")
Back to Journals » OncoTargets and Therapy » Volume 9
BET inhibitors in the treatment of hematologic malignancies: current insights and future prospects
Authors Abedin S, Boddy C, Munshi H
Received 7 July 2016
Accepted for publication 25 August 2016
Published 28 September 2016 Volume 2016:9 Pages 5943—5953
DOI https://doi.org/10.2147/OTT.S100515
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
Sameem M Abedin,1 Craig S Boddy,1 Hidayatullah G Munshi1–3
1Department of Medicine, Feinberg School of Medicine, Northwestern University, 2Medicine Service, Jesse Brown VA Medical Center, 3The Robert H. Lurie Comprehensive Cancer Center, Northwestern University, Chicago, IL, USA
Abstract: The bromodomain and extra-terminal (BET) family of proteins are important epigenetic regulators involved in promoting gene expression of critical oncogenes. BET inhibitors have been demonstrated to repress c-Myc expression, and were initially shown to have efficacy in a number of c-Myc-dependent hematologic malignancies. Recent studies have now revealed a broader role for BET inhibitors in hematologic malignancies. In this review, we summarize the efficacy of BET inhibitors in preclinical models of acute leukemia, lymphoma, and multiple myeloma. We also summarize recent results of clinical trials utilizing BET inhibitors in hematologic malignancies, characterize potential resistance mechanisms to BET inhibitors, and discuss potential combination therapies with BET inhibitors in patients with hematologic malignancies.
Keywords: leukemia, lymphoma, multiple myeloma, toxicity, resistance mechanisms
Introduction
Epigenetic dysregulation plays an important role in driving the aberrant gene expression patterns seen in a variety of hematologic malignancies. As many epigenetic alterations are reversible, these factors have drawn considerable attention as potential antineoplastic targets. One particular target of significant clinical interest is the bromodomain and extra-terminal (BET) family of proteins, which includes BRD2, BRD3, BRD4, and the testis-specific BRDT.1,2 Bromodomains (BRDs) are protein domains that possess a high affinity for binding to acetylation motifs, including acetylated histone proteins within chromatin.1,2 The BET family of proteins bind to acetylated chromatin and regulate gene transcription.1,2 Selective inhibition of the interaction between BET proteins and acetylated chromatin has resulted in significant activity in preclinical models of acute leukemia, lymphoma, and multiple myeloma (MM).3,4 Initially, this effect was largely attributed to inhibition of c-Myc expression, but repression of a variety of oncogenic factors is being discovered. This has led to early-phase clinical trials utilizing BET inhibitors in a number of hematologic malignancies, with encouraging clinical results.5,6 In this review, we discuss the evidence for the efficacy of BET inhibitors in select hematologic malignancies. We also discuss the use of current BET inhibitors in clinical trials along with results from a recently completed Phase I study utilizing BET inhibitors in advanced hematologic malignancies. Finally, we discuss proposed combination therapies with BET inhibitors and mechanisms of resistance to BET inhibitors.
Mechanism of action of BET inhibitors
The BET family of proteins is a structurally homologous group composed of two tandem N-terminal BRD modules, an extra-terminal domain, various conserved motifs, and a C-terminal motif (CTM) that is present in BRD4 and BRDT.2,7 The BRD modules on BET proteins facilitate binding of BET proteins to sites of active transcription through a hydrophobic pocket that associates with acetylated lysine residues on histones.7 Upon binding, BET proteins then recruit and stabilize transcription effectors and thereby promote transcription.7,8 Additionally, the CTM of BRD4 and BRDT interacts with the P-TEFb, resulting in the release of the inhibitory protein HEXIM1 and subsequent P-TEFb activation.7,8 Overall, BET proteins play a central role in facilitating effective transcription. Figure 1 illustrates, using BRD4 as an example, the central role that BET proteins play in transcription.
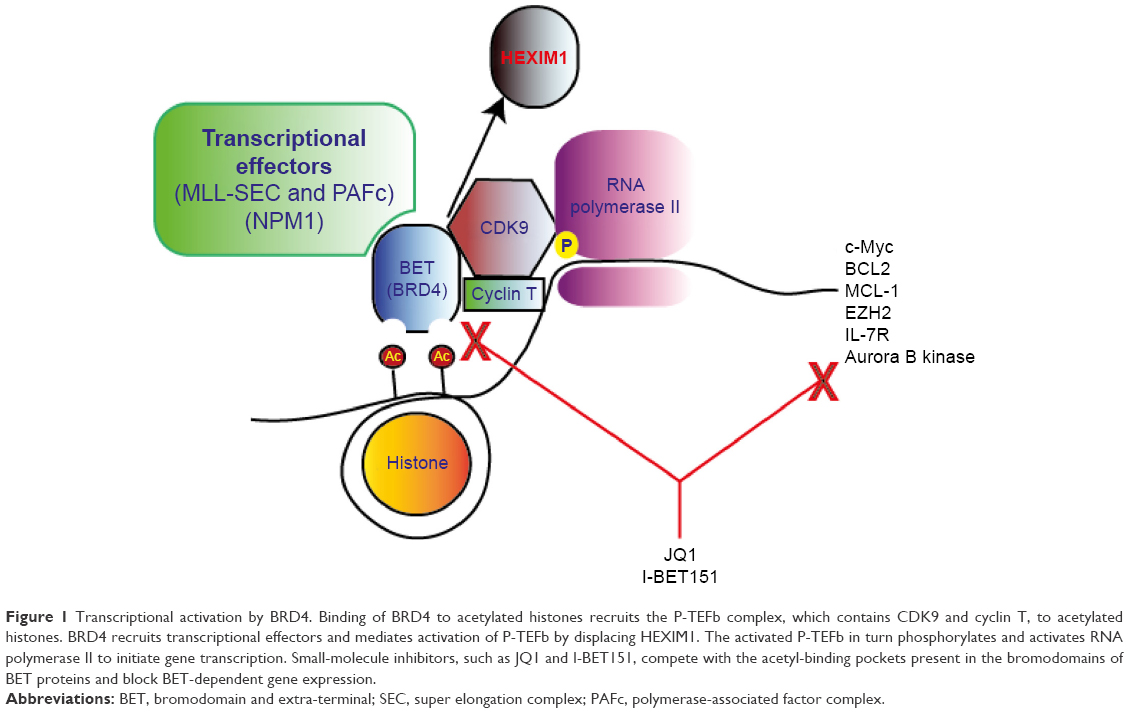 | Figure 1 Transcriptional activation by BRD4. Binding of BRD4 to acetylated histones recruits the P-TEFb complex, which contains CDK9 and cyclin T, to acetylated histones. BRD4 recruits transcriptional effectors and mediates activation of P-TEFb by displacing HEXIM1. The activated P-TEFb in turn phosphorylates and activates RNA polymerase II to initiate gene transcription. Small-molecule inhibitors, such as JQ1 and I-BET151, compete with the acetyl-binding pockets present in the bromodomains of BET proteins and block BET-dependent gene expression. |
Recent studies have demonstrated that while BET proteins bind to enhancer sites throughout the genome, they are preferentially found at enhancer sites of oncogenes and critical lineage-specific genes, deemed as super-enhancer sites.9–11 For example, when the binding of BRD4 was assessed across nearly 18,000 enhancer sites of various genes in lymphoma, nearly 30% of BRD4 binding occurred at only 285 (1.4%) enhancer sites.12 When these few sites were examined, BRD4 was found to preferentially bind in the proximity of critical lymphoma-related oncogenes, such as c-Myc and CD79B, and at enhancers essential for B-cell fate determination, such as PAX5 and IRF8.12 This finding indicated that targeting BET proteins could specifically target transcription of oncogenes and genes critical to disease development and progression.
Given the critical role of BET proteins in transcription and in malignancy, significant efforts were made to develop inhibitors to the BET family of proteins. In the late 2000s, after characterization of the hydrophobic interaction between BRDs and acetyl-lysine, triazolothienodiazepine compounds were synthesized which disrupted the interaction between the BRD of BET proteins and acetyl-lysine.7 Two small-molecule compounds, JQ1 and I-BET762, were synthesized and found to selectively disrupt the interaction between the BET BRD and acetyl-lysine, resulting in the dissociation of BET proteins from chromatin.7 These compounds were initially studied in myeloma and demonstrated repression of c-Myc expression.1 Since the initial discovery of these compounds, several other compounds with similar effects have been developed. Through these inhibitors, BET inhibition has been explored in a variety of hematologic malignancies. In this review, we discuss these significant findings that make BET inhibition an attractive strategy in leukemias, lymphomas, and multiple myelomas (MMs).
BET inhibitors in acute leukemia
Treatment of acute myeloid leukemia
Among the hematologic malignancies, leukemia represents the deadliest group of diseases accounting for an estimated 24,000 deaths in the US annually.13 Acute myeloid leukemia (AML) is the commonest form of acute leukemia in adults, and current therapy only affords approximately a 40% chance of survival for 5 years.13 The major limitation in AML therapy is that while the majority of patients diagnosed with AML are older, current AML treatment appears to be less effective in older patients.13 Above the age of 40 years, with each subsequent decade, a lower number of patients achieve remission with conventional therapy, and a higher rate of relapse occurs among patients who have achieved remission. Additionally, in patients with relapsed or refractory disease, treatment options are mostly ineffective, and long-term survival is only approximately 10%.14
Despite the limitations of current AML therapy, recent advances in the understanding of the molecular mechanisms of AML promise to improve outcomes. Over the past decade, several genetic abnormalities in AML have proved to be important prognostic markers. One example is the NPM1 gene mutation, which, in the absence of other mutations, has been found to confer a more favorable prognosis.15 Another example is that internal tandem duplications (ITDs) involving the FLT3 have been shown to portend a poorer prognosis, with higher rates of relapses in patients with this abnormality.16 Finally, translocations that involve the MLL gene have been found to occur in a higher frequency in patients with therapy-related AML, and these patients also tend to have a worse overall prognosis.17 Practice changes based on these findings include early referral for allogeneic stem cell transplantation (SCT) if a poor prognostic mutation is discovered and remission is achieved. Beyond prognostication, these mutations represent targetable abnormalities. Genetic abnormalities involving NPM1 and MLL likely result in aberrant expression of critical oncogenes. Further, mutations involving FLT3 likely result in constitutive activation of multiple differentiation and survival pathways. In this review, we discuss about how the BET family of proteins may be involved in the pathogenesis of AML, particularly in NPM1 and MLL leukemias. We also discuss about how the BET family of proteins likely promotes oncogenesis through a number of shared pathways activated in FLT3 AML. Finally, we discuss about how BET inhibition may be effective in these subsets of AMLs. Table 1 summarizes potential targets in leukemia sensitive to BET inhibition.
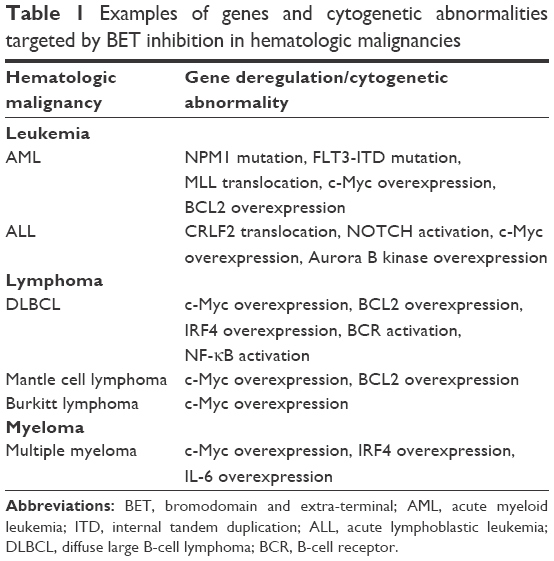 | Table 1 Examples of genes and cytogenetic abnormalities targeted by BET inhibition in hematologic malignancies |
NPM1-mutated AML
The NPM1 mutation is present in approximately 35% of AML cases, representing one of the commonest distinctions in AML.15 NPM1-mutant AML is generally considered a more favorable subtype; however, increasing evidence demonstrates that these patients represent a heterogeneous population, and prognosis varies depending on other mutations that accompany the NPM1 mutation.16 Functionally, NPM1 serves multiple functions including a role in ribosomal assembly, as a nucleolar histone chaperone, and a regulator of the ARF-p53 pathway.18 These roles rely on shuttling of NPM1 between the nucleus and cytoplasm. The most common NPM1 mutations result in a mutated protein lacking a folded C-terminal domain, leading to cytoplasmic localization of the NPM1 protein.19 Recent studies on NPM1-mutant AML have identified nuclear NPM1 as an important regulator of transcription through BRD4.18 In NPM1 wild-type patients, NPM1 is thought to associate with BRD4 in the nucleus to repress BRD4-dependent transcription. However, in NPM1-mutant AML, transcriptional repression of BRD4 by NPM1 is lost due to cytoplasmic mislocalization, resulting in an increased gene expression of critical oncogenes, including c-Myc and BCL2.18 Therefore, inhibition of BRD4 appeared to be a rational target in NPM1-mutant AML. To this extent, treatment of the NPM1-mutant AML cell line OCI-AML with I-BET151, a BET inhibitor with activity against BRD2, BRD3, and BRD4 was examined.18 BET inhibition in this cell line resulted in decreased proliferation and increased apoptosis, and mechanistically, downregulation of BCL2 and c-Myc was observed.18 Subsequently, various patient samples containing an NPM1 mutation were treated with I-BET151, and downregulation of BCL2 and c-Myc was confirmed in these samples.18 Finally, in vivo, I-BET151 was administered to an immunodeficient mouse model (NOD mouse) xenografted with OCI-AML, and tumor growth was impaired leading to an improvement in overall survival.18 These findings provide rationale for BET inhibition in NPM1-mutant AML.
MLL-translocated AML
Chromosomal translocations involving the MLL gene, located at chromosome band 11q23, frequently occur in AML, particularly in therapy-related AML.17 Rearrangements involving MLL also occur in approximately 10% of acute lymphoblastic leukemia (ALL) cases.17 Typically, these findings are associated with a poor prognosis.17 MLL encodes for a histone methyltransferase involved in transcriptional regulation.20 Translocations involving MLL commonly result in the fusion of MLL with important regulators of transcriptional elongation, including the super elongation complex (SEC).21 The result is deregulated transcription driving leukemogenesis. Both BRD3 and BRD4 are components of the SEC. Additionally, BRD3 and BRD4 are components of the polymerase-associated factor complex (PAFc) which plays a role in MLL localization to promote transcription.21 Given these functions, BET inhibition was identified as a potential therapy in MLL leukemia. BET inhibitors I-BET151 and JQ1 have both been tested in MLL-fusion leukemia cell lines and in MLL-driven mouse models.21 MLL leukemia was found to be sensitive to BET inhibition, and NOD mice xenografted with MLL leukemia had a prolonged survival with BET inhibitor treatment.21 Both inhibitors mechanistically were shown to decrease binding of the SEC and PAFc to critical gene loci, leading to inhibition of transcription and downregulation of important oncogenes, including c-Myc and BCL2.21 Overall, these data support the use of BET inhibitors in acute leukemia containing translocations involving the MLL gene.
FLT3-mutated AML
FLT3 mutations occur in approximately 30% of patients with AML, and FLT3-mutant AML represents an aggressive form of AML with an inferior leukemia-free survival compared to patients without the mutation.16 The most common FLT3 mutation encountered is ITD mutation, resulting in constitutive FLT3 activation and subsequent downstream activation of important proliferation, differentiation, and survival pathways.22 Increased STAT5, AKT, and ERK phosphorylation is observed as a result of constitutive FLT3 activation.22 Recently, several FLT3 tyrosine kinase inhibitors (FLT3-TKIs) have been introduced in clinical trial for FLT3-mutant AML, and while these inhibitors in some cases have resulted in remissions, typically, these remissions are short lived, and resistance and relapse often occur.22 One hypothesis for resistance is through activation of various transcriptional mediators which result in reactivation of various proliferative pathways.22 Given this potential resistance mechanism, the BET inhibitor JQ1 was evaluated with FLT3-TKIs including quizartinib.23 Combination therapy resulted in further attenuation of p-STAT5, p-AKT, and p-ERK1/2 compared to FLT3 inhibition alone, and additionally increased expression of the apoptotic proteins p21 and BIM.23 Decreased viability and increased apoptosis were demonstrated in both FLT3-ITD cell lines MOLM13 and MV4-11 and in primary AML samples in patients harboring an FLT3-ITD mutation.23 BET inhibition was also evaluated in the TKI-resistant cell line MOLM13-TKIR.23 Phenotypically, compared to TKI-sensitive MOLM13 cells, MOLM13-TKIR cells demonstrated markedly increased c-Myc and BCL2 expression, and increased expression of proteins belonging to the polycomb repressive complex 2, EZH2, and SUZ12.23 JQ1 treatment increased apoptosis of MOLM13-TKIR cells.23 Interestingly, apoptosis was seen to a greater extent in the MOLM13-TKIR cells compared to JQ1-induced apoptosis seen in MOLM13 cells.23 JQ1 treatment further was shown to result in downregulation of c-Myc, BCL2, EZH2, and SUZ12 in MOLM13-TKIR cells.23 As EZH2 and SUZ12 were hypothesized to confer a survival benefit in MOLM13-TKIR cells, BET inhibition has been proposed as a therapeutic option for TKI-resistant FLT3-ITD AML.
Treatment of ALL
B-cell ALL
B-cell ALL in adults carries a similar prognosis compared with AML with an estimated 40% survival for 5 years.13 Recent genetic characterization of this disease has resulted in an improved understanding regarding prognosis in this disease, and provides new targets to treat and potentially improve outcomes. One relevant genetic alteration recently identified involves rearrangements in the gene for CRLF2. CRLF2 alterations have been identified in approximately 15% of adult B-cell ALL patients, and are associated with a high rate of relapse and poor overall survival.24 When the BET inhibitor JQ1 was evaluated in a series of B-cell ALL cell lines to determine its therapeutic potential, it was found to potently reduce viability in B-cell ALL lines harboring CRLF2 alterations.25 CRLF2 heterodimerizes with the IL-7 receptor, leading to activation of JAK1, JAK2, and STAT5.25 BET inhibition, primarily through BRD4 inhibition, resulted in downregulation of the IL-7R gene, and in B-cell ALL cell lines harboring CRLF2 alterations, JQ1 treatment resulted in significantly diminished JAK2 and STAT5 phosphorylation by 24 hours.25 Further, c-Myc downregulation was observed with decreased c-Myc target gene expression.25 Accordingly, these cell lines demonstrated significant sensitivity to JQ1 treatment, resulting in inhibition of proliferation and induction of apoptosis. Another potential effect seen with BET inhibition in B-cell ALL was downregulation of Aurora B kinase.26 Aurora B kinase plays a role in directing mitosis, is upregulated by c-Myc, and can be overexpressed in B-cell ALL.26 Importantly, treatment of mice xenografted with primary B-cell ALL blasts with JQ1 resulted, in addition to downregulation of c-Myc, in significant downregulation of Aurora B kinase in the bone marrow of these mice.26
T-cell ALL
T-cell ALL represents approximately 25% of total ALL cases and generally has a more favorable prognosis compared to B-cell ALL.13 Through various genetic abnormalities, including mutations involving PTEN/PI3K/AKT signaling pathway, NOTCH, and FBXW7, over 50% of T-cell ALL cases are found to have c-Myc upregulation and activation.27 Due to the central role of c-MYC in T-cell ALL, JQ1 was also examined in T-cell ALL cell lines. Across all T-ALL cell lines, JQ1 was found to inhibit proliferation and viability.27 Further, c-Myc mRNA and protein expression were both strongly diminished in T-cell ALL cell lines upon treatment with JQ1.27 The efficacy of JQ1 was also evaluated in vivo in NOD mice xenografted with patient-derived T-cell ALL leukemic blasts. Two of four mice demonstrated inhibition of tumor growth and decreased cell proliferation upon treatment with JQ1.27 Finally, as vincristine is commonly used in the treatment of T-ALL, JQ1 was combined with vincristine to assess for potential synergy of combining BET inhibitors with chemotherapy. Both T-ALL cell lines and NOD mice xenografted with patient T-ALL leukemic blasts revealed a synergistic decrease in proliferation and impaired tumor growth when JQ1 was combined with vincristine.27
BET inhibitors and lymphoma
Treatment of diffuse large B-cell lymphoma
Annually, there are approximately 70,000 patients newly diagnosed with non-Hodgkin lymphoma (NHL) in the US While the 5-year survival in NHL now approaches 70%, over 20,000 patients still die annually from NHL, highlighting an ongoing need for new therapies in lymphoma.13
Diffuse large B-cell lymphoma (DLBCL) represents the most common form of NHL, representing approximately 30%–40% of all cases.13 DLBCL is curable, and frontline immunochemotherapy results in a complete remission in approximately 65%–75% of patients, with a 10-year overall survival rate of approximately 50%.28,29 However, in patients with relapsed and refractory DLBCL, outcomes are less favorable. Salvage regimens can result in a remission in approximately 40% of cases, but these remissions tend to be less durable.30 Recently, based on an improved understanding of the molecular basis, DLBCL can be categorized into distinct subtypes which have prognostic significance and potential therapeutic implications. The major classification of DLBCL is based on cell origin, which is determined through cytogenetic and molecular testing. DLBCL can be classified as either germinal center B-cell (GCB) subtype or activated B-cell-like (ABC) subtype.31 Clinically, the ABC subtype has been found to represent a more aggressive variant, with an inferior response to initial therapy, and a lower overall survival compared to GCB DLBCL.31 Biologically, ABC DLBCL is characterized by constitutive activation of the NF-κB which promotes lymphomagenesis.31 Conversely, GCB DLBCL is felt to be more responsive to standard immunochemotherapy.31 Biologically, c-Myc plays a significant role in the pathogenesis in many GCB cases.31 Another relevant distinction made is a determination of whether a DLBCL is a “double-hit” lymphoma. These lymphomas are characterized by a chromosomal translocation involving the c-Myc gene, and are sometimes considered a subset of GCB DLBCL, but additionally possess a chromosomal translocation of either BCL2 or BCL6 genes.32 This confers a more aggressive course leading to an inferior response to traditional therapy and decreased overall survival. Here, we discuss about how the BET family of proteins may contribute to the pathogenesis of ABC DLBCL and c-Myc-mediated DLBCL, and the potential role of BET inhibitors in these lymphomas. Table 1 summarizes potential targets in lymphoma sensitive to BET inhibition.
Activated B-cell-like DLBCL
ABC DLBCL likely originates from a post-germinal B-cell based on gene expression profiling. Clinically, ABC DLBCL is more aggressive, with an inferior response to rituximab-based initial therapy compared to GCB-origin lymphomas.31 As mentioned earlier, the NF-κB pathway is constitutively active in ABC DLBCL through various mechanisms. Two relevant mechanisms of NF-κB activation are through mutations leading to B-cell receptor (BCR) activation and signaling, and through mutations that target MyD88, which is an adaptor protein within the Toll-like receptor pathway that modulates activation of NF-κB.33 Both the BCR- and MyD88-related signaling converges to activate IKK, which ultimately leads to NF-κB nuclear localization and transcription of NF-κB target genes.33 BET inhibition has been shown to have activity against super enhancer-mediated transcription, including NF-κB-mediated transcription, and therefore was studied in ABC DLBCL. Treatment of four ABC DLBCL cell lines with JQ1 resulted in decreased NF-κB activity in each of these cell lines, along with decreased cell viability and increased apoptosis.33 Interestingly, knockdown of BRD2 or BRD4 or BET inhibition resulted in decreased phosphorylation of IKK-β, which led to IKK inhibition.33 IKK inhibition inhibited BCR-, MyD88-, and NF-κB-related gene expression.33 Further, BET inhibition was examined in combination with the Bruton’s tyrosine kinase (BTK) inhibitor, ibrutinib. Inhibition of BTK led to inhibition of BCR signaling, and therefore, ibrutinib and JQ1 were combined to determine whether they potentially could synergistically inhibit BCR signaling.33 Combination of JQ1 and ibrutinib was found to strongly inhibit tumor growth in ABC DLBCL mouse xenograft models to an extent greater than either inhibitor alone.33 Overall, these preclinical findings provide rationale for BET inhibitor clinical trials to include patients with ABC DLBCL.
c-Myc-dependent and double-hit lymphoma
Chromosomal translocations involving c-Myc are common to GCB DLBCL and essential for classification of double-hit lymphoma. Overall, up to 32% of DLBCL cases demonstrate c-Myc overexpression, and clinically, high-level c-Myc protein expression is an adverse prognostic factor.34 Given the important role of the c-Myc oncogene in DLBCL, JQ1 was examined in DLBCL in order to determine if c-Myc expression can be inhibited, thereby inhibiting cell proliferation and survival. In one study, a panel of eleven human DLBCL cell lines, including seven GCB DLBCL cell lines, were treated with JQ1 for 3 days, and all cell lines demonstrated decreased viability in a dose-dependent manner.35 Both cell-cycle arrest and apoptosis were seen in lymphoma cell lines treated with JQ1.35 Further, JQ1 significantly reduced c-Myc protein expression in ten of eleven cell lines.35 This included five cell lines with c-Myc translocations, and two cell lines with c-Myc amplifications.35 Finally, in an NOD mouse model xenografted with a c-Myc-translocated DLBCL cell line, JQ1 was found to suppress tumor growth and improve survival.32 Another BET inhibitor, OTX015, has also been evaluated in a panel of B-cell neoplasms, including GCB and ABC DLBCL, and was found to have antiproliferative effects through c-Myc target gene downregulation.4 These findings were replicated using a third BET inhibitor, I-BET151.12 Therefore, preclinical evidence demonstrates that BET inhibition can specifically downregulate c-Myc, leading to decreased proliferation and survival in c-Myc-dependent DLBCL. Overall, through inhibition of IKK and subsequently NF-κB or c-Myc, BET inhibition has been observed to have preclinical efficacy in a range of DLBCL subtypes warranting clinical trials examining BET inhibitors in lymphomas.
Treatment of Burkitt lymphoma
Burkitt lymphoma (BL) represents a highly aggressive lymphoma, characterized by acute onset and rapid doubling time. BL clinically presents either as an endemic form, typically in African children, or sporadically, which is most commonly seen in developed nations affecting mainly children, and less commonly adults.36 Genetically, BL is characterized by translocations involving the c-Myc gene on chromosome 8. The most commonly observed translocation is t(8;14), but translocations t(2;8) and t(8;22) are also seen in BL.36 These result in a fusion of an immunoglobulin heavy- or light-chain gene promoter to c-Myc, leading to c-Myc overexpression and lymphomagenesis. Treatment for BL involves aggressive chemotherapy, and almost 80% achieve long-term remission.36 However, when BL is found to be refractory to initial therapy, or there is relapse, salvage chemotherapy is ineffective, and prognosis is extremely poor.
BET inhibition has been studied in BL due to both the critical role of c-Myc and the need for more effective therapies in the relapsed/refractory setting. Treatment of three BL cell lines with JQ1 inhibited proliferation.3 JQ1 potently decreased c-MYC expression in the BL cell line Raji in vitro and inhibited tumor growth and significantly increased survival in a mouse model xenografted with the Raji cell line.3 Similar to findings reported in DLBCL cell lines, various other BET inhibitors, including OTX015 and I-BET151, also demonstrated antiproliferative effects when applied to various BL cell lines.4,35 These preclinical results all support clinical investigation of BET inhibitors in patients with BL.
Treatment of mantle cell lymphoma
Mantle cell lymphoma (MCL) represents a mature B-cell NHL and accounts for approximately 7% of all NHLs.37 MCL typically has a characteristic translocation, t(11;14), which fuses the cyclin D1 gene to the immunoglobulin heavy locus gene, resulting in cyclin D1 overexpression.37 Similar to other B-cell lymphomas, MCL has been found to have various genetic alterations resulting in increased BCR and NF-κB activity.37 BRD4 has been implicated in the regulation of cyclin D1 transcription, and transcription of BCR- and NF-κB-related genes. Therefore, JQ1 and I-BET151 were studied in MCL. Treatment with BET inhibitors resulted in increased apoptosis in all cell lines.38 Further, when cell-cycle effect was evaluated, treatment of two MCL cell lines with JQ1 resulted in cell-cycle arrest.38 Mechanistically, treatment with JQ1 was found to result in depletion of c-Myc and BCL2 gene expression, and depletion of NF-κB target gene expression including expression of BTK in MCL cells.38 Finally, due to observed BTK depletion, JQ1 and ibrutinib were evaluated in MCL for synergy.38 Cotreatment of MCL cells with JQ1 and ibrutinib synergistically induced apoptosis greater than either inhibitor alone.38 Further, in NOD mice xenografted with the MCL cell line Mino, cotreatment with JQ1 and ibrutinib impaired tumor proliferation greater than either inhibitor alone, and also significantly improved survival over single-agent treatment.38 These findings provide rationale for BET inhibitors in MCL. Additionally, due to synergy with ibrutinib, BET inhibitors potentially offer an option in the setting of ibrutinib resistance in MCL.
Treatment of less common lymphomas
Beyond these commonly occurring lymphomas, several less common lymphomas reportedly are sensitive to BET inhibition. These include primary effusion lymphoma (PEL), which is associated with Kaposi sarcoma-associated herpesvirus (KSHV/HHV-8) and carries a poor prognosis. KSHV-encoded latent proteins trigger c-Myc deregulation, resulting in PEL proliferation.39 PEL cell lines treated with JQ1 and I-BET151 were highly sensitive to BET inhibition, with decreased proliferation and cell survival.39 Additional studies also point to a potential role for BET inhibition in anaplastic T-cell lymphoma, marginal zone lymphoma, and small lymphocytic lymphoma/chronic lymphocytic leukemia.4,40,41
BET inhibitors and multiple myeloma
MM is a malignancy characterized by an abnormal proliferation of dysfunctional plasma cells in the bone marrow, often leading to increase in immunoglobulin expression and potential organ dysfunction.42 MM represents the second commonest hematologic malignancy in the US; over 20,000 patients are newly diagnosed annually, and 10,000 patients die each year.42 Significant therapeutic discoveries have occurred for the treatment of MM leading to improved survival; however, MM is still considered incurable. The molecular pathogenesis of MM is heterogeneous and complex. Similar to NHL, activation of c-Myc and NF-κB target gene expression play an important role in the pathogenesis of MM.42 Additionally, activation of the transcription factor IRF4 is thought to play an important role in the development of MM.42 Beyond aberrant gene expression, IL-6 cytokine signaling is also felt to play an important role in the growth and survival of MM.42 Table 1 summarizes potential targets in MM sensitive to BET inhibition.
Given that c-Myc transcription plays an important role in the pathogenesis of MM, and BET inhibition was initially evaluated in MM cell lines in order to inhibit c-Myc transcription. Treatment of three MM cell lines with JQ1 resulted in significant downregulation of c-Myc-regulated genes.1 Further, the c-Myc gene itself was inhibited by JQ1.1 The antiproliferative effect of JQ1 was next assessed against a panel of 25 MM cell lines, and JQ1 demonstrated uniform inhibition of proliferation.1 These results were confirmed in primary MM samples. Finally, JQ1 was assessed in vivo in an MM tumor-bearing mouse model. Treatment of these xenografted mice with JQ1 resulted in significantly decreased tumor burden, decreased immunoglobulin expression, and a significant improvement in overall survival.1 A second mouse model representative of extramedullary MM, or plasmacytoma, also demonstrated a significant response to JQ1.1
Beyond therapeutic targeting of c-Myc, BET inhibition plays a possible role in suppressing IL-6. Because IL-6 is thought to play an important role in MM pathogenesis, JQ1 was applied to MM cells to determine if IL-6 secretion was suppressed.43 JQ1 significantly reduced lipopolysaccharide-induced IL-6 secretion in primary MM cells. JQ1 treatment also inhibited IL-6 transcription.43 Finally, gene expression profiling of the MM cell line KMS12BM demonstrated that BET inhibition also downregulated IRF4-dependent transcription.44 These data provide additional mechanisms for BET inhibition in MM beyond repression of c-Myc.
BET inhibitors in allogeneic SCT
SCT is an important treatment option in aggressive lymphomas and acute leukemia due to a potential graft-versus-tumor affect, which can provide long-term disease control.45 However, allogeneic SCT is associated with significant morbidity and mortality due to graft-versus-host disease (GVHD).45 Acute GVHD occurs when donor T-cells are primed by recipient antigens subsequently triggering an inflammatory reaction against the host. Both dendritic cells (DCs) and T-cells play an important role in mediating this inflammatory reaction in acute GVHD, as priming triggers these cells to secrete inflammatory cytokines, such as IL-6 and TNF-α.45 Given the broad transcriptional regulatory role of BRD4, particularly regulation of transcription of inflammatory factors, the ability of BET inhibitors to reduce DC and T-cell function was evaluated. Treatment with I-BET151 resulted in significant reduction in the secretion of IL-6, TNF-α, and IL-12 by stimulated DCs.46 Additionally, various DC surface markers were downregulated on stimulated DCs by I-BET151.46 Overall, DC function was felt to be significantly reduced by BET inhibition. Additionally, I-BET151 significantly reduced T-cell proliferation.46 In terms of mechanism, BET inhibition was found to disrupt the interaction between BRD4 and acetyl-310 RelA in both DCs and T-cells.46 This resulted in a decrease in acetylation-dependent transcription events. When tested in vivo in GVHD mouse models, I-BET151-treated mice lived significantly longer, with significantly reduced GVHD severity, and without any changes in donor chimerism or engraftment or tumor-related mortality.46 Therefore, BET inhibitors may serve as a prophylactic therapy against acute GVHD without affecting the graft-versus-tumor effect.
BET inhibitors in clinical trials for hematologic malignancies
At present, extensive efforts have led to the development of several BET inhibitors currently in clinical trial for both leukemias and lymphomas. Table 2 provides a summary of current clinical trials utilizing BET inhibitors for various hematologic malignancies. Recently, two completed Phase I trials utilized the BET inhibitor OTX015 either in leukemia or in lymphoma and MM.5,6 These published results give a window into the toxicity and initial response to BET inhibition.
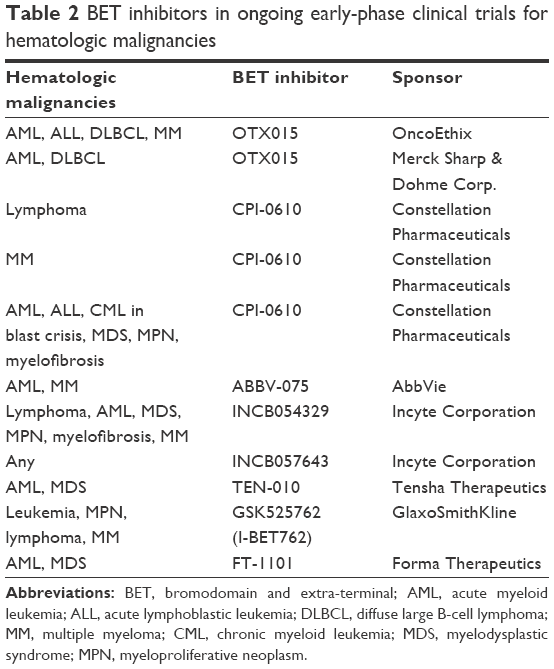 | Table 2 BET inhibitors in ongoing early-phase clinical trials for hematologic malignancies |
Based on preclinical data, OTX015 was evaluated in a Phase Ib trial in relapsed/refractory leukemia patients.5 In total, 36 patients with AML, with a median age of 70 years, with most having received at least two lines of prior therapy, were enrolled.5 In addition, three patients with ALL, one patient with acute undifferentiated leukemia, and one patient with myelodysplastic syndrome were also enrolled. The primary end point was determining the dose safe for further studies, which was found to be 80 mg once daily given 14 days on and 7 days off.5 At all doses, the main side effects observed were gastrointestinal (diarrhea, nausea, vomiting), fatigue, and hyperbilirubinemia.5 Most gastrointestinal side effects were grade 1 or 2 in severity, whereas several patients had grade ≥3 fatigue or hyperbilirubinemia. Overall, there were no treatment-related deaths and no patients discontinued treatment due to adverse effects.5 Treatment response assessments revealed that three patients achieved either a complete remission or complete remission with incomplete platelet recovery.5 An additional two patients had partial blast clearance, and several other patients had a transient decrease in blast count and increased neutrophil count.
OTX015 was also investigated in a Phase Ib trial in relapsed/refractory lymphoma and myeloma patients.6 A total of 45 patients, 33 with lymphoma and 12 with myeloma, with a median age of 66 years, having received a median of four prior lines of chemotherapy, were enrolled.6 Similar to the leukemia study utilizing this drug, the primary objective was dose determination, and this study also recommended a dose of 80 mg/day on a 14-day on and a 7-day off schedule.6 In terms of toxicities, gastrointestinal side effects were still present, but cytopenias were more prominent, and grade 3 thrombocytopenia was seen in 58% of patients.6 Three patients with DLBCL achieved durable CR, and six additional patients with lymphoma demonstrated evidence of clinical activity, defined as any measurable disease change.6
Overall, both studies demonstrate that OTX015 could be safely administered to older patients with advanced hematologic malignancies. Phase II studies are required to further evaluate treatment response, and to determine if any biomarkers can predict clinical efficacy.
Mechanisms of BET inhibitor resistance in hematologic malignancies
The promising therapeutic potential of BET inhibitors has led to recent efforts on identifying potential mechanisms by which resistance develops to BET inhibitors. In leukemia models, two groups identified that resistant cells can restore c-Myc expression mediated through Wnt signaling.47,48 One study utilized MLL-AF9-transduced progenitor cells, and developed cell clones resistant, in vitro and in vivo, to the BET inhibitor I-BET151.47 Transcriptional profiling of the resistant clones was performed, and significant upregulation of the Wnt/β-catenin and TGF-β pathways was seen.47 When the Wnt/β-catenin pathway was antagonized, the resistant clones regained sensitivity to I-BET151 inhibition.45 Conversely, when the Wnt/β-catenin pathway was stimulated in BET inhibitor-sensitive cells, this conferred rapid resistance to I-BET151. A second study demonstrated that in leukemia cells, acquired suppression of the PRC2, which typically functions as a histone methyltransferase that represses gene expression, resulted in increased Wnt signaling and restoration of c-Myc transcription.48 Overall, these studies demonstrate that resistant leukemia can utilize alternate pathways, namely Wnt signaling, to restore c-Myc expression and c-Myc-driven proliferation. Consideration of mechanisms to inhibit Wnt signaling may ultimately improve clinical efficacy of BET inhibitors.
Combination strategies utilizing BET inhibitors
Growing evidence suggests that selective targeting of a single oncogenic target is unlikely to result in sustained or sufficient clinical responses. Thus, there are increasing efforts to identify rational targeted drug combinations for synergy and to overcome potential resistance mechanisms against targeted therapies.
In leukemia, several combinations have been explored with BET inhibitors. In AML, the backbone of conventional intensive chemotherapy is ara-C. Therefore, JQ1 has been combined with ara-C to determine whether synergistic growth inhibition could be observed. When JQ1 and ara-C were combined in AML cell lines HL60 and KG1, a synergistic effect was observed with greater inhibition of proliferation compared to either drug alone.49 Azacitidine is another approved chemotherapy agent for AML and has been combined with the BET inhibitor OTX015 to determine whether synergy could be observed. Both simultaneous and sequential treatment of the AML cell line Kasumi with azacitidine and OTX015 resulted in synergistic growth proliferation.50 Beyond conventional therapies for AML, BET inhibitors have been combined with FLT3 inhibitors in FLT3 AML and demonstrated synergistic antiproliferative effects.23 Finally, the combination of the HDAC inhibitor panobinostat with JQ1 synergistically attenuated c-Myc and BCL2, resulting in greater apoptosis in AML cell lines and significantly improved survival in NOD mice xenografted with AML cells.51
In lymphoma, preclinical data suggest that BET inhibitors have the potential to synergize with several targeted agents presently used in practice. In one study, the BET inhibitor OTX015 was examined as a single agent, or in combination with target agents, against established cell lines for DLBCL. Antiproliferative synergism was demonstrated in DLBCL when OTX015 was combined with rituximab, the mTOR inhibitor everolimus, the PI3K-δ inhibitor idelalisib, the HDAC inhibitor vorinostat, the hypomethylating agent decitabine, and the immunomodulant lenalidomide.4 In addition, among ABC DLBCL cell lines, the combination of OTX015 and ibrutinib was demonstrated to have synergistic antiproliferative effects.4 Combination therapy with OTX015 and bendamustine, doxorubicin, and the HDAC inhibitor romidepsin was demonstrated to have an additive antiproliferative effect.4 Further, combination therapy with BET inhibitors was examined in MCL. As mentioned earlier, one study demonstrated that ibrutinib and JQ1 were synergistically lethal in MCL.38 Another study demonstrated that in bortezomib-resistant MCL, the combination of JQ1 with lenalidomide was effective and inhibited proliferation synergistically.52 Finally, BET inhibition in combination with bortezomib, a proteasome inhibitor used in MM, was evaluated in MM cell lines with acquired resistance to bortezomib and melphalan. The BET inhibitor CPI203, in combination with bortezomib, synergistically reduced viability in the resistant MM cell lines, and additionally reduced viability in a bortezomib-resistant patient sample.53
Overall, in both leukemias and lymphomas, BET inhibition preclinically appears to enhance the antiproliferative effects of various therapies in current use for these diseases. Development of combination strategies using targeted agents will likely be important to adequately treat hematologic malignancies in the future, similar to the success that was seen when combination chemotherapy regimens were developed. With an acceptable toxicity profile demonstrated in the Phase I trials utilizing OTX015, and preclinical evidence that BET inhibitors synergize with a number of targeted therapies currently in clinical use, summarized in Table 3, it is possible that BET inhibitors will play an important role when combination strategies are devised in the future.
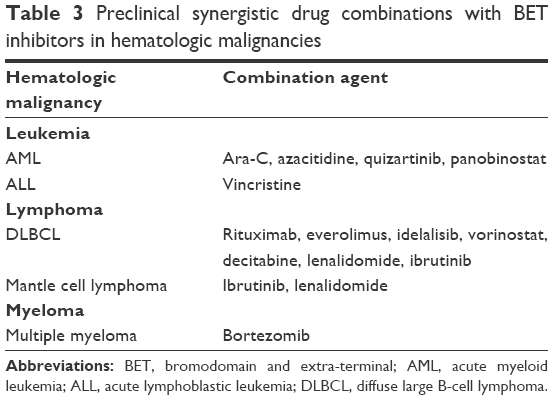 | Table 3 Preclinical synergistic drug combinations with BET inhibitors in hematologic malignancies |
Conclusion
A growing body of preclinical evidence demonstrates that BET inhibition, and subsequent transcriptional inhibition, is an important therapeutic approach in a number of hematologic malignancies. Beyond c-Myc, BET inhibition has been demonstrated to repress aberrant oncogenic transcription driven by genetic abnormalities in AML, including by NPM1 mutations and MLL translocations. Further, in lymphomas, repression of transcriptional activation due to BCR activation or NF-κB activation makes BET inhibition broadly effective in a diverse range of pathologies. The immunomodulatory effect of BET inhibition, particularly on natural killer cells, may make BET inhibition an attractive strategy after SCT. Significant clinical efforts are underway to determine the safety of BET inhibitors in clinical practice. Initial reports indicate that BET inhibition is likely safe. To maximize the potential of BET inhibition, future studies should focus on the development of combinations that address resistance to BET inhibition, or synergistic combinations that may ultimately be able to address relapsed or refractory cases of leukemia, or lymphoma, for which currently few options exist.
Acknowledgment
This work was supported by grant R01CA186885 (to HG Munshi).
Disclosure
The authors declare no conflicts of interest in this work.
References
Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–917. | ||
Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073. | ||
Mertz JA, Conery AR, Bryant BM, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108(40):16669–16674. | ||
Boi M, Gaudio E, Bonetti P, et al. The BET bromodomain inhibitor OTX015 affects pathogenetic pathways in preclinical B-cell tumor models and synergizes with targeted drugs. Clin Cancer Res. 2015;21(7):1628–1638. | ||
Berthon C, Raffoux E, Thomas X, et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol. 2016;3(4):e186–e195. | ||
Amorim S, Stathis A, Gleeson M, et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016;3(4):e196–e204. | ||
Wang CY, Filippakopoulos P. Beating the odds: BETs in disease. Trends Biochem Sci. 2015;40(8):468–479. | ||
Sahai V, Redig AJ, Collier KA, Eckert FD, Munshi HG. Targeting bet bromodomain proteins in solid tumors. Oncotarget. 2016;7(33):53997–54009. | ||
Loven J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–334. | ||
Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;152(2):307–319. | ||
Hnisz D, Abraham BJ, Lee TI, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–947. | ||
Chapuy B, McKeown MR, Lin CY, et al. Discovery and characterization of super-enhancer associated dependencies in diffuse large B-cell lymphoma. Cancer Cell. 2013;24(6):777–790. | ||
Howlader N, Noone AM, Krapcho M, et al. SEER cancer statistics review, 1975–2013. Bethesda, MD: National Cancer Institute. Available from: http://seer.cancer.gov/csr/1975_2013/. Accessed June 16, 2016. | ||
Rowe JM, Li X, Cassileth PA, et al. Very poor survival of patients with AML who relapse after achieving a first complete remission: the Eastern Cooperative Oncology Group experience. Blood. 2005;106(11):546. | ||
Döhner K, Schlenk RF, Habdank M, et al. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005;106(12):3740–3746. | ||
Schlenk RF, Döhner K, Krauter J, et al; German-Austrian Acute Myeloid Leukemia Study Group. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358(18):1909–1918. | ||
Thirman MJ, Gill HJ, Burnett RC, et al. Rearrangement of the MLL gene in acute lymphoblastic and acute myeloid leukemias with 11q23 chromosomal translocations. N Engl J Med. 1993;329(13):909–914. | ||
Dawson MA, Gudgin EJ, Horton SJ, et al. Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia. 2014;28(2):311–320. | ||
Falini B, Bolli N, Shan J, et al. Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc + AML. Blood. 2006;107(11):4514–4523. | ||
Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7(11):823–833. | ||
Dawson MA, Prinjha RK, Dittmann A, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478(7370):529–533. | ||
Kindler T, Lipka DB, Fischer T. FLT3 as a therapeutic target in AML: still challenging after all these years. Blood. 2010;116(24):5089–5102. | ||
Fiskus W, Sharma S, Qi J, et al. BET protein antagonist JQ1 is synergistically lethal with FLT3 tyrosine kinase inhibitor (TKI) and overcomes resistance to FLT3-TKI in AML cells expressing FLT-ITD. Mol Cancer Ther. 2014;13(10):2315–2327. | ||
Roll JD, Reuther GW. CRLF2 and JAK2 in B-progenitor acute lymphoblastic leukemia: a novel association in oncogenesis. Cancer Res. 2010;70(19):7347–7352. | ||
Ott CJ, Kopp N, Bird L, et al. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood. 2012;120(14):2843–2852. | ||
Picaud S, Costa DD, Thanasopoulou A, et al. PFI-1 – a highly selective protein interaction inhibitor targeting BET bromodomains. Cancer Res. 2013;73(11):3336–3346. | ||
Loosveld M, Castellano R, Gon S, et al. Therapeutic targeting of c-Myc in T-cell acute lymphoblastic leukemia, T-ALL. Oncotarget. 2014;5(10):3168–3172. | ||
Coiffier B, Thieblemont C, Van Den Neste E, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d’Etudes des Lymphomes de l’Adulte. Blood. 2010;116(12):2040–2045. | ||
Pfreundschuh M, Kuhnt E, Trümper L, et al; MabThera International Trial (MInT) Group. CHOP-like chemotherapy with or without rituximab in young patients with good-prognosis diffuse large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera International Trial (MInT) Group. Lancet Oncol. 2011;12(11):1013–1022. | ||
Gisselbrecht C, Glass B, Mounier N, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010;28(27):4184–4190. | ||
Scott DW, Mottok A, Ennishi D, et al. Prognostic significance of diffuse large B-cell lymphoma cell of origin determined by digital gene expression in formalin-fixed paraffin-embedded tissue biopsies. J Clin Oncol. 2015;33(26):2848–2856. | ||
Aukema SM, Siebert R, Schuuring E, et al. Double-hit B-cell lymphomas. Blood. 2011;117(8):2319–2331. | ||
Ceribelli M, Kelly PN, Shaffer AL, et al. Blockade of oncogenic IκB kinase activity in diffuse large B-cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proc Natl Acad Sci U S A. 2014;111(31):11365–11370. | ||
Horn H, Ziepert M, Becher C, et al; German High-Grade Non-Hodgkin Lymphoma Study Group. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood. 2013;121(12):2253–2263. | ||
Trabucco SE, Gerstein RM, Evens AM, et al. Inhibition of bromodomain proteins for the treatment of human diffuse large B-cell lymphoma. Clin Cancer Res. 2015;21(1):113–122. | ||
Blum KA, Lozanski G, Byrd JC. Adult Burkitt leukemia and lymphoma. Blood. 2004;104(10):3009–3020. | ||
Pérez-Galán P, Dreyling M, Wiestner A. Mantle cell lymphoma: biology, pathogenesis, and the molecular basis of treatment in the genomic era. Blood. 2011;117(1):26–38. | ||
Sun B, Shah B, Fiskus W, et al. Synergistic activity of BET protein antagonist-based combinations in mantle cell lymphoma cells sensitive or resistant to ibrutinib. Blood. 2015;126(13):1565–1574. | ||
Tolani B, Gopalakrishnan R, Punj V, Matta H, Chaudhary PM. Targeting Myc in KSHV-associated primary effusion lymphoma with BET bromodomain inhibitors. Oncogene. 2014;33(22):2928–2937. | ||
Boi M, Bonetti P, Ponzoni M, et al. The Brd-inhibitor OTX015 shows pre-clinical activity in anaplastic large T-cell lymphoma (ALCL). Blood. 2012;120(21):4872. | ||
Larsson CA, Kojima K, Wang Y, et al. BET bromodomain inhibition reduces leukemic burden and prolongs survival in the Eμ-TCL1 transgenic mouse model of chronic lymphocytic leukemia (CLL) independent of TP53 mutation status. Blood. 2013;122(21):876. | ||
Mahindra A, Hideshima T, Anderson KC. Multiple myeloma: biology of the disease. Blood Rev. 2010;24 Suppl 1:S5–S11. | ||
Ghurye RR, Stewart HJS, Chevassut TJ. Bromodomain inhibition by JQ1 suppresses lipopolysaccharide-stimulated interleukin-6 secretion in multiple myeloma cells. Cytokine. 2015;71(2):415–417. | ||
Chaidos A, Caputo V, Gouvedenou K, et al. Potent antimyeloma activity of the novel bromodomain inhibitors I-BET151 and I-BET762. Blood. 2014;123(5):697–705. | ||
Antin JH, Raley DY. Manual of Stem Cell and Bone Marrow Transplantation. New York, NY: Cambridge University Press; 2009. | ||
Sun Y, Wang Y, Toubai T, et al. BET bromodomain inhibition suppresses graft-versus-host disease after allogeneic bone marrow transplantation in mice. Blood. 2015;125(17):2724–2748. | ||
Fong CY, Gilan O, Lam EYN, et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature. 2015;525(7570):538–542. | ||
Rathert P, Roth M, Neumann T, et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature. 2015;525(7570):543–547. | ||
Herrmann H, Blatt K, Shi J, et al. Small-molecule inhibition of BRD4 as a new potent approach to eliminate leukemic stem- and progenitor cells in acute myeloid leukemia (AML). Oncotarget. 2012;3(12):1588–1599. | ||
Coudé M-M, Braun T, Berrou J, et al. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget. 2015;6(19):17698–17712. | ||
Fiskus W, Sharma S, Qi J, et al. Highly active combination of BRD4 antagonist and histone deacetylase inhibitor against human acute myelogenous leukemia cells. Mol Cancer Ther. 2014;13(5):1142–1154. | ||
Moros A, Rodríguez V, Saborit-Villarroya I, et al. Synergistic antitumor activity of lenalidomide with the BET bromodomain inhibitor CPI203 in bortezomib-resistant mantle cell lymphoma. Leukemia. 2014;28(10):2049–2059. | ||
Siegel MB, Liu SQ, Davare MA, et al. Small molecule inhibitor screen identifies synergistic activity of the bromodomain inhibitor CPI203 and bortezomib in drug resistant myeloma. Oncotarget. 2015;6(22):18921–18932. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.