")
Back to Journals » The Application of Clinical Genetics » Volume 8
Bilateral granulosa cell tumors: a novel malignant manifestation of multiple endocrine neoplasia 1 syndrome found in a patient with a rare menin in-frame deletion
Authors Hall M, Innocent J, Rybak C, Veloski C, Scott W, Wu H, Ridge J, Hoffman J, Borghaei H, Turaka A , Daly M
Received 4 August 2014
Accepted for publication 7 October 2014
Published 17 February 2015 Volume 2015:8 Pages 69—73
DOI https://doi.org/10.2147/TACG.S72223
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Martin Maurer
Michael J Hall,1 Julie Innocent,2 Christina Rybak,1 Colleen Veloski,3 Walter J Scott,4 Hong Wu,5 John A Ridge,4 John P Hoffman,4 Hossein Borghaei,2 Aruna Turaka,6 Mary B Daly1
1Department of Clinical Genetics, 2Department of Medical Oncology, 3Department of Internal Medicine, 4Department of Surgical Oncology, 5Department of Pathology, 6Department of Radiation Oncology, Fox Chase Cancer Center, Philadelphia, PA, USA
Introduction: Multiple endocrine neoplasia 1 (MEN1) is a cancer syndrome resulting from mutations of the MEN1 gene. The syndrome is characterized by neoplasia of the parathyroid and pituitary glands, and malignant tumors of the endocrine pancreas. Other manifestations include benign lipomas, angiofibromas, and carcinoid tumors commonly originating in the colon, thymus, and lung. This is the first report of MEN1 syndrome manifesting as bilateral granulosa cell ovarian tumors, and which is associated with a rare intronic mutation of the MEN1 gene.
Case report: A 41-year-old woman presented with abdominal pain, increasing abdominal girth, and dysmenorrhea. Ultrasound demonstrated enlarged ovaries and uterine fibroids. After an exploratory laparotomy, she subsequently underwent bilateral salpingo–oophorectomy with hysterectomy where the pathology revealed bilateral cystic granulosa cell tumors of the ovaries. Additional workup including computed tomography imaging discovered a thymic mass, which the pathology showed was malignant, along with a pancreatic mass suspicious for a neuroendocrine tumor. Hyperparathyroidism was also discovered and was found to be secondary to a parathyroid adenoma. Genetic testing revealed an exceedingly rare mutation in the MEN1 gene (c.654 + 1 G>A).
Discussion: Mutations of the menin gene leading to MEN1 syndrome are classically nonsense or missense mutations producing a dysfunctional protein product. Recently, researchers described a novel mutation of MEN1 (c.654 + 1 G>A) in a male proband meeting the criteria for clinical MEN1 syndrome. Functional analysis performed on the stable mutant protein showed selective disruption of the transforming growth factor beta signaling pathway, yet it maintained its wild-type ability to inhibit nuclear factor kappa B and to suppress JunD transcriptional activity.
Conclusion: To our knowledge, this is the first report of MEN1 syndrome associated with bilateral granulosa cell malignancy. We postulate that this presentation may be due to the novel menin gene mutation recently described.
Keywords: menin gene, ovarian tumors, hyperparathyroidism
Introduction
Multiple endocrine neoplasia (MEN)1 is an adult-onset cancer syndrome characterized by a constellation of clinical abnormalities within the organs of the endocrine system. The MEN1 syndrome is inherited in an autosomal-dominant manner.1 The syndrome is classically characterized by nonmalignant neoplasia of the parathyroid and pituitary glands, and malignant tumors of the endocrine pancreas. Other manifestations include multiple benign lipomas and angiofibromas, uterine fibromyomas (ie, fibroids), and malignant carcinoid tumors, commonly originating in the colon, thymus, and lung. Over 98% of patients with MEN1 will develop hyperparathyroidism during their lifetime.1 This is the first report of MEN1 syndrome manifesting as bilateral granulosa cell ovarian tumors, and which is associated with a rare intronic mutation of the MEN1 gene.
Case report
A 41-year-old woman of Mexican descent presented to her gynecologist with abdominal pain, increasing abdominal girth, and dysmenorrhea. Ultrasound demonstrated markedly enlarged ovaries, as well as asymptomatic uterine fibroids. She underwent an exploratory laparotomy and, subsequently, a bilateral salpingo–oophorectomy with hysterectomy in July 2010. The pathology revealed bilateral cystic granulosa cell tumors of the ovaries (Figure 1), as well as a uterine atypical smooth muscle tumor of uncertain malignant potential (Figure 2). Radiographic preoperative imaging of the chest (computed tomography [CT]) also identified a 4.0×4.0 cm anterior mediastinal mass, and subsequent resection revealed both a well-differentiated neuroendocrine carcinoma of the thymus and a type B3 thymoma.
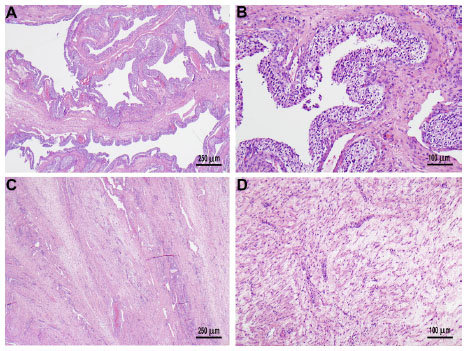 | Figure 1 Sectioning of the bilaterally enlarged ovaries revealed multiloculated cysts containing clear serous fluid. |
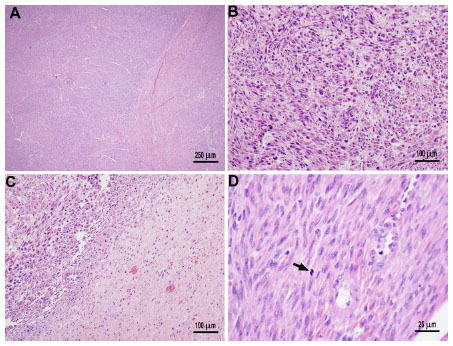 | Figure 2 The uterus had multiple intramural leiomyomas. |
Her prior medical history was remarkable for the resection of several subcutaneous lipomas over the past 4–5 years. Concurrent with the diagnosis of a mediastinal neuroendocrine carcinoma, primary hyperparathyroidism was observed following hypercalcemia (initial serum calcium level: 11.6 mg/dL; laboratory normal range: 8.4–10.2 mg/dL) in the setting of elevated parathyroid hormone levels (initial level: 109 pg/mL; laboratory normal range: 12–88 pg/mL). Ultrasound evaluation of the thyroid and parathyroid glands revealed a suspicious thyroid lesion, and the patient was referred to a head and neck surgeon. The pathology from a total thyroidectomy, central node dissection, and subtotal (3.5 glands) parathyroidectomy in November 2012 was consistent with a stage III papillary thyroid carcinoma with local lymph node involvement, as well as a right parathyroid adenoma. Finally, during the course of her evaluation, an enhancing hypervascular mass measuring 1.7×2.4 cm was identified in the body of the pancreas on contrast-enhanced CT scan. While currently unbiopsied, this mass has been evaluated by a pancreatobiliary surgeon and is considered to be radiographically highly suspicious for a pancreatic neuroendocrine tumor. Serum gastrin, chromogranin A, and glucagon levels were within normal limits. Of important note, magnetic resonance imaging of the brain revealed a normal pituitary gland; serum prolactin and other pituitary hormones were normal.
At the time of the initial evaluation by the clinical genetics service in May 2012, the patient described no additional manifestations of MEN1 syndrome (see Table 1). She was married with two healthy children, and worked full time as a Spanish interpreter. She denied any past or current tobacco use or known chemical exposures. Her family history was significant for a father and paternal uncle with prostate cancer at ages 65 years and 50 years, respectively. On the paternal side, a cousin had been diagnosed with acute lymphocytic leukemia at the age of 6 years. On the maternal side, she could recall only a cousin with a “stomach mass”. She has more than 50 first cousins, many of whom lived in Mexico, whose medical histories were unknown to her.
 | Table 1 Review of pertinent positive and negative symptoms in this patient’s personal and family history |
Due to her constellation of findings, blood samples were sent for genetic testing to Ambry Genetics (Aliso Viejo, CA, USA). Through gene sequence and deletion/duplication analysis of the MEN1 gene, the patient was found to carry a mutation in the MEN1 gene (c.654 + 1 G>A). The c.654 +1 G>A mutation is located within intron 3 of MEN1. The mutant sequence differs from the wild type by an in-frame deletion of 35 amino acids.2 The mutation has been reported only once previously in a family where the proband presented with hyperparathyroidism and a gastrinoma. A paternal history of hyperparathyroidism was also reported in this family.
Discussion
The MEN syndromes consist of two subtypes: MEN type 1 (MEN1) and MEN type 2 (MEN2). The MEN1 syndrome is inherited in an autosomal-dominant fashion and results from germ-line mutations of the MEN1 gene. It is estimated that up to one-third of cases of MEN1 syndrome may be due to new (de novo) mutations in MEN1, and are therefore not associated with a family history of endocrine disorders or tumors. The MEN1 gene is located on the long arm of chromosome 11 (11q13) and encodes for the 610 amino acid menin protein.3 In addition to being associated with neoplasia of the parathyroid glands, pituitary glands, and malignant tumors of the endocrine pancreas, cases of patients with MEN1 with coincident papillary thyroid carcinoma have also been reported.4 MEN2 syndrome is caused by mutations in the RET gene and is subcategorized into MEN2A, MEN2B, and familial medullary thyroid cancer. Hallmarks of MEN2A include medullary thyroid cancer, pheochromocytoma, and hyperparathyroidism. MEN2B is associated with the earlier onset of medullary thyroid cancer, pheochromocytoma, the absence of hyperparathyroidism, and the presence of multiple mucosal neuromas. Familial medullary thyroid cancer confers a strong predisposition to medullary thyroid cancer, but it is not associated with other endocrine neoplasms.
The menin protein plays a critical role as a tumor suppressor. It is involved in the regulation of gene transcription, apoptosis, and genome stability. The function of menin is complex, and discovering its mechanism of action and its role in tumorigenesis in the MEN1 syndrome has largely been through the study of its interaction with nuclear and cytosolic proteins.5,6 There are several key menin-interacting proteins through which menin regulates cellular proliferation – one being transforming growth factor beta (TGF-β). TGF-β is one of a super-family of transcription factors that regulates cellular growth through a SMAD-mediated signaling pathway that is linked to nuclear factor kappa B (NF-kb) and JunD, a member of the activator protein-1 family and a transcription factor, which has tumor suppressor properties when bound to menin.2 Mutations of MEN1 have been found to disrupt these important menin/protein interactions; however, of the hundreds of known MEN1 gene mutations, only the newly discovered mutant by Canaff et al2 has been found to maintain some function of the wild-type gene.
Mutations of menin
There are over 500 known mutations of the MEN1 gene, the majority of which are nonsense mutations resulting in a truncated protein product.2,7 Approximately 20% are missense mutations, and the mutant proteins are targeted for degradation.7 In 2012, Canaff et al2 described a novel intronic mutation of MEN1 (c.654 + 1 G>A) in a male proband meeting criteria for clinical MEN1 syndrome. Surprisingly, functional analysis performed on the mutant protein showed that it maintained its wild-type ability to inhibit NF-kb and to suppress JunD transcriptional activity.2 However, further studies by this group showed that the novel menin mutant lacked the ability to bind to the SMAD3 transcription factor, leading to the specific disruption of the TGF-β signaling pathway and the downstream downregulation of cellular proliferation. We postulate that aberrant TGF-β signaling also underlies the novel clinical phenotype of the bilateral granulosa cell tumors seen in our patient.
TGF-β and its association with granulosa cell tumors
TGF-β inhibits the growth of many epithelial, endothelial, fibroblast, neuronal, lymphoid, and hematopoietic cells. The SMAD family proteins are intracellular proteins that transduce extracellular signals from TGF-β ligands to the nucleus, where they activate downstream gene transcription.8 TGF-β has also been shown to inhibit parathyroid cellular proliferation and parathyroid hormone production as part of homeostatic regulation.9 Kaji et al9 demonstrated that menin interacts with the TGF-β pathway by binding to SMAD3. This interaction was found to result in a decrease in cellular proliferation. Experiments in cultured human parathyroid cells demonstrated an increase in cellular proliferation and a loss of TGF-β-mediated inhibition when the mutant menin protein did not bind SMAD3,9,10 providing additional evidence of the role of TGF-β in the pathophysiology of MEN1 syndrome.
Aberrations of the TGF-β signaling pathway have been identified in tumors from a variety of tissues including breast and gastrointestinal tumors, yet no such finding to date has been discovered in granulosa cell tumors.11 Granulosa cell tumors are rare sex cord–stromal neoplasms.12,13 They comprise approximately 2%–5% of ovarian tumors and are divided between adult and juvenile forms, with the average ages of onset being 50 years and 10–20 years, respectively. Bilateral disease is seen in only 2%–8% of cases.12,13 The role of TGF-β in the pathogenesis of granulosa cell tumors has yet to be fully elucidated. However, research from Middlebrook et al12 showed that double-knockout mice for two SMAD transcription factors developed granulosa cell tumors. Immunohistochemical staining of these tumors showed evidence of disruption of TGF-β signaling.12 Hence, these findings suggest that the TGF-β signaling pathway may be important to the pathogenesis of granulosa cell tumors.
Conclusion
The rare bilateral ovarian granulosa cell tumors diagnosed in our patient with florid MEN1 syndrome due to a rare intronic mutation of the MEN1 gene (c.654 + 1 G>A) represents a novel phenotypic manifestation of the disease. To our knowledge, this is the first report of a young female patient with MEN1 syndrome associated with bilateral granulosa cell malignancy, and it is only the second case where this mutation has been identified. Further support for the pathogenicity of this mutation comes from the American College of Medical Genetics guidelines, which suggest that all nonconservative missense mutations are more likely to be disease-causing, secondary to erroneous splicing.14 This case serves to further demonstrate the complexity of the phenotypic variation of the MEN1 syndrome. Based on elegant work by Canaff et al2 and Middlebrook et al,12 we hypothesize that the rarity of our patient’s MEN1 mutation and its atypical impact on menin protein function explain the unprecedented clinical presentation. Further research and investigation of our patient’s granulosa cell tumor tissue is needed to understand whether selective disruption of TGF-β signaling is driving the aggressive malignant phenotype observed in this case.
Acknowledgments
This research was supported by the Fox Chase Cancer Center Core Grant (P30-006927-45). Dr Hall is a recipient of a Mentored Research Scholar Grant from the American Cancer Society (MRSG-07-232-01-CPHPS).
Disclosure
The authors report no conflicts of interest in this work.
References
Lemmens I, Van de Ven WJ, Kas K, et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Hum Mol Genet. 1997;6(7):1177–1183. | |
Canaff L, Vanbellinghen JF, Kaji H, Goltzman D, Hendy GN. Impaired transforming growth factor-β (TGF-β) transcriptional activity and cell proliferation control of a menin in-frame deletion mutant associated with multiple endocrine neoplasia type 1 (MEN1). J Biol Chem. 2012;287(11):8584–8597. | |
Guru SC, Goldsmith PK, Burns AL, et al. Menin, the product of the MEN1 gene, is a nuclear protein. Proc Natl Acad Sci U S A. 1998;95(4):1630–1634. | |
Kim HJ, Park JS, Kim CS, et al. A case of multiple endocrine neoplasia type 1 combined with papillary thyroid carcinoma. Yonsei Med J. 2008;49(3):503–506. | |
Balogh K, Rácz K, Patócs A, Hunyady L. Menin and its interacting proteins: elucidation of menin function. Trends Endocrinol Metab. 2006;17(9):357–364. | |
Poisson A, Zablewska B, Gaudray P. Menin interacting proteins as clues toward the understanding of multiple endocrine neoplasia type 1. Cancer Lett. 2003;189(1):1–10. | |
Yaguchi H, Ohkura N, Takahashi M, Nagamura Y, Kitabayashi I, Tsukada T. Menin missense mutants associated with multiple endocrine neoplasia type 1 are rapidly degraded via the ubiquitin-proteasome pathway. Mol Cell Biol. 2004;24(15):6569–6580. | |
Massagué J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. | |
Kaji H, Canaff L, Lebrun JJ, Goltzman D, Hendy GN. Inactivation of menin, a Smad3-interacting protein, blocks transforming growth factor type beta signaling. Proc Natl Acad Sci U S A. 2001;98(7):3837–3842. | |
Sowa H, Kaji H, Kitazawa R, et al. Menin inactivation leads to loss of transforming growth factor beta inhibition of parathyroid cell proliferation and parathyroid hormone secretion. Cancer Res. 2004;64(6):2222–2228. | |
Massagué J. TGFbeta in Cancer. Cell. 2008;134(2):215–230. | |
Middlebrook BS, Eldin K, Li X, Shivasankaran S, Pangas SA. Smad1-Smad5 ovarian conditional knockout mice develop a disease profile similar to the juvenile form of human granulosa cell tumors. Endocrinology. 2009;150(12):5208–5217. | |
Schumer ST, Cannistra SA. Granulosa cell tumor of the ovary. J Clin Oncol. 2003;21(6):1180–1189. | |
Richards CS, Bale S, Bellissimo DB, et al; Molecular Subcommittee of the ACMG Laboratory Quality Assurance Committee. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008;10(4):294–300. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.