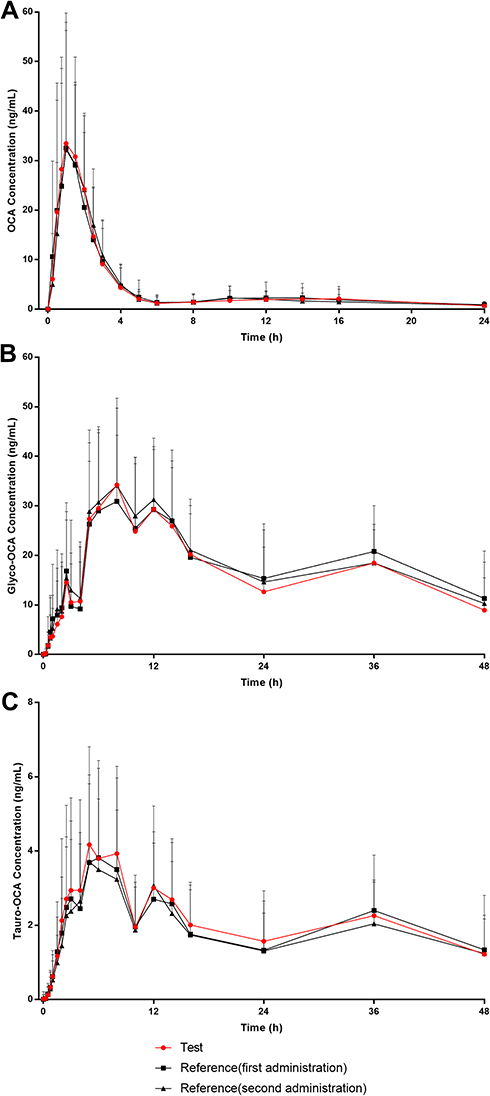 |
Figure 2 Mean plasma concentration-time curves of OCA, Glyco-OCA and Tauro-OCA after a single 10 mg dose of the test/reference drug in healthy subjects under fed conditions. (A) OCA. (B) Glyco-OCA. (C) Tauro-OCA. Abbreviations: OCA, obeticholic acid; Glyco-OCA, glyco-obeticholic acid; Tauro-OCA, tauro-obeticholic acid. |
The SWR of Cmax, AUC0-t, and AUC0-∞ of OCA in healthy subjects after a single oral administration of 10 mg test drug or reference drug under fasting condition were 0.445, 0.370, and 0.448 respectively, which were all higher than 0.294. Using the RSABE for BE assessment, the point estimation values of GMR (test drug/reference drug) of Cmax, AUC0-t and AUC0-∞ were 1.009, 1.015, and 0.983 respectively, which all fell within the interval (0.80–1.25), and the cutoff standards were −0.110,-0.078, −0.115 respectively, which were all less than 0 (Table 4).
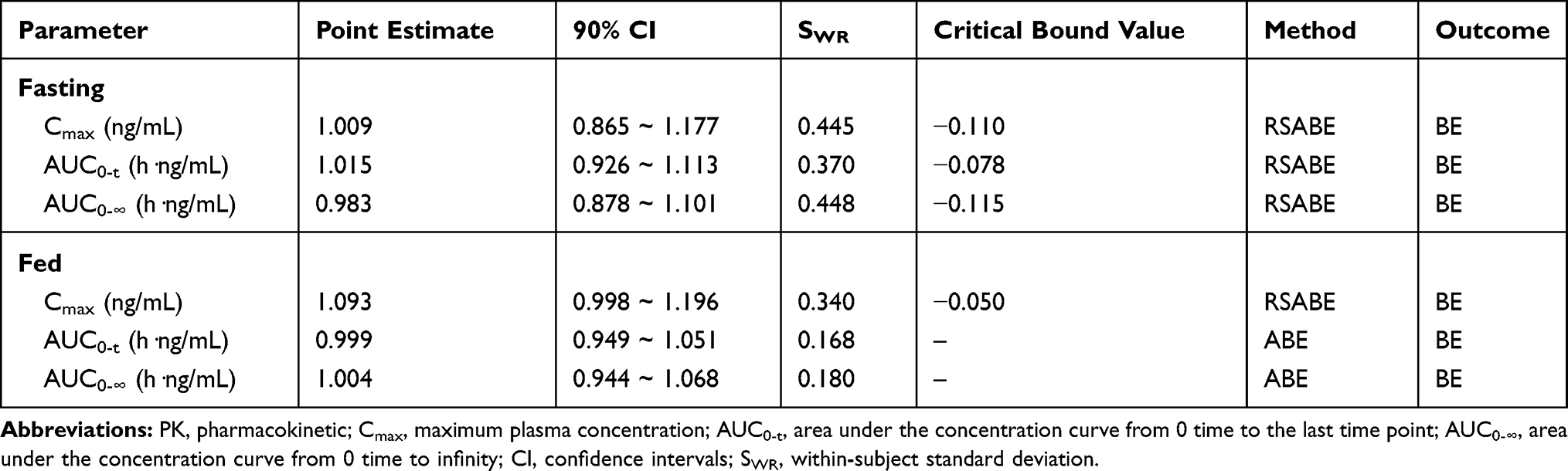 |
Table 4 BE Statistics Under Fasting and Fed Conditions by Using RSABE or ABE |
After a single 10 mg dose of the test drug or reference drug under the fed condition, the SWR of Cmax, AUC0-t, and AUC0-∞ were 0.340, 0.168, and 0.180, respectively, in which the SWR of the Cmax was higher than 0.294. The RSABE method was used to evaluate BE of the Cmax, the point estimation of the GMR (test drug/reference drug) of Cmax was 1.093, which fell within the range of 0.80–1.25, and the cutoff standard was −0.050, which was less than 0. The ABE method was used to evaluate BE of the AUC0-t and AUC0-∞, the GMR (90% CI) for AUC0-t and AUC0-∞ were 99.86% (94.85–105.13%) and 100.41 (94.39–104.81%) between the test drug and reference drug in the fed group. The corresponding 90% CI were completely contained within the equivalence margin of 80–125% (Table 4).
The ANOVA results showed that the AUC0-t of the test drug and the AUC0-t, AUC0-∞ of the reference drug were significantly different between fasting and fed conditions. In addition, food had no significant effect on other PK parameters (Table 5).
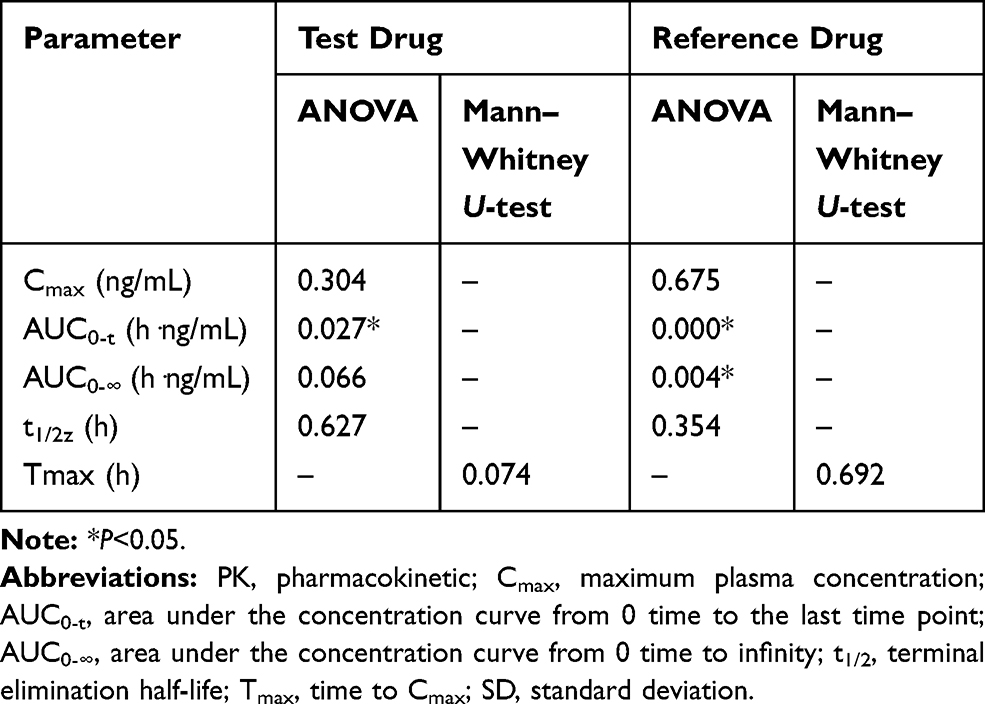 |
Table 5 Effects of Fasting and Fed Conditions on the PK Profile of Obeticholic Acid |
Discussion
This random, three-period, three-sequence, partial replicated crossover, Phase I study under fasting and fed conditions was performed to evaluate the similarity of the PK, tolerability and safety between OCA and re-OCA formulations with a 10-mg oral administration dose. OCA passes through the enterohepatic circulation and is a highly variable drug, intra-individual variability in the Cmax, AUC0-t may exceed 30%, therefore the partial replicated crossover design was selected.10,13 Subjects were given the reference drug for two cycles to obtain an accurate intra-individual coefficient of variation. As for the number of subjects, we assumed that the geometric mean ratio of AUC0-t and Cmax was from 0.95 to 1.05, setting the power (1-β) at 90%, the individual CV%=50%, and the one-sided test level α=0.05. Considering the 20% shedding rate, a total of 48 subjects were enrolled in each group.14 According to the label of the branded formulation of OCA,15 it can be taken before and after meals, so fasting and fed groups were set up for the BE study.
OCA is mainly metabolized by OCA combined with glycine or taurine in the liver and excreted into the bile. These conjugates are absorbed through the small intestine and then circulate enterohepatically.16 The main active metabolites are Glyco-OCA and Tauro-OCA.17 The AUC0-∞ of OCA could not be calculated completely because OCA is subject to enterohepatic circulation, the PK parameters of the active metabolites, Glyco-OCA and Tauro-OCA were used as supporting data in this study. The PK results showed that the test drug had similar PK characteristics to the reference drug, and overlapped with the drug-time curve of the reference drug, with similar trends under fasting and fed conditions. The BE results proved that the test drug was equivalent to the reference drug in fasting group and fed group. Moreover, food had an influence on the PK of the OCA. The AUC0-t of the test drug, and AUC0-t, AUC0-∞ of the reference drug in the fed group were higher than that of the fasting group, and the Tmax of OCA were lower, which explained why food may increase the absorption of OCA.
As for safety, the main ADRs occurred in the subjects taking 10 mg of OCA tablets (test or reference drug) under fasting condition were pruritus (7 cases), low high-density lipoprotein cholesterol (6 cases), while the main ADRs in the fed group were low high-density lipoprotein cholesterol (9 cases) and elevated bilirubin (5 cases). These main ADRs were consistent with those reported in previous studies.18–20 Comparing the incidence of ADRs between reference drug and test drug in the fasting and fed groups, there was no significant difference in the incidence between the two drugs, indicating that the safety of the two drugs was similar.
Conclusion
In this paper, we successfully verified that the test preparation is BE to the reference preparation in Chinese healthy subjects under both fasting and fed conditions. We also determined that food may affect the PK profiles of the two drugs. In addition, both preparations were safe with no serious or unexpected ADRs.
Data Sharing Statement
The raw data of this study will not be shared because of agreements.
Ethical Approval
This study was approved by the Medical Ethics Committee of the Third Xiangya Hospital of Central South University and was performed according to the ethical standards laid down in the 1964 Declaration of Helsinki.
Informed Consent
Informed consent was obtained by all individual participants included in the study.
Acknowledgments
This study was supported by the Nanjing Zhengda Tianqing Pharmaceutical Co., Ltd., the National Natural Science Foundation of China (No.81673519 and No. 81803639) and National major new drug creation project of China (No.2020ZX09201010).
Author Contributions
All authors contributed to data analysis, drafting and revising of the article, gave final approval of the version to be published, agreed on the journal to which the article was submitted and agreed to be accountable for all aspects of the work.
Disclosure
Hai-Tao Yu is an employee of Nanjing Chia Tai Tianqing Pharmaceutical Co., Ltd. The authors report no other potential conflicts of interest in this work.
References
1. Carey EJ, Ali AH, Lindor KD. Primary biliary cirrhosis. Lancet. 2015;386(10003):1565–1575. doi:10.1016/s0140-6736(15)00154-3
2. Invernizzi P, Gershwin ME. Primary biliary cirrhosis: bad genes, bad luck. Dig Dis Sci. 2012;57(3):599–601. doi:10.1007/s10620-011-1993-3
3. Gulamhusein AF, Hirschfield GM. Primary biliary cholangitis: pathogenesis and therapeutic opportunities. Nat Rev Gastroenterol Hepatol. 2020;17(2):93–110. doi:10.1038/s41575-019-0226-7
4. Tanaka A, Leung PSC, Gershwin ME. Pathogen infections and primary biliary cholangitis. Clin Exp Immunol. 2019;195(1):25–34. doi:10.1111/cei.13198
5. Hirschfield GM, Dyson JK, Alexander GJM, et al. The British Society of Gastroenterology/UK-PBC primary biliary cholangitis treatment and management guidelines. Gut. 2018;67(9):1568–1594. doi:10.1136/gutjnl-2017-315259
6. Wang L, Gershwin ME, Wang FS. Primary biliary cholangitis in China. Curr Opin Gastroenterol. 2016;32(3):195–203. doi:10.1097/mog.0000000000000257
7. Camilleri M. Bile Acid diarrhea: prevalence, pathogenesis, and therapy. Gut Liver. 2015;9(3):332–339. doi:10.5009/gnl14397
8. Pathak P, Xie C, Nichols RG, et al. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology. 2018;68(4):1574–1588. doi:10.1002/hep.29857
9. Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol. 2014;11(1):55–67. doi:10.1038/nrgastro.2013.151
10. Markham A, Keam SJ. Obeticholic acid: first global approval. Drugs. 2016;76(12):1221–1226. doi:10.1007/s40265-016-0616-x
11. Silveira MG, Lindor KD. Obeticholic acid and budesonide for the treatment of primary biliary cirrhosis. Expert Opin Pharmacother. 2014;15(3):365–372. doi:10.1517/14656566.2014.873404
12. Davit BM, Chen ML, Conner DP, et al. Implementation of a reference-scaled average bioequivalence approach for highly variable generic drug products by the US Food and Drug Administration. AAPS J. 2012;14(4):915–924. doi:10.1208/s12248-012-9406-x
13. US Food and Drug Administration. Guidance for industry bioavailability and bioequivalence studies submitted in NDAs orINDs-general considerations. Available from: https://www.fda.gov/media/88254/download.
14. Tothfalusi L, Endrenyi L. Sample sizes for designing bioequivalence studies for highly variable drugs. J Pharm Pharm Sci. 2012;15(1):73–84. doi:10.18433/j3z88f
15. Intercept Pharmaceuticals. OCALIVA (obeticholic acid) US prescribing information. Available from: http://www.interconnectsupport.com/ocaliva_pi.pdf.
16. Edwards JE, LaCerte C, Peyret T, et al. Modeling and experimental studies of obeticholic acid exposure and the impact of cirrhosis stage. Clin Transl Sci. 2016;9(6):328–336. doi:10.1111/cts.12421
17. Gai K, Huang Y, Liu B, Zhang Y. Synthesis of obeticholic acid, a farnesoid X receptor agonist, and its major metabolites labeled with deuterium. J Labelled Comp Radiopharm. 2018;61(10):799–804. doi:10.1002/jlcr.3643
18. Pate J, Gutierrez JA, Frenette CT, et al. Practical strategies for pruritus management in the obeticholic acid-treated patient with PBC: proceedings from the 2018 expert panel. BMJ Open Gastroenterol. 2019;6(1):e000256. doi:10.1136/bmjgast-2018-000256
19. Kowdley KV, Vuppalanchi R, Levy C, et al. A randomized, placebo-controlled, Phase II study of obeticholic acid for primary sclerosing cholangitis. J Hepatol. 2020;73(1):94–101. doi:10.1016/j.jhep.2020.02.033
20. Nevens F, Andreone P, Mazzella G, et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. 2016;375(7):631–643. doi:10.1056/NEJMoa1509840
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.