")
Back to Journals » Cancer Management and Research » Volume 11
Beyond Ruxolitinib: Fedratinib and Other Emergent Treatment Options for Myelofibrosis
Authors Bewersdorf JP , Jaszczur SM, Afifi S , Zhao JC , Zeidan AM
Received 12 November 2019
Accepted for publication 13 December 2019
Published 24 December 2019 Volume 2019:11 Pages 10777—10790
DOI https://doi.org/10.2147/CMAR.S212559
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Yong Teng
Jan Philipp Bewersdorf,1 Sara Mohamed Jaszczur,2 Salma Afifi,2 Jennifer C Zhao,2 Amer M Zeidan1,3
1Department of Internal Medicine, Section of Hematology, Yale School of Medicine, New Haven, CT, USA; 2Department of Pharmacy, Yale New Haven Hospital, New Haven, CT, USA; 3Cancer Outcomes, Public Policy, and Effectiveness Research (COPPER) Center, Yale University, New Haven, CT, USA
Correspondence: Amer M Zeidan
Section of Hematology, Department of Internal Medicine, Yale University, 37 College Street, PO Box 208028, New Haven, CT 06520-8028, USA
Tel +1 203-737-7103
Fax +1 203-785-7232
Email [email protected]
Abstract: Myelofibrosis (MF) is a myeloproliferative neoplasm characterized by clonal proliferation of differentiated myeloid cells leading to bone marrow fibrosis, cytopenias and extramedullary hematopoiesis. In late 2019, the FDA approved the highly selective JAK2 inhibitor, fedratinib, for intermediate-2 or high-risk primary or secondary MF, making it the second drug approved for MF after ruxolitinib, a JAK1/2 inhibitor, which was approved for MF in 2011. The approval of fedratinib was based on phase II trials and the phase III JAKARTA trial, in which the drug significantly reduced splenomegaly and symptom burden compared to placebo, including some patients previously treated with ruxolitinib. The main side effects of fedratinib include anemia, gastrointestinal symptoms, and elevations in liver transaminases. Fedratinib also has ablack box warning for encephalopathy, although this occurred only in about 1% of the treated patients, most of which were ultimately felt not to represent Wernicke’s encephalopathy. Nonetheless, monitoring of thiamine levels and supplementation are recommended especially in high-risk patients. This concern has led to a prolonged clinical hold and delayed the drug approval by several years during which the drug exchanged manufacturers, highlighting the need for meticulous investigation and adjudication of serious, but rare, adverse events in drug development that could end up preventing drugs with favorable risk/benefit ratio from being approved. In this review, we discuss the pharmacokinetic data and efficacy, as well as the toxicity results of clinical trials of fedratinib. We also review ongoing trials of JAK inhibitors in MF and explore future treatment options for MF patients who are refractory to ruxolitinib.
Keywords: fedratinib, ruxolitinib, myelofibrosis, MF, JAK2
Introduction
Myeloproliferative neoplasms (MPNs) are clonal, BCR-ABL1 negative hematopoietic diseases that are characterized by abnormal proliferation of terminally differentiated myeloid cells and comprise essential thrombocytosis (ET), polycythemia vera (PV), and primary myelofibrosis (PMF).1–3 These entities occur on a wide spectrum of clinical presentations ranging from asymptomatic elevations of hemoglobin/hematocrit and platelet count to progressive bone marrow failure and a variable risk of transformation to acute myeloid leukemia (AML).2 While the life-expectancy in patients with ET can be normal in (very) low-risk disease, many ET and PV patients have an inferior survival compared to age-matched and sex-matched controls mainly due to thromboembolic events.4,5 Outcomes in patients with myelofibrosis are significantly poorer with progressive bone marrow failure and progression to AML as drivers for morbidity and mortality.6–8 In a large cohort study of 1054 patients with myelofibrosis, the median OS was 69 months but varied substantially based on patient (age, constitutional symptoms) and disease factors (hemoglobin, circulating blast, and leukocyte levels).9
Clonal expansion of hematopoietic stem cells driven by somatic mutations in Janus kinase 2 (JAK2), calreticulin (CALR), and myeloproliferative leukemia virus oncogene (MPL) have been classically associated with MPNs.3 However, patients with a triple-negative status can be seen, which has been associated with an adverse prognosis in patients with PMF.8,10 Mutational status in these genes has also been shown to be a prognostic marker for AML progression and overall prognosis.8,11 With advances in diagnostic techniques, additional high-risk mutations with prognostic significance such as ASXL1, SRSF2, EZH2, and IDH1/2 have been identified especially in PMF and might have a role in risk stratification and treatment selection in MPN patients.12–15
The JAK/signal transducer and activation of transcription (STAT) pathway is a key regulator of cytokine receptor signaling and plays a critical role in hematopoiesis and immune responses.16,17 The JAK2 V617F variant, which is located on exon 14 and induces constitutive activation of STAT, is identified in 95% of patients with PV (and post-PV MF) and 50–60% of patients with primary MF and ET (and post-ET MF) making it the most prevalent mutation in MPNs.18,19 This gain-of-function mutation leads to the constitutive activation of the tyrosine kinase domain of JAK2 which underlies the hypersensitivity of erythroid precursor cells to hematopoiesis-stimulating cytokines and erythropoietin-independent proliferation.16 In PV patients, the presence of a JAK2 V617F mutation has been linked to a higher rate of thromboembolic and hemorrhagic complications as well as progression to secondary myelofibrosis and AML.19 Of note, small clonal populations harboring JAK2 V617F mutations are frequently encountered in healthy individuals, but variant allele frequencies of 50% or greater are typical for MPNs.3,11,20 In up to 10% of PV patients, JAK2 mutations affecting exon 12 are encountered which presents with a different phenotype (higher hemoglobin and lower platelet levels) but does not appear to have an adverse prognosis with regard to thromboembolic complications or progression to myelofibrosis and AML.21,22
Given its high prevalence and pathogenetic significance, aberrant JAK2 signaling has been a promising target for drug development. Ruxolitinib, an oral JAK1/2 inhibitor, was the first agent approved for the treatment of MF after it was shown to decrease spleen size and disease-related symptoms compared to placebo in the double-blind COMFORT-I trial of 309 patients with intermediate-2 or high-risk myelofibrosis.23 Significant improvements in symptom burden and splenomegaly for treatment with ruxolitinib compared to physician choice have also been reported (COMFORT-II).24 Five-year follow-up data of these trials have not only shown durable responses to ruxolitinib but even a significant survival benefit with about 30% relative risk reduction for death compared to best available treatment after 5 years of follow-up (HR, 0.69; 95% CI, 0.50–0.96; P = 0.025).25,26 However, thrombocytopenia and anemia can be dose- or even treatment-limiting adverse events and patients who discontinue ruxolitinib have dismal outcomes, making this situation an area of significant unmet need.23,24
Fedratinib is a selective oral JAK2-kinase inhibitor that was recently FDA approved for adults with intermediate-2 or high-risk primary or secondary MF based on favorable results from placebo-controlled, randomized phase II and III clinical trials showing significant symptom improvement and reduction in spleen size.27–30 This makes fedratinib the second drug to be approved in this disease state with additional clinical efficacy in patients who were ruxolitinib-resistant or intolerant.27 In this article, we review the pharmacokinetics, safety, and efficacy of fedratinib in MF and discuss treatment options for ruxolitinib-refractory patients. Other JAK1/2 inhibitors such as pacritinib and momelotinib are also in advanced clinical development and could add to the treatment armamentarium for MF should they be approved as well.31,32 Table 1 summarizes major clinical trials of JAK1/2 inhibitors in MF.
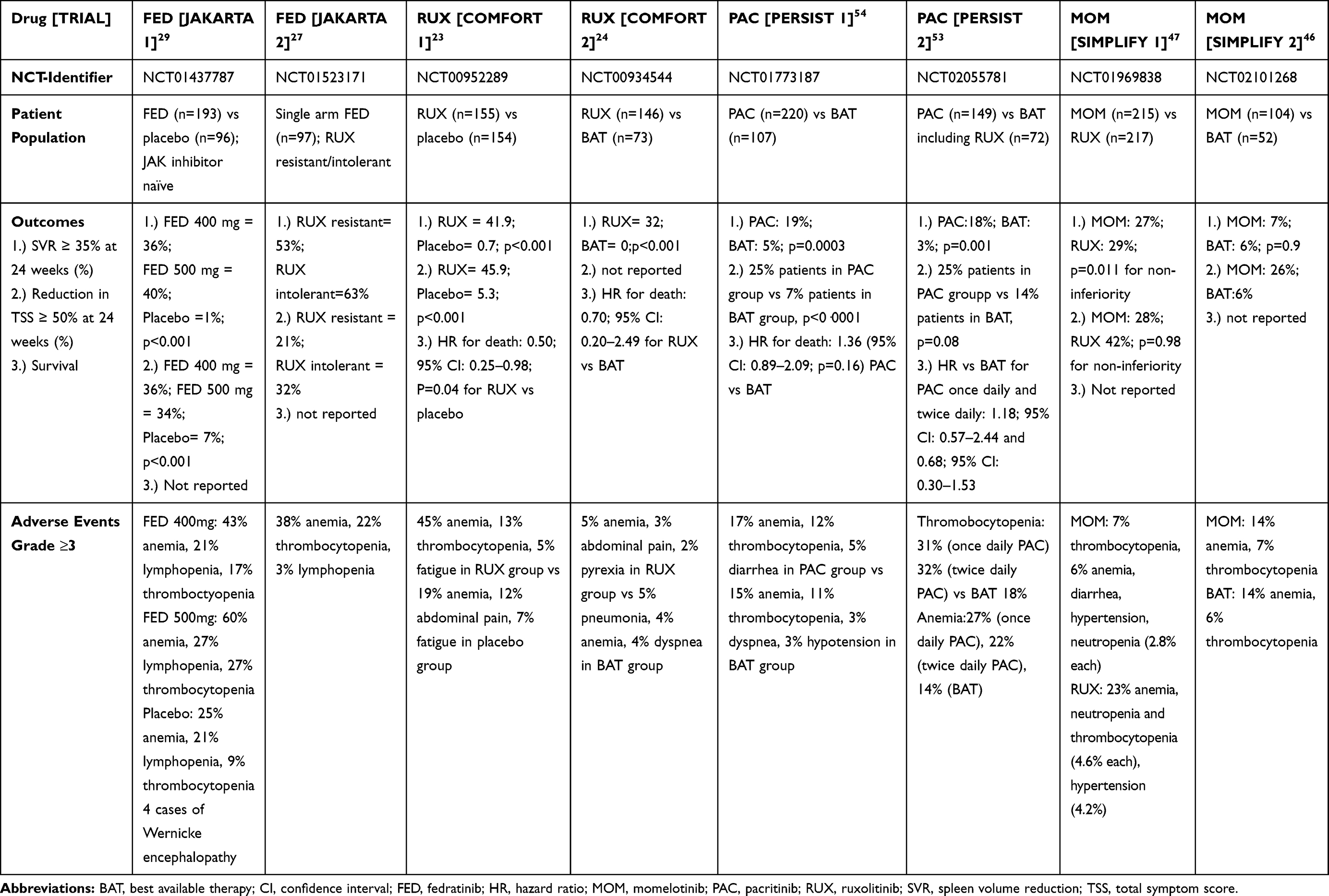 |
Table 1 Comparison of Selected Clinical Trials on JAK Inhibitors for Treatment of Myelofibrosis |
Pharmacokinetics and Pharmacodynamics
Fedratinib competes with wild type JAK2 as well as the mutated form JAK2 V617F for ATP binding, which results in inhibition of JAK2 activation and inhibition of the JAK-STAT signaling pathway. This pathway becomes overactive in patients with MF due to JAK2, CALR or MPL mutations. Fedratinib has also been noted to inhibit FMS-like tyrosine kinase 3 (FLT3) and is reducing B- and T-lymphocyte-mediated cytokine production.27–30,33
Pharmacokinetics (PK) of fedratinib have been characterized in both healthy volunteers and patients with MF.28,30,34,35 It has been shown to have rapid absorption after oral administration with peak plasma concentrations reached within 0.5–4 hrs.28,30 Fedratinib’s PK can be described by a two-compartment model with first-order absorption incorporating a lag time and first-order elimination.36 Serum levels of fedratinib increased linearly for doses of 200 mg and above. Plasma fedratinib levels reached steady state within 15 days of once-daily dosing.28 Mean terminal half-life of fedratinib was 62–78 hrs at a single dose of 300–680 mg in healthy patients. Following a single 300 mg fedratinib dose, the area under the curve (AUC) increased by 1.5-fold in subjects with moderate renal function impairment (creatinine clearance [CrCl] 30 to 59 mL/min) and 1.9-fold in subjects with severe (CrCl 15 to 29 mL/min) impairment, compared to that in subjects with normal renal function (CrCl ≥90 mL/min). Therefore, dose adjustments based on renal function are recommended in patients with a CrCl <30 ml/min. Food intake had minimal impact on the PKs of fedratinib including terminal half-life and AUC. In addition, the tolerability (ie, gastrointestinal toxicities) of this drug was improved when fedratinib was taken following a high-fat breakfast.35 No clinically meaningful effect on fedratinib PK was observed with regard to body weight, age, race, sex and mild/moderate hepatic impairment.36
Although various differences have been noted among patients with MPNs in various studies, phosphorylation of STAT3 and STAT5 has been linked to the presence but not to the allele burden in JAK2 V617F-positive patients and to correlate with disease severity.37–39 In a phase 2 trial, levels of phosphorylated STAT3 (pSTAT3) were correlated with fedratinib doses and pSTAT3 levels were reduced relative to baseline independent of the fedratinib dose.28 The greatest reductions on day 1 occurred 2 hrs post-dose, corresponding to the tmax of fedratinib. Inhibition of pSTAT3 then decreased at 6 and 24 hrs, concurrently with reduced fedratinib exposure. This supports that fedratinib acts via suppression of STAT3 signaling and patients with greater levels of pSTAT3 inhibition were more likely to achieve a spleen response.28
In the phase I trial, evaluating fedratinib use in MF, although the maximum tolerated dose (680 mg/day) was the most efficacious dose, it was also associated with the highest incidence of adverse events.30 Therefore, a lower starting dose (400–500 mg) was used in phase II and III trials to provide an optimal risk/benefit balance.
Efficacy
Phase II Clinical Trial Data
One phase II dose-ranging study included adult patients with PMF, post-PV MF or post-ET MF who were intermediate-2 or high risk.28 Patients were enrolled regardless of JAK2 mutational status. Patients who received prior treatment with a JAK2 inhibitor or any chemotherapy were excluded. Patients were randomized 1:1:1 into 3 dose cohorts (300 mg, 400 mg, and 500 mg). Those in the 300 mg group were permitted to undergo dose escalation to 500 mg if there was a lack of efficacy response and no safety concerns were identified. The primary efficacy endpoint was spleen volume reduction (SVR) by ≥35% at 12 weeks (after 3 cycles) on MRI compared to baseline. Thirty-one patients were enrolled and the rate of SVR at 12 weeks was 30.3%, 33.1%, and 43.3% in the 300, 400 and 500 mg dose groups, respectively. Results were similar between 12 and 24 weeks and durable with a median duration of response of ~250 days independent of the fedratinib dose.28 However, by 24 weeks, spleen size reduction was greater in both the 400 mg and 500 mg groups, compared to 300 mg although the small sample size precluded any formal statistical testing.28 Additionally, fedratinib led to an improvement in symptom burden as measured by the modified Myelofibrosis Symptom Assessment Form (MPN-SAF) at 12 and 24 weeks with no apparent relationship to the fedratinib dose.28 In subsets of patients with baseline leukocytosis and thrombocytosis, treatment with fedratinib led to a normalization of peripheral cell counts in up to 44% of patients.28 Based on these results, the 400 mg and the 500 mg doses were chosen to be further evaluated in the phase III trial.
The single-arm, open-label, non-randomized, multi-center phase II trial [JAKARTA-2] of 97 patients with intermediate-1, intermediate-2, or high-risk PMF, post-PV MF, or post-ET MF who were either resistant to or unable to tolerate ruxolitinib evaluated fedratinib 400 mg once daily for six 28-day cycles.27 In order to enroll in the trial, patients had to be previously treated with ruxolitinib for at least 14 days. Among 83 evaluable patients, 46 patients (55%) achieved a spleen response (≥35% based on CT-/MRI-imaging) with comparable efficacy in ruxolitinib-refractory (29 out of 55 patients; 53%) and ruxolitinib-intolerant patients (17 out of 27 patients; 63%).27 Ninety patients were evaluated for symptom response and 26% (23/90) achieved a 50% or greater reduction in total symptom score (TSS) after 6 cycles of treatment. While ruxolitinib-resistant patients seem to have a lower response rate to fedratinib, it is important to note that the study was not powered to detect differences between ruxolitinib-refractory and ruxolitinib-intolerant patients.27 A caveat in this trial is that the definitions of ruxolitinib failure or intolerance were not prespecified in the study protocol. In the absence of standardized criteria, patients who discontinued ruxolitinib at any dose after at least 2 weeks of treatment were eligible for the trial. However, both the median duration of prior ruxolitinib treatment of 10.25 months and the fact that 71% of patients had initially received the target dose of 30–40 mg daily suggest that the majority of patients in JAKARTA-2 had indeed received a sufficient therapeutic trial and ruxolitinib was not discontinued prematurely.27
As one of the major criticisms of the JAKARTA-2 trial was the definition of ruxolitinib resistance and intolerance, a recently published update from this trial using more stringent definitions as inclusion criteria has been presented recently.40 Relapsed/refractory disease was defined as at least 3 months of prior treatment with ruxolitinib with an initial response, while ruxolitinib intolerance was defined as the development of RBC transfusion requirements or grade ≥3 cytopenias after at least 28 days of prior ruxolitinib treatment.40 In this patient cohort, fedratinib achieved a spleen volume reduction in about 30% of patients with relapsed/refractory disease or ruxolitinib intolerance.40
Since thrombocytopenia can be a dose-limiting adverse event to treatment with ruxolitinib, evaluating fedratinib as an alternative in patients with baseline thrombocytopenia is of significant clinical interest. Fifteen percent of patients in the JAKARTA-I and 34% of patients in the JAKARTA-II trial had baseline platelet counts of 50 to <100×109/L.41 At least 50% symptom reduction was achieved in 31% and 39% in the patient cohorts with baseline platelet counts of 50 to <100×109/L in JAKARTA-I and JAKARTA-II, respectively. SVR ≥35% were seen in 36% in both trials.41 While grade ≥3 bleeding events were more common in the patients with baseline platelet counts of 50 to <100×109/L, no new safety signals were reported.41 Both publications suggest that fedratinib can be a safe and effective alternative in patients who failed or were unable to tolerate ruxolitinib.
Data from Phase III Clinical Trials
Based on the clinical benefit demonstrated in those early phase trials, the safety and efficacy of fedratinib in patients with primary or secondary (post PV or ET) MF was evaluated in a multi-center, randomized, double-blind, placebo-controlled phase III clinical trial (JAKARTA-).29 Patients were randomized to receive oral fedratinib 400 mg, 500 mg or placebo once daily for at least six consecutive 28-day treatment cycles with continuation until disease progression/relapse or excess toxicity. Of the total 289 patients who were enrolled at the data analysis cutoff date, 64 (67%), 59 (61%) and 1 (1%) of patients in the fedratinib 400 mg, 500 mg, and placebo groups were still receiving treatment as originally assigned, respectively. A total of 70 patients crossed over from placebo to fedratinib after 24 weeks of treatment or at the time of disease progression.29
The primary endpoint was the proportion of patients who achieved at least a 35% reduction in spleen volume on CT or MRI imaging from baseline to the end of cycle 6 (week 24) and confirmed 4 weeks later. Secondary endpoints included the proportion of patients with at least a 50% reduction in TSS based on six key symptoms using the modified MPN-SAF as well as 35% SVR regardless of confirmation. The primary endpoint was reached in 35 patients (36%) in the fedratinib 400 mg group and 39 patients (40%) in the fedratinib 500 mg group. This was significantly higher than the placebo group, in which a spleen response was achieved in 1 patient (1%) [p <0.001].29 A spleen response without confirmation at 4 weeks was seen in 47%, 49% and 1% of the fedratinib 400 mg, 500 mg and placebo group, respectively.29 Responses were higher in the fedratinib groups compared with placebo irrespective of patient characteristics including baseline platelet count, disease subtype, risk category and JAK 2 mutation status.29 A reduction of at least 50% in TSS from baseline to week 24 was noted in 36% in the fedratinib 400 mg group, 34% in the 500 mg group and 7% in the placebo group, respectively.29 Based on the efficacy results from these phase II and phase III trials, fedratinib became the second FDA approved treatment for intermediate-2 or high-risk primary or secondary MF. Table 1 is comparing reported results to date from landmark clinical trials for ruxolitinib, momelotinib, pacritinib and fedratinib in patients with MF.
Safety and Adverse Event Profile of Fedratinib
Phase II studies showed an adverse effect (AE) profile similar to that seen in phase I trials.27,28,30 The most common AEs reported in the phase II dose-ranging study were gastrointestinal (GI) events, fatigue, peripheral edema, dyspnea, and treatment-related anemia.28 However, no grade 3/4 amylase elevations were observed at the 400 or 500 mg dose, while they were previously observed at the 800 mg dosing during phase I. All patients had at least one treatment-emergent adverse event (TEAE). All patients in the 500 mg group had grade 3/4 TEAEs. The most common nonhematologic TEAEs were GI disorders, fatigue, peripheral edema, dyspnea, and pain in extremity. Grade 3/4 asymptomatic lipase elevations were seen in 6 patients but were reversible after dose reduction. While grade 3/4 amylase elevations were observed at a dose of 800 mg in the phase I trial, they were not seen with the 400 or 500 mg dose.28,30 The prevalence of GI TEAEs decreased over time. The most common hematologic AE was anemia with grade 3/4 seen in up to 58% of patients.27–29 Treatment discontinuation secondary to adverse events was necessary in 8–20% mostly due to gastrointestinal side effects and thrombocytopenia in the various clinical trials.27–29
Wernicke’s Encephalopathy
A concerning adverse event seen in clinical trials of fedratinib is Wernicke’s encephalopathy (WE), which is caused by thiamine (vitamin B1) deficiency. Of a total of more than 600 patients with MF, PV, or solid tumors who received fedratinib, eight cases of severe neurologic adverse events were suspicious for WE.29 Due to this risk, the clinical development of fedratinib was placed on hold in 2013. However, after further analysis, only one of those cases was confirmed to be WE and the FDA lifted the hold on fedratinib development.
The underlying pathophysiology of how fedratinib may cause WE has been proposed to be that it may exacerbate malnutrition in patients due to its common GI adverse effects such as nausea, vomiting, and diarrhea. Preclinical data with Caco-2 cells showed that fedratinib may inhibit thiamine transporter (THTR) in a protein-free culture media environment suggesting impaired cellular thiamine uptake as a potential mechanism of WE.42 Studies in rats treated with fedratinib at doses comparable to those used for the treatment of myelofibrosis in humans did not show any neurologic deficits.43 This supports the finding that fedratinib, even at much higher doses than those used in the phase III study, does not affect thiamine levels or cause thiamine deficiency-related disorders.42,43
Given the concern for WE, 81 patients during the extended safety follow-up of 90 days after study discontinuation in the JAKARTA-2 trial received thiamine supplementation. No cases of encephalopathy or heart failure were reported during the extended safety follow-up.27 However, in the fedratinib 500 mg cohort of JAKARTA-1, a total of 4 cases of WE were confirmed by an independent expert safety panel based on either clinical features and imaging (3 patients) or clinical features alone (1 patient). These symptoms developed 6 to 44 weeks after initiation of treatment and were thought to be in the setting of elevated mean drug levels in two patients based on pharmacokinetic analysis, which was not performed for the other 2 patients, as they were placebo crossovers. Fedratinib was permanently discontinued in all 4 patients and intravenous thiamine was administered with residual cognitive deficits remaining in all patients at the time of the study report. Of note, no cases of WE were seen in the 400-mg fedratinib group. This led to the early termination of the JAKARTA-1 study and clinical development of fedratinib.29
Despite this conflicting evidence, fedratinib is FDA-approved at a dose of 400 mg for the treatment of myelofibrosis but encephalopathy remains a rare, but serious concern and is a listed black box warning. Especially, in patients with baseline risk factors for WE such as chronic diarrhea, weight loss, malnutrition, or chronic alcohol use, fedratinib use should be monitored very closely and thiamine supplementation can be considered.
Other JAK Inhibitors in Clinical Trials
Currently, fedratinib is the only FDA-approved second-line JAK inhibitor and presents a new option for patients who have experienced a treatment failure with ruxolitinib. The National Comprehensive Cancer Network (NCCN) designates fedratinib as a category 2A option for ruxolitinib-refractory/-intolerant patients and as a category 2B recommendation for upfront use in ruxolitinib naïve patients with intermediate-2 or high-risk MF.44 However, both momelotinib and pacritinib have been successfully in clinical trials as well but their uptake has been hampered by concerns about associated adverse events.2,32,45
Momelotinib is a selective JAK1/2 inhibitor that has been compared in phase III clinical trials to both ruxolitinib in treatment-naïve patients with myelofibrosis (SIMPLIFY 1; NCT01969838) and to best available therapy (BAT) in patients previously treated with ruxolitinib (SIMPLIFY 2; NCT02101268).46 In both trials, a 35% reduction in spleen volume after 24 weeks of treatment was chosen as the primary endpoint. Results of both trials have had lackluster results with momelotinib being not superior to BAT in terms of SVR (7% in momelotinib vs 6% in BAT group) but higher rates of at least 50% reduction in TSS compared to BAT (26% with momelotinib vs 6% with BAT; p=0.0006) in SIMPLIFY-2.46 Comparison with ruxolitinib in the SIMPLIFY-1 trial did not show benefits to treatment with momelotinib with regard to spleen size reduction and symptom improvement (spleen response: 26.5% in momelotinib group and 29% of the ruxolitinib group (p=0.011 for non-inferiority); ≥50% reduction in total symptom score: 28.4% for momelotinib vs 42.2% for ruxolitinib [noninferiority not met; p=0.98]).46,47 Adverse event profiles were comparable for momelotinib, ruxolitinib, and BAT with peripheral neuropathy (in up to 50% of patients) and myelosuppression being the most frequent adverse events seen with momelotinib.46,48,49 In single-arm studies of mostly treatment-naïve patients, momelotinib achieved clinical/symptom responses in up to 57.6% and spleen responses in 45% of patients, respectively, and no survival benefit compared to risk-matched patients not receiving momelotinib was seen.48–50 However, momelotinib unexpectedly improved anemia in patients with myelofibrosis, which can be a dose- or even treatment-limiting side effect with other JAK inhibitors.46,47 In the SIMPLIFY-1 and -2 trials, rates of RBC transfusion independence were higher with momelotinib in both treatment-naïve and ruxolitinib-pretreated patients.46,47 While the exact mechanism has not been fully elucidated, animal models have suggested that momelotinib reduces hepcidin production in the liver by inhibiting the ACVR1 pathway leading to greater iron availability for hematopoiesis.51 Based on the favorable effect on anemia, momelotinib will be tested against danazol in the randomized, double-blind phase III MOMENTUM trial for the treatment of anemia in myelofibrosis (NCT04173494). Patient recruitment for this trial has not begun yet.
Pacritinib is another oral multikinase inhibitor that inhibits not only JAK2 but is also targeting FLT3, IRAK1, and CSF1R.52 In the randomized phase III trial of pacritinib vs BAT including ruxolitinib (45% of patients) for thrombocytopenic patients with myelofibrosis (PERSIST-2; NCT02055781), twice-daily treatment with pacritinib led to significant improvements in both spleen volume reduction by ≥35% and ≥50% reduction in TSS over BAT (SVR: 16 patients [22%] vs 2 patients [3%]; p=0.001; ≥50% reduction in TSS: 24 patients [32%] vs 10 patients [14%]; p=0.01).53 In PERSIST-1 (NCT01773187) patients were randomized to either pacritinib or BAT other than ruxolitinib with higher rates of spleen responses seen in the pacritinib treated group (42 patients [19%] vs 5 patients [5%]; p=0.0003).54 The most common adverse events with pacritinib were thrombocytopenia and anemia which seemed to be less profound than with other treatment modalities.55 Although the exact mechanism why pacritinib appears to be less myelosuppressive than ruxolitinib is not known, the lack of JAK1 inhibition with pacritinib has been proposed as a potential explanation.45,53,56 While prior treatment with a JAK inhibitor was allowed in PERSIST-2, patients in PERSIST-1 had to be JAK inhibitor-naïve in order to be eligible for trial enrollment.53,54 Similar to fedratinib, development was briefly placed on hold in February 2016 after reports of patients dying of heart failure and intracranial hemorrhage in the PERSIST trials.57 However, after an additional review of those cases, the development hold on pacritinib was lifted.45
As the PERSIST-2 trial showed the efficacy of pacritinib also in patients previously treated with ruxolitinib, the phase II PAC203 trial was designed and randomized patients who failed or were intolerant of treatment with ruxolitinib to various doses of pacritinib. One hundred and sixty-four patients were included in this trial with 68% being intolerant of and 73% having failed treatment with ruxolitinib with a median treatment duration of 1.4 years preceding trial enrollment.58 Abstract data from this trial demonstrated that SVR ≥35% and TSS improvement of ≥50% were observed in 9.3% and 7.4% of patients, respectively, at a dose of 200 mg pacritinib twice daily with lower response rates seen with lower doses.58 Thrombocytopenia (32%), anemia (22%) and gastrointestinal adverse events were the most common treatment-emergent adverse events.58
Several clinical trials studying pacritinib in the pre-transplant setting (NCT03645824) and in patients previously treated with ruxolitinib (NCT03165734) are currently ongoing. Finally, the phase III PACIFICA trial that randomizes thrombocytopenic (platelet count < 50,000/mL) patients with primary or secondary myelofibrosis to pacritinib or physician’s choice (low-dose [≤5 mg] ruxolitinib, lenalidomide, corticosteroids, hydroxyurea) is anticipated to start enrollment shortly.59
Novel JAK Inhibitor-Based Combination Treatments
As anemia is a common dose-limiting side effect with ruxolitinib, combinations of ruxolitinib with erythropoiesis-stimulating agents such as the immunomodulators thalidomide and pomalidomide as well as the activin receptor ligand trap sotatercept, which inhibits signaling via the transforming growth factor (TGF)-β pathway, are currently ongoing. Especially the immunomodulator pomalidomide with or without prednisone has been successfully tested to treat cytopenias (anemia, thrombocytopenia) in patients with MPN-associated myelofibrosis.60–62 While the clinical trial (NCT01375140) of ruxolitinib in combination with lenalidomide had to be terminated early due to lack of therapeutic benefit and significant side effects necessitating dose interruptions in all 20 trial patients, preliminary data showed mixed results with such combinations but data should be interpreted cautiously until fully published.63–65
Epigenetic changes such as abnormal DNA methylation patterns have been identified in patients with myelofibrosis leading to clinical trials combining the hypomethylating agents (HMAs) azacitidine and decitabine with ruxolitinib.3,66,67 While both azacitidine and decitabine have only limited clinical activity if used as monotherapies, the combination therapy of ruxolitinib and azacitidine has led to ORR of 72% in a phase II study (NCT01787487).68 Notably, 95% of the responding patients maintained a spleen response (>50% reduction in spleen length) by week 48 of the trial. Furthermore, 57% of patients had improvements in the extent of bone marrow reticulin fibrosis which may suggest a disease-modifying effect of this combination.68 Combining ruxolitinib with HMAs might be especially effective in patients with MDS/MPN-overlap since HMA remains the standard of care for MDS patients especially in the setting of high-risk features such as elevated bone marrow blast percentage.69 This is further supported by a recent phase I study of ruxolitinib and decitabine in MPNs in accelerated or blast phase.70 In this trial median OS among the 21 patients was 7.9 months (95%-CI: 4.1 months – not reached) with an ORR (CR, CRi, partial remission) of 42.9% (9 out of 21 patients) in the intention-to-treat analysis.70 Based on these results, the combination of decitabine and ruxolitinib has been studied in a phase II trial (NCT02076191).
The hedgehog pathway is involved in the early stages of hematopoiesis and combinations of ruxolitinib and hedgehog inhibitors such as vismodegib and glasdegib have been shown to have synergistic effects in in-vitro experiments.71,72 However, results from a phase Ib/II clinical trial (NCT02226172) of glasdegib and ruxolitinib in 21 patients with JAK inhibitor-refractory MF have been disappointing. None of the trial patients achieved the secondary endpoint of SVR≥35% and only 2 patients had at least 50% improvement in symptom burden leading to the early termination of the study.73 Slightly better responses have been reported from a phase Ib trial (NCT02593760) combining vismodegib with ruxolitinib in 8 patients with MF that showed spleen responses in 3 patients and symptom improvement in 5 patients.74 Although the combination was safe, it will not be further developed for MF.74
After the B-cell lymphoma 2 (BCL-2) inhibitor venetoclax has shown impressive results in combination with HMA and low-dose cytarabine in AML, inhibition of the anti-apoptotic effects of BCL-2 by navitoclax,75,76 a related orally bio-available small-molecule BCL-2 inhibitor, has been tested in combination with ruxolitinib in a recent phase II trial in patients with myelofibrosis (NCT03222609).77 Abstract data for the week 24 endpoint showed that 29% and 20% of the 24 evaluable patients achieved an SVR ≥35% and symptomatic improvement, respectively.77 However, all patients had treatment-emergent adverse events with 77% experiencing grade ≥3 adverse events with thrombocytopenia (82%), diarrhea (62%), fatigue (53%), anemia (27%), and nausea (27%) being most common.77 Notably, the combination of navitoclax and ruxolitinib also led to a decrease in the VAF of driver mutations and improvements in bone marrow fibrosis, suggesting a potential disease-modifying effect of the combination.77
Another potential candidate for combination therapy with ruxolitinib is CPI-0610, a Bromodomain and Extraterminal Domain (BET) inhibitor, which modulates NFκB and TGF-β signaling pathways and has been shown to have synergistic effects with ruxolitinib in vitro.78 The MANIFEST study uses a two-arm design with one arm being CPI-0610 monotherapy in ruxolitinib-refractory/intolerant patients, and the other arm of CPI-0610 in combination with ruxolitinib (NCT02158858). Preliminary data showed that 94% of the 31 patients (both arms combined) achieved an SVR (median best change: −17% [range: −50.7, 10.2]; rate of ≥35% SVR not reported) and 39% had a ≥50% symptom reduction.79 Treatment was well tolerated with anemia (8.3%) and thrombocytopenia (8.3%) being the most common grade ≥3 treatment-emergent adverse events.79
Finally, in-vitro studies have shown synergistic effects for the combination of ruxolitinib with the tyrosine kinase inhibitor nilotinib and prednisone as well as the telomerase inhibitor imetelstat.80,81 While imetelstat has been successfully tested as monotherapy in MF even in JAK inhibitor-refractory patients,82 no clinical trial data for either combination are available yet and a study combining nilotinib and ruxolitinib in patients with chronic myelogenous leukemia has been temporarily suspended (NCT02973711). Other current phase II clinical trials in JAK inhibitor-refractory patients with myelofibrosis are testing the oral MDM2 inhibitor KRT-232 (NCT03662126) and selinexor (NCT03627403; ESSENTIAL trial) but neither of them has published results yet. Ongoing clinical trials of JAK inhibitor-based combination therapies are outlined in Table 2.
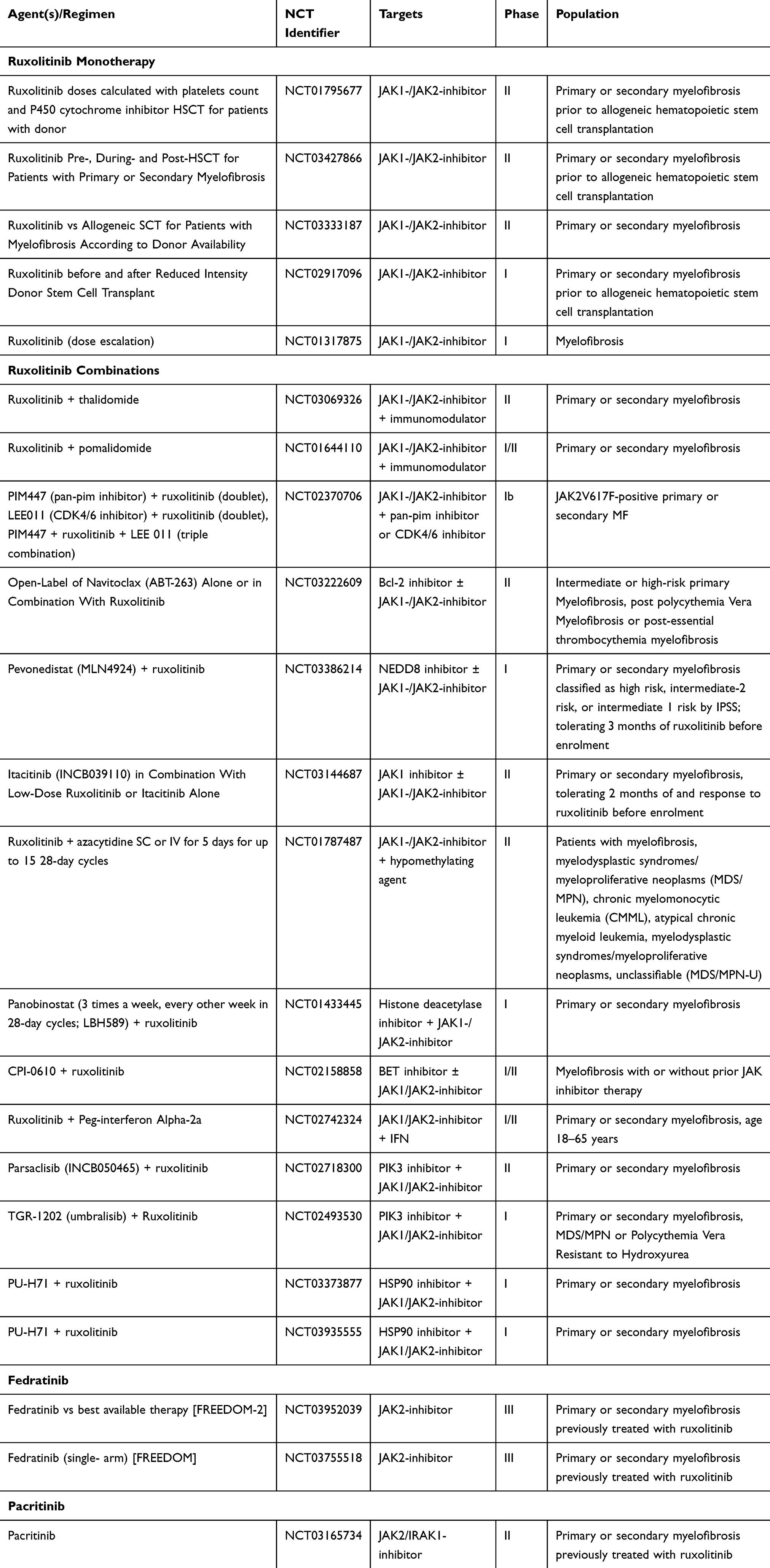 |
Table 2 Ongoing Clinical Trials of JAK Inhibitors in Myelofibrosis |
Conclusion and Future Directions
Ruxolitinib continues to be the first-line option for the treatment of symptomatic intermediate- or high-risk MF. Major challenges remain in the treatment of patients after ruxolitinib failure, for which there is not yet a clear-cut definition. Generally, spleen response or progressive increase of blast cells are used in clinical practice to define ruxolitinib-resistance. In the COMFORT-I and COMFORT-II trials, 50% and 75% of ruxolitinib treated patients experienced treatment failure or unacceptable adverse effects at 3 and 5 years, respectively.23,26 In this setting, clinical trials, allogeneic hematopoietic stem cell transplant (HSCT) or alternative JAK2 inhibitors remain viable options in eligible patients.1 Reasons for ruxolitinib resistance are poorly understood but include the variable JAK2 V617F VAF among patients, the use of ruxolitinib too late in the disease course, alternative mechanisms of JAK-STAT signaling pathway activation that are not inhibited by ruxolitinib, and a response modifying effect of concurrent mutations such as ASXL1 or SRSF2.57,83–85 Furthermore, 25–35% of patients in the COMFORT trials discontinued ruxolitinib due to adverse events with anemia, infectious complications, and diarrhea being the most commonly observed side effects in about a third of patients each.23,25 Prognosis of patients after ruxolitinib failure is poor and additional therapeutic options are highly warranted.
Currently, fedratinib presents the only FDA-approved option for patients who have experienced a treatment failure with ruxolitinib. However, both momelotinib and pacritinib have been successfully tested and could be potential therapeutic options for selected patients. While cytopenias have been a limiting factor for the treatment with ruxolitinib, momelotinib has been shown to reduce transfusion requirements and improve anemia which makes it a potential option for anemic patients.47 Similarly, pacritinib seems to be safe and effective in patients with treatment-limiting thrombocytopenia but further studies are needed to evaluate safety and efficacy.55 Of note, none of these second-line JAK inhibitors has shown a survival benefit and the concern about serious treatment-related side effects has stalled further clinical trials. Fedratinib is currently being studied in a phase 3b trial in 110 patients with intermediate to high-risk MF who have previously received ruxolitinib (FREEDOM; NCT03755518). This is a single-arm, open-label trial examining the efficacy and safety of fedratinib in patients with DIPSS (Dynamic International Prognostic Scoring System)-Intermediate or High-Risk PMF or post PV/post ET MF previously treated with ruxolitinib. The ongoing, randomized phase 3, open-label FREEDOM2 trial (NCT03952039) is comparing fedratinib to the best supportive care and started patient enrollment in September 2019. The primary objective of both studies is to evaluate the percentage of subjects with at least a 35% reduction of spleen volume. Secondary outcome measures include ≥50% reduction in spleen size, durability of symptom and spleen volume response and to evaluate the safety of fedratinib with a focus on GI adverse events, occurrence of confirmed WE events and monitoring and correction of thiamine levels. Results are expected for 2022 and will provide additional information on the role of fedratinib in myelofibrosis. Table 2 provides an overview of currently active clinical trials of JAK inhibitors as monotherapy or combination treatments in myelofibrosis.
The role of Fedratinib in the frontline setting of higher risk MF is less clear. While it is approved for this indication, there have been no published randomized clinical trials to date that compared it head-to-head to ruxolitinib which has been in the market for almost 8 years and has a prolonged clinical experience.
As outlined above future directions in myelofibrosis management include investigation of alternate pathways such as targeting bone marrow fibrosis, and consideration of JAK inhibitor-based combination therapies with other therapeutic classes such as hedgehog inhibitors, immunomodulators, epigenetic agents, and PI3K pathway inhibitors.32,86 One promising agent is the orally available LSD1 inhibitor IMG-7289 (bomedemstat) that had shown promising effects in animal models and is currently being tested in a phase I/II study in myelofibrosis patients intolerant of or refractory to ruxolitinib (NCT03136185).87,88 Preliminary results from this trial showed an SVR in 50% of patients (not defined as ≥35% as in other trials) and ≥50% symptom reduction in 21% of patients.88 However, it remains to be seen what the final results using more stringent response criteria show before the role of IMG-7289 in the treatment landscape of myelofibrosis can be evaluated. LCL161 is another compound that is tested as a single agent in a clinical trial in patients with myelofibrosis (NCT02098161). LCL161 is a second mitochondria-derived activator of caspases (SMAC) mimetic that induces apoptosis in cancer cells, leading to a phase II study that enrolled 43 patients with myelofibrosis. Using less stringent response criteria, 5 and 10 patients experienced anemia and symptom improvement, respectively, while only 1 patient had a spleen response.89 While additional clinical trials of these single agents and combination therapies are necessary, the currently available data suggest high ORR especially for the combination of ruxolitinib with hypomethylating agents with an acceptable risk profile.
In conclusion, fedratinib has shown improvement in both spleen size and MF-related symptom burden in the JAKARTA trials with efficacy seen in both frontline and in ruxolitinib-refractory patients. Providers should pay close attention to early detection and management, including pre-emptive interventions especially in high-risk patients, of rare but serious side effects such as WE. Additional studies are ongoing to inform the role of fedratinib in the treatment landscape for MF and studies to assess synergistic effects with other drug classes are needed.
Acknowledgments
AMZ is a Leukemia and Lymphoma Society Scholar in Clinical Research and is also supported by an NCI’s Cancer Clinical Investigator Team Leadership Award (CCITLA). This research was partly funded by the Dennis Cooper Hematology Young Investigator Award (AZ) and was in part supported by the National Cancer Institute of the National Institutes of Health under Award Number P30 CA016359. The conduction of the study and interpretation of the data are the sole responsibility of the authors.
Disclosure
SMJ has been employed by Celgene since 10/18/2019. All sections contributed to this manuscript were written and completed during prior employment at Yale New Haven Hospital-Smilow Cancer Center. No payment or services have been received pertaining to any aspect of the submitted work. SA has been employed by Sanofi Genzyme since 7/01/2019. No payment or services have been received pertaining to any aspect of the submitted work. AMZ received research funding (institutional) from Celgene, Acceleron, Abbvie, Otsuka, Pfizer, Medimmune/AstraZeneca, Boehringer-Ingelheim, Trovagene, Incyte, Takeda, and ADC Therapeutics. AMZ had a consultancy with and received honoraria from AbbVie, Otsuka, Pfizer, Celgene, Ariad, Agios, Boehringer-Ingelheim, Novartis, Acceleron, Astellas, Daiichi Sankyo, Trovagene, BeyondSpring, and Takeda. AMZ reports personal fees from Celgene, Acceleron,Abbvie, Otsuka, Pfizer, Medimmune/AstraZeneca, Boehringer-Ingelheim, Trovagene, Incyte, Takeda, ADC Therapeutics, Ariad, Agios, Novartis, Astellas, Daiichi Sankyo, and BeyondSpring, outside the submitted work. The other authors have no conflicts of interest to declare.
References
1. Passamonti F, Maffioli M. The role of JAK2 inhibitors in MPNs 7 years after approval. Blood. 2018;131(22):2426–2435. doi:10.1182/blood-2018-01-791491
2. Vannucchi AM, Harrison CN. Emerging treatments for classical myeloproliferative neoplasms. Blood. 2017;129(6):693–703. doi:10.1182/blood-2016-10-695965
3. Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. 2017;129(6):667–679. doi:10.1182/blood-2016-10-695940
4. Tefferi A, Vannucchi AM, Barbui T. Essential thrombocythemia treatment algorithm 2018. Blood Cancer J. 2018;8(1):2. doi:10.1038/s41408-017-0041-8
5. Szuber N, Mudireddy M, Nicolosi M, et al. 3023 mayo clinic patients with myeloproliferative neoplasms: risk-stratified comparison of survival and outcomes data among disease subgroups. Mayo Clin Proc. 2019;94(4):599–610. doi:10.1016/j.mayocp.2018.08.022
6. Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;27(9):1874–1881. doi:10.1038/leu.2013.163
7. Passamonti F, Cazzola M. Cytoreductive therapy for patients with essential thrombocythemia at high risk of thromboembolic complications. The difficult choice of the optimal drug. Haematologica. 2004;89(11):1284.
8. Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood. 2014;124(16):2507–2513. quiz 615. doi:10.1182/blood-2014-05-579136
9. Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the international working group for myelofibrosis research and treatment. Blood. 2009;113(13):2895–2901. doi:10.1182/blood-2008-07-170449
10. Kim SY, Im K, Park SN, Kwon J, Kim JA, Lee DS. CALR, JAK2, and MPL mutation profiles in patients with four different subtypes of myeloproliferative neoplasms: primary myelofibrosis, essential thrombocythemia, polycythemia vera, and myeloproliferative neoplasm, unclassifiable. Am J Clin Pathol. 2015;143(5):635–644. doi:10.1309/AJCPUAAC16LIWZMM
11. Rumi E, Pietra D, Ferretti V, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood. 2014;123(10):1544–1551. doi:10.1182/blood-2013-11-539098
12. Senin A, Fernandez-Rodriguez C, Bellosillo B, et al. Non-driver mutations in patients with JAK2V617F-mutated polycythemia vera or essential thrombocythemia with long-term molecular follow-up. Ann Hematol. 2018;97(3):443–451. doi:10.1007/s00277-017-3193-5
13. Tefferi A, Lasho TL, Guglielmelli P, et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016;1(1):21–30. doi:10.1182/bloodadvances.2016000216
14. Tefferi A, Lasho TL, Finke CM, et al. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016;1(2):105–111. doi:10.1182/bloodadvances.2016000208
15. Tefferi A. Primary myelofibrosis: 2019 update on diagnosis, risk-stratification and management. Am J Hematol. 2018;93(12):1551–1560. doi:10.1002/ajh.v93.12
16. James C, Ugo V, Le Couédic J-P, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. doi:10.1038/nature03546
17. Ward AC, Touw I, Yoshimura A. The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood. 2000;95(1):19–29. doi:10.1182/blood.V95.1.19
18. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. doi:10.1016/S0140-6736(05)71142-9
19. Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. doi:10.1056/NEJMoa051113
20. Bewersdorf JP, Ardasheva A, Podoltsev NA. et al. From clonal hematopoiesis to myeloid leukemia and what happens in between: will improved understanding lead to new therapeutic and preventive opportunities? Blood Rev;2019. 100587. doi:10.1016/j.blre.2019.100587
21. Passamonti F, Elena C, Schnittger S, et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood. 2011;117(10):2813–2816. doi:10.1182/blood-2010-11-316810
22. Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356(5):459–468. doi:10.1056/NEJMoa065202
23. Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. doi:10.1056/NEJMoa1110557
24. Harrison C, Kiladjian -J-J, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–798. doi:10.1056/NEJMoa1110556
25. Verstovsek S, Mesa RA, Gotlib J, et al. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017;10(1):55. doi:10.1186/s13045-017-0417-z
26. Verstovsek S, Gotlib J, Mesa RA, et al. Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J Hematol Oncol. 2017;10(1):156. doi:10.1186/s13045-017-0527-7
27. Harrison CN, Schaap N, Vannucchi AM, et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): a single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017;4(7):e317–e24. doi:10.1016/S2352-3026(17)30088-1
28. Pardanani A, Tefferi A, Jamieson C, et al. A phase 2 randomized dose-ranging study of the JAK2-selective inhibitor fedratinib (SAR302503) in patients with myelofibrosis. Blood Cancer J. 2015;5:e335. doi:10.1038/bcj.2015.63
29. Pardanani A, Harrison C, Cortes JE, et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: a randomized clinical trial. JAMA Oncol. 2015;1(5):643–651. doi:10.1001/jamaoncol.2015.1590
30. Pardanani A, Gotlib JR, Jamieson C, et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol. 2011;29(7):789–796. doi:10.1200/JCO.2010.32.8021
31. Bose P, Alfayez M, Verstovsek S. New concepts of treatment for patients with myelofibrosis. Curr Treat Options Oncol. 2019;20(1):5. doi:10.1007/s11864-019-0604-y
32. Bose P, Verstovsek S. JAK2 inhibitors for myeloproliferative neoplasms: what is next? Blood. 2017;130(2):115–125. doi:10.1182/blood-2017-04-742288
33. Singer JW, Al-Fayoumi S, Taylor J, Velichko S, O’Mahony A. Comparative phenotypic profiling of the JAK2 inhibitors ruxolitinib, fedratinib, momelotinib, and pacritinib reveals distinct mechanistic signatures. PLoS One. 2019;14(9):e0222944–e. doi:10.1371/journal.pone.0222944
34. Zhang M, Xu CR, Shamiyeh E, et al. A randomized, placebo-controlled study of the pharmacokinetics, pharmacodynamics, and tolerability of the oral JAK2 inhibitor fedratinib (SAR302503) in healthy volunteers. J Clin Pharmacol. 2014;54(4):415–421. doi:10.1002/jcph.v54.4
35. Zhang M, Xu C, Ma L, et al. Effect of food on the bioavailability and tolerability of the JAK2-selective inhibitor fedratinib (SAR302503): results from two phase I studies in healthy volunteers. Clin Pharmacol Drug Dev. 2015;4(4):315–321. doi:10.1002/cpdd.v4.4
36. Ogasawara K, Zhou S, Krishna G, Palmisano M, Li Y. Population pharmacokinetics of fedratinib in patients with myelofibrosis, polycythemia vera, and essential thrombocythemia. Cancer Chemother Pharmacol. 2019;84(4):891–898. doi:10.1007/s00280-019-03929-9
37. Abba C, Campanelli R, Catarsi P, et al. Constitutive STAT5 phosphorylation in CD34+ cells of patients with primary myelofibrosis: correlation with driver mutation status and disease severity. PLoS One. 2019;14(8):e0220189. doi:10.1371/journal.pone.0220189
38. RISUM M, MADELUNG A, BONDO H, et al. The JAK2V617F allele burden and STAT3- and STAT5 phosphorylation in myeloproliferative neoplasms: early prefibrotic myelofibrosis compared with essential thrombocythemia, polycythemia vera and myelofibrosis. APMIS. 2011;119(8):498–504. doi:10.1111/apm.2011.119.issue-8
39. Teofili L, Martini M, Cenci T, et al. Different STAT-3 and STAT-5 phosphorylation discriminates among Ph-negative chronic myeloproliferative diseases and is independent of the V617F JAK-2 mutation. Blood. 2007;110(1):354–359. doi:10.1182/blood-2007-01-069237
40. Harrison C, Schaap N, Vannucchi A, et al. Fedratinib induces spleen responses in patients with Myeloproliferative Neoplasm (MPN)-associated intermediate- or high-risk myelofibrosis (MF) resistant or intolerant to ruxolitinib: an updated analysis of the phase II JAKARTA2 study. Clin Lymphoma Myeloma Leuk. 2019;19:S356. doi:10.1016/j.clml.2019.07.375
41. Harrison C, Schaap N, Vannucchi A, et al. Fedratinib induces spleen responses and reduces symptom burden as first-line or salvage therapy in patients with myeloproliferative neoplasm-associated intermediate- or high-risk myelofibrosis (MF) and low platelet counts. Clin Lymphoma Myeloma Leuk. 2019;19:S355. doi:10.1016/j.clml.2019.07.374
42. Zhang Q, Zhang Y, Diamond S, et al. The Janus kinase 2 inhibitor fedratinib inhibits thiamine uptake: a putative mechanism for the onset of Wernicke’s encephalopathy. Drug Metab Dispos. 2014;42(10):1656–1662. doi:10.1124/dmd.114.058883
43. Hazell AS, Afadlal S, Cheresh DA, Azar A. Treatment of rats with the JAK-2 inhibitor fedratinib does not lead to experimental Wernicke’s encephalopathy. Neurosci Lett. 2017;642:163–167. doi:10.1016/j.neulet.2017.01.041
44. NCCN. NCCN guidelines version 3.2019: myeloproliferative neoplasms; 2019 [
45. Stahl M, Zeidan AM. Improved JAK inhibition in myelofibrosis—the long road ahead. JAMA Oncol. 2018;4(5):659–660. doi:10.1001/jamaoncol.2017.5802
46. Harrison CN, Vannucchi AM, Platzbecker U, et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): a randomised, open-label, phase 3 trial. Lancet Haematol. 2018;5(2):e73–e81. doi:10.1016/S2352-3026(17)30237-5
47. Mesa RA, Kiladjian JJ, Catalano JV, et al. SIMPLIFY-1: a phase III randomized trial of momelotinib versus ruxolitinib in janus kinase inhibitor-naive patients with myelofibrosis. J Clin Oncol. 2017;35(34):3844–3850. doi:10.1200/JCO.2017.73.4418
48. Pardanani A, Gotlib J, Roberts AW, et al. Long-term efficacy and safety of momelotinib, a JAK1 and JAK2 inhibitor, for the treatment of myelofibrosis. Leukemia. 2018;32(4):1035–1038. doi:10.1038/leu.2017.330
49. Tefferi A, Barraco D, Lasho TL, et al. Momelotinib therapy for myelofibrosis: a 7-year follow-up. Blood Cancer J. 2018;8(3):29. doi:10.1038/s41408-018-0067-6
50. Gupta V, Mesa RA, Deininger MWN, et al. A phase 1/2, open-label study evaluating twice-daily administration of momelotinib in myelofibrosis. Haematologica. 2017;102(1):94–102. doi:10.3324/haematol.2016.148924
51. Asshoff M, Petzer V, Warr MR, et al. Momelotinib inhibits ACVR1/ALK2, decreases hepcidin production, and ameliorates anemia of chronic disease in rodents. Blood. 2017;129(13):1823–1830. doi:10.1182/blood-2016-09-740092
52. Diaz AE, Mesa RA. Pacritinib and its use in the treatment of patients with myelofibrosis who have thrombocytopenia. Future Oncol. 2018;14(9):797–807. doi:10.2217/fon-2017-0494
53. Mascarenhas J, Hoffman R, Talpaz M, et al. Pacritinib vs best available therapy, including ruxolitinib, in patients with myelofibrosis: a randomized clinical trial. JAMA Oncol. 2018;4(5):652–659. doi:10.1001/jamaoncol.2017.5818
54. Mesa RA, Vannucchi AM, Mead A, et al. Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST-1): an international, randomised, phase 3 trial. Lancet Haematol. 2017;4(5):e225–e36. doi:10.1016/S2352-3026(17)30027-3
55. Mascarenhas J, Virtgaym E, Stal M, et al. Outcomes of patients with myelofibrosis treated with compassionate use pacritinib: a sponsor-independent international study. Ann Hematol. 2018;97(8):1369–1374. doi:10.1007/s00277-018-3309-6
56. Singer JW, Al-Fayoumi S, Ma H, Komrokji RS, Mesa R, Verstovsek S. Comprehensive kinase profile of pacritinib, a nonmyelosuppressive Janus kinase 2 inhibitor. J Exp Pharmacol. 2016;8:11–19. doi:10.2147/JEP.S110702
57. Pardanani A, Tefferi A. How I treat myelofibrosis after failure of JAK inhibitors. Blood. 2018;132(5):492–500. doi:10.1182/blood-2018-02-785923
58. Gerds AT, Savona MR, Scott BL, et al. Results of PAC203: a randomized phase 2 dose-finding study and determination of the recommended dose of pacritinib. Blood. 2019;134(Supplement_1):667.
59. Harrison CN, Gerds AT, Kiladjian -J-J, et al. Pacifica: a randomized, controlled phase 3 study of pacritinib vs. physician’s choice in patients with primary myelofibrosis, post polycythemia vera myelofibrosis, or post essential thrombocytopenia myelofibrosis with severe thrombocytopenia (platelet count <50,000/mL). Blood. 2019;134(Supplement_1):4175.
60. Schlenk RF, Stegelmann F, Reiter A, et al. Pomalidomide in myeloproliferative neoplasm-associated myelofibrosis. Leukemia. 2017;31(4):889–895. doi:10.1038/leu.2016.299
61. Tefferi A, Verstovsek S, Barosi G, et al. Pomalidomide is active in the treatment of anemia associated with myelofibrosis. J Clin Oncol. 2009;27(27):4563–4569. doi:10.1200/JCO.2008.21.7356
62. Tefferi A, Al-Ali HK, Barosi G, et al. A randomized study of pomalidomide vs placebo in persons with myeloproliferative neoplasm-associated myelofibrosis and RBC-transfusion dependence. Leukemia. 2017;31(4):896–902. doi:10.1038/leu.2016.300
63. Daver N, Cortes J, Newberry K, et al. Ruxolitinib in combination with lenalidomide as therapy for patients with myelofibrosis. Haematologica. 2015;100(8):1058–1063. doi:10.3324/haematol.2015.126821
64. Bose P, Daver N, Jabbour EJ, et al. Phase-2 study of sotatercept (ACE-011) in myeloproliferative neoplasm-associated myelofibrosis and anemia. Blood. 2016;128(22):478. doi:10.1182/blood.V128.22.478.478
65. Stegelmann F, Bangerter M, Heidel FH, et al. A phase-Ib/II study of ruxolitinib plus pomalidomide in myelofibrosis. Blood. 2015;126(23):826. doi:10.1182/blood.V126.23.826.826
66. Jacquelin S, Straube J, Cooper L, et al. Jak2V617F and Dnmt3a loss cooperate to induce myelofibrosis through activated enhancer-driven inflammation. Blood. 2018;132(26):2707–2721. doi:10.1182/blood-2018-04-846220
67. Rampal R, Ahn J, Abdel-Wahab O, et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc Natl Acad Sci U S A. 2014;111(50):E5401–E5410. doi:10.1073/pnas.1407792111
68. Masarova L, Verstovsek S, Hidalgo-Lopez JE, et al. A phase 2 study of ruxolitinib in combination with azacitidine in patients with myelofibrosis. Blood. 2018;132(16):1664–1674. doi:10.1182/blood-2018-04-846626
69. Greenberg PL, Stone RM, Al-Kali A, et al. Myelodysplastic syndromes, version 2.2017, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2017;15(1):60–87. doi:10.6004/jnccn.2017.0007
70. Rampal RK, Mascarenhas JO, Kosiorek HE, et al. Safety and efficacy of combined ruxolitinib and decitabine in accelerated and blast-phase myeloproliferative neoplasms. Blood Adv. 2018;2(24):3572–3580. doi:10.1182/bloodadvances.2018019661
71. Tibes R, Mesa RA. Targeting hedgehog signaling in myelofibrosis and other hematologic malignancies. J Hematol Oncol. 2014;7:18. doi:10.1186/1756-8722-7-18
72. Shallis RM, Bewersdorf JP, Boddu PC, Zeidan AM. Hedgehog pathway inhibition as a therapeutic target in acute myeloid leukemia. Expert Rev Anticancer Ther. 2019;19(8):717–729. doi:10.1080/14737140.2019.1652095
73. Gerds AT, Tauchi T, Ritchie E, et al. Phase 1/2 trial of glasdegib in patients with primary or secondary myelofibrosis previously treated with ruxolitinib. Leuk Res. 2019;79:38–44. doi:10.1016/j.leukres.2019.02.012
74. Couban S, Benevolo G, Donnellan W, et al. A phase Ib study to assess the efficacy and safety of vismodegib in combination with ruxolitinib in patients with intermediate- or high-risk myelofibrosis. J Hematol Oncol. 2018;11(1):122. doi:10.1186/s13045-018-0661-x
75. DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7–17. doi:10.1182/blood-2018-08-868752
76. Wei AH, Strickland SA
77. Harrison CN, Garcia JS, Mesa RA, et al. Results from a phase 2 study of navitoclax in combination with ruxolitinib in patients with primary or secondary myelofibrosis. Blood. 2019;134(Supplement_1):671.
78. Saenz DT, Fiskus W, Qian Y, et al. Novel BET protein proteolysis-targeting chimera exerts superior lethal activity than bromodomain inhibitor (BETi) against post-myeloproliferative neoplasm secondary (s) AML cells. Leukemia. 2017;31(9):1951–1961. doi:10.1038/leu.2016.393
79. Mascarenhas J, Kremyanskaya M, Hoffman R, et al. MANIFEST, a phase 2 study of CPI-0610, a Bromodomain and Extraterminal Domain Inhibitor (BETi), as monotherapy or “add-on” to ruxolitinib, in patients with refractory or intolerant advanced myelofibrosis. Blood. 2019;134(Supplement_1):670.
80. Cortes AA, Diaz RA, Hernandez-Campo P, et al. Ruxolitinib in combination with prednisone and nilotinib exhibit synergistic effects in human cells lines and primary cells from myeloproliferative neoplasms. Haematologica. 2019;104(5):937–946. doi:10.3324/haematol.2018.201038
81. Wang X, Hu CS, Petersen B, et al. Imetelstat, a telomerase inhibitor, is capable of depleting myelofibrosis stem and progenitor cells. Blood Adv. 2018;2(18):2378–2388. doi:10.1182/bloodadvances.2018022012
82. Tefferi A, Lasho TL, Begna KH, et al. A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N Engl J Med. 2015;373(10):908–919. doi:10.1056/NEJMoa1310523
83. Barosi G, Klersy C, Villani L, et al. JAK2V617F allele burden ≥50% is associated with response to ruxolitinib in persons with MPN-associated myelofibrosis and splenomegaly requiring therapy. Leukemia. 2016;30(8):1772–1775. doi:10.1038/leu.2016.45
84. Patel KP, Newberry KJ, Luthra R, et al. Correlation of mutation profile and response in patients with myelofibrosis treated with ruxolitinib. Blood. 2015;126(6):790–797. doi:10.1182/blood-2015-03-633404
85. Koppikar P, Bhagwat N, Kilpivaara O, et al. Heterodimeric JAK–STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature. 2012;489(7414):155–159. doi:10.1038/nature11303
86. Stahl M, Zeidan AM. Management of myelofibrosis: JAK inhibition and beyond. Expert Rev Hematol. 2017;10(5):459–477. doi:10.1080/17474086.2017.1317590
87. Jutzi JS, Kleppe M, Dias J, et al. LSD1 inhibition prolongs survival in mouse models of MPN by selectively targeting the disease clone. Hemasphere. 2018;2(3):e54. doi:10.1097/HS9.0000000000000054
88. Pettit K, Gerds AT, Yacoub A, et al. A phase 2a study of the LSD1 inhibitor Img-7289 (bomedemstat) for the treatment of myelofibrosis. Blood. 2019;134(Supplement_1):556.
89. Pemmaraju N, Carter BZ, Kantarjian HM, et al. LCL161, an oral smac mimetic/IAP antagonist for patients with myelofibrosis (MF): novel translational findings among long-term responders in a phase 2 clinical trial. Blood. 2018;132(Supplement 1):687. doi:10.1182/blood-2018-99-119753
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.