")
Back to Journals » International Journal of General Medicine » Volume 7
Benefits of chronic blood transfusion in hemoglobin E/β thalassemia with pulmonary arterial hypertension
Authors Chueamuangphan N, Patumanond J, Wongtheptien W, Nawarawong W, Sukonthasarn A , Chuncharunee S, Tawichasri C
Received 21 April 2014
Accepted for publication 1 July 2014
Published 19 August 2014 Volume 2014:7 Pages 411—416
DOI https://doi.org/10.2147/IJGM.S66610
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Nonlawan Chueamuangphan,1,3 Jayanton Patumanond,2 Wattana Wongtheptien,3 Weerasak Nawarawong,4 Apichard Sukonthasarn,4 Suporn Chuncharunee,5 Chamaiporn Tawichasri6
1Clinical Epidemiology Program, Faculty of Medicine, Chiang Mai University, Chiang Mai, 2Clinical Epidemiology Program, Faculty of Medicine, Thammasat University, Bangkok, 3Department of Medicine, Chiang Rai Hospital, Chiang Rai, 4Department of Medicine Faculty of Medicine, Chiang Mai University, Chiang Mai, 5Department of Medicine, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Bangkok, 6Clinical Epidemiology Society at Chiang Mai, Chiang Mai, Thailand
Objective: The aim of the research reported here was to compare pulmonary artery systolic pressure (PASP) and 6-minute walk distance after 1 year of follow-up in hemoglobin E/β thalassemia (E/β-Thal) with pulmonary arterial hypertension (PAH) patients who received chronic blood transfusions versus those who received occasional transfusions.
Methods: A nonrandomized clinical trial was conducted at the Hematological Outpatient Clinic of Chiang Rai Hospital, Thailand. All adult cases of E/β-Thal with PAH (defined as PASP >35 mmHg by Doppler echocardiography) were evaluated and followed for the next 12 months. The patients were classified into two groups by patient preference. Group 1 patients received chronic blood transfusions – one to two units of leukocyte-poor packed red cells every 2–4 weeks – over 1 year to maintain pre-transfusion hemoglobin levels of ≥7.0 g/dL. Group 2 patients received occasional transfusions over the course of 1 year, with more than 4 weeks between transfusions. All patients were treated with iron chelation when serum ferritin levels were ≥1,000 µg/dL. PASP and the 6-minute walk distance were evaluated at baseline and at 6 and 12 months. Propensity score adjustment was used to control for confounding by indication and contraindication. Multivariable regression analysis was used to evaluate the effects of chronic blood transfusion.
Results: There were 16 (53.3%) patients in Group 1 and 14 (46.7%) in Group 2. At 12 months, patients in Group 1 had a greater reduction in PASP than those in Group 1 (adjusted mean difference, −16.83; 95% confidence interval, −26.35 to −7.32; P=0.001). The 6-minute walk distance at 12 months in Group 1 patients was greater than that in Group 2 patients (adjusted mean difference, 46.55; 95% confidence interval, 18.08 to 75.02; P=0.001).
Conclusion: This study found evidence that chronic blood transfusions may have beneficial effects in PAH in thalassemia patients over 1 year.
Keywords: pulmonary artery systolic pressure, Thailand, 6-minute walk distance, leukocyte-poor packed red cells
Introduction
“Pulmonary arterial hypertension” (PAH) is a common cardiovascular complication in thalassemic patients. It is a progressive disease that causes exercise limitation, right ventricular failure, and death. One study found that almost 60% of cases in a large cohort of 110 thalassemia intermedia (TI) patients had developed PAH.1 Its prevalence in Thailand has been found to be 43%.2 Another study has reported thrombi in small pulmonary arteries in 44% of hemoglobin E/β thalassemia (E/β-Thal) autopsies.3 PAH is due to increased pulmonary vascular resistance.1 In thalassemia, PAH could be due to an increased pulmonary blood flow from chronic anemia, and the oxidative stress resulting from chronic hemolysis is enhanced by iron overload. The possible mechanisms leading to PAH in thalassemia may involve platelet activation and the coagulation cascade.4–10 Therapeutic strategies for dealing with PAH in thalassemia are controversial. Some authors have suggested that the PAH development can be prevented by starting transfusion and chelation therapy early in life for patients with TI.1,11
In the study reported here, we compared pulmonary artery systolic pressure (PASP) and 6-minute walk distance after 1 year of follow-up in E/β-Thal with PAH patients who received chronic blood transfusions versus those who received occasional transfusions.
Materials and methods
A nonrandomized clinical trial was conducted at the Hematological Outpatient Clinic at Chiang Rai Hospital, Thailand. All adult (≥15 years) cases of E/β-Thal with PAH (defined as PASP >35 mmHg by Doppler echocardiography) attending the clinic between June 2011 and November 2012 were evaluated and followed for the next 12 months. Inclusion criteria consisted of TI and serum ferritin level <2,500 μg/dL in patients capable of being followed-up who signed informed consent to participate in the study. Exclusion criteria were patients who had clinical evidence of other secondary causes of PAH, including human immunodeficiency virus infection, collagen vascular diseases, cirrhosis, and chronic obstructive airway diseases, acquired heart disease associated with pulmonary venous hypertension; mitral valve disease; congenital heart disease; and hyperthyroidism.
After the physician advised patients that they may gain some benefit from chronic blood transfusions; patients were classified into one of two groups according to their preference. Group 1 patients were those with a preference for chronic blood transfusions. Patients in this group would receive one to two units of leukocyte-poor packed red cells every 2–4 weeks to maintain the pre-transfusion hemoglobin level of ≥7.0 g/dL over 1 year. Group 2 patients, comprising those patients who indicated a preference for occasional blood transfusions, would receive occasional transfusions not more often than every 4 weeks. All patients were treated with medications following standard guidelines, including intensive chelation therapy when their serum ferritin level was above 1,000 μg/dL.
Project exclusion rule
The patients who developed an alloantibody to red blood cells and for whom the blood bank could not regularly supply compatible blood and/or who had serious blood transfusion complications such as anaphylaxis, acute pulmonary edema, or a serum ferritin level >2,500 μg/dL (serum ferritin monitoring was undertaken every 3 months) were excluded from the trial and were managed by the hospital’s standard guidelines.
Blood transfusion
All transfusions were undertaken following the standard operating procedures of our blood bank, which adhere to the Thai Red Cross Society’s National Blood Center.12 To prevent alloimmunization, transfusions were of blood phenotypically matched for Rhesus (Rh) and other alloantibodies, with leukocyte-poor (depleted) packed red cells at 250–350 cc/unit, and the standard complication monitoring system for blood transfusions was observed.
Indicators
PASP and 6-minute walk distance13 were measured at baseline and at 6 and 12 months. The clinical outcomes consisted of functional class status, clinical right heart failure, and oxygen saturation. Functional class status was defined by the New York Heart Association (NYHA) Functional Classification14 (I: no symptoms with ordinary physical activity; II: symptoms with ordinary activity and slight limitation of activity; III: symptoms with less than ordinary activity, marked limitation of activity; IV: symptoms with any activity or even at rest). Echocardiography was performed by the same cardiologist. The patients’ history, including blood transfusion frequency and splenectomy status, was blinded during echocardiographic examinations.
The study protocol was approved by the Institutional Research Ethics Committee, Faculty of Medicine, Chiang Mai University and Chiang Rai Hospital.
Study size estimation
The sample size was estimated from a pilot study comparing the mean PASP difference between two independent groups. A mean PASP of the patients was recorded before and after treatments for 1 year. The calculation was based on a type I error at 0.05 and type II error at 0.1. When using a standard statistical package to calculate the number of samples needed with the ratio of the patients of the two groups being 1:1, six subjects per treatment group were required. We aimed to collect the data from at least 14 patients per treatment group.
Echocardiographic procedure
Complete two-dimensional, M-mode, and Doppler (pulsed wave, continuous wave, and color) echocardiography was performed at rest. In the study patients, a tricuspid regurgitation jet was sought from all available mid-precordial and apical positions until a flow signal with the maximum spectral representation of the highest velocities was obtained. Peak velocity was recorded from a holosystolic regurgitant jet. From the maximum velocity (V) of the regurgitant jet, the systolic pressure gradient (ΔP) between the right ventricle and the right atrium was calculated by the modified Bernoulli equation (ΔP =4V2).15 Right atrial pressure was estimated by the response of the inferior vena cava diameter to inspiration. Right atrial pressure was assumed to be 5 mmHg if the inferior vena cava completely collapsed with inspiration, 10 mmHg if the inferior vena cava diameter decreased more than 50% during inspiration, and 15 mmHg if it decreased less than 50%. If the inferior vena cava diameter was larger than 2.5 cm and reduced by less than 50% during inspiration, right atrial pressure was assumed to be 20 mmHg.16 Adding the transtricuspid gradient to the mean right atrial pressure provided the right ventricular systolic pressure or peak systolic pulmonary arterial pressure in the absence of right ventricular outflow tract obstruction. PAH was defined as a PASP >35 mmHg.17 In cases with an absence of tricuspid regurgitation, the end of the diastolic pulmonic regurgitant flow velocity was calculated using the pressure gradient between the pulmonary artery and right ventricular end diastolic pressure. Pulmonary artery pressure =4 (VPR)2 + right atrial pressure.
Statistical analysis
The baseline characteristics were compared using exact probability tests for categorical variables; Student’s t-test or Wilcoxon rank-sum test was used to compare the mean difference of continuous variables. Propensity score adjustment was used to control confounding by chronic blood transfusion indication and contraindication. Propensity scores for receiving chronic blood transfusions versus occasional transfusions were calculated from a logistic regression model that estimated the likelihood of receiving chronic blood transfusions based on the observed patient characteristics. Gaussian and an exponential risk regression were carried out. Uni-variable and multivariable regression analyses were used to evaluate the effects of chronic blood transfusions. Data are presented by frequency, percentage, mean, median, standard deviation (SD), range, beta coefficient, 95% confidence interval, and P-value. All statistical analyses were two-tailed. A P-value of <0.05 was considered statistically significant.
Results
A total of 30 patients with E/β-Thal were enrolled in the study, 16 (53.3%) in the chronic blood transfusion group (Group 1) and 14 (46.7%) in the occasional transfusion group (Group 2). The mean age of the patients was 36.1±14.6 and 34.1±15.2 years. Patients’ mean PASP was 59.7±15.1 and 49.0±13.8 mmHg, while eight (50.0%) and four (28.6%) patients were post-splenectomy in Group 1 and Group 2, respectively. The baseline characteristics of the patients did not differ significantly between the two groups, except in terms of NYHA classification and PAH severity (Table 1). More patients in Group 1 had a functional NYHA class of III than those in Group 2 (5 [31.3%] vs 0 [%] patients, P=0.045) and PAH severity was greater in Group 1 than in Group 2 (mild: 4 [25.0%] vs 10 [71.4%] patients, P=0.049; moderate: 8 [50.0%] vs 2 [14.3%]; severe: 4 [25.0%] vs 2 [14.3%]).
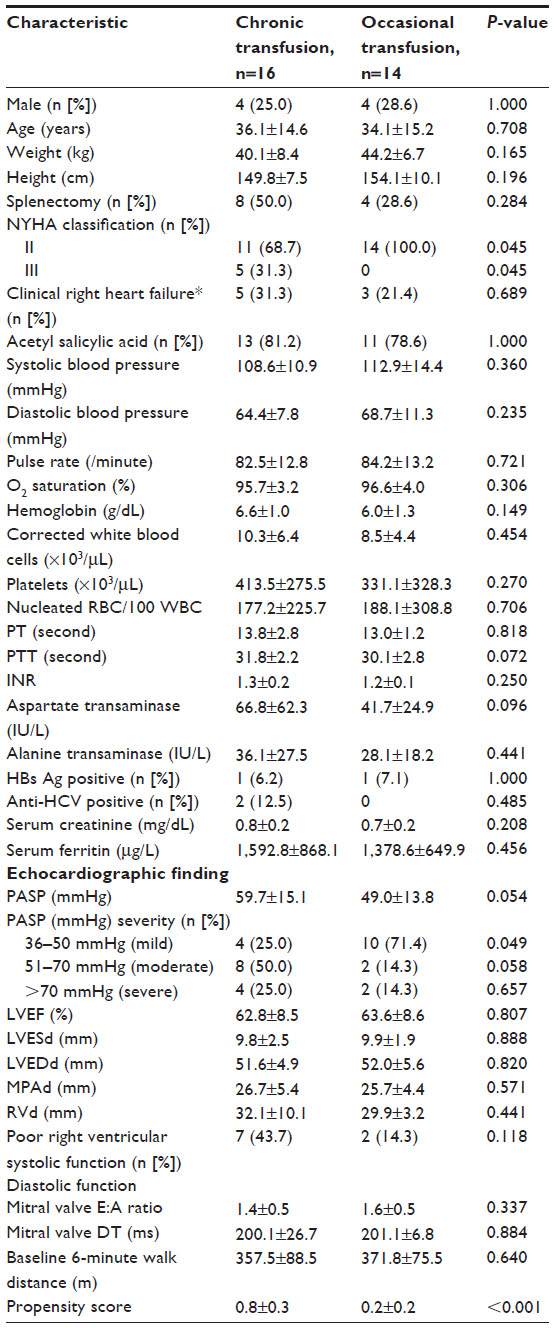 | Table 1 Baseline characteristics of the patientsa |
During the follow-up period, the mean PASP was 50.4±14.6 versus 49.4±16.9 at 6 months and 48.8±18.8 versus 54.1±21.4 mmHg at 12 months, in Groups 1 and 2, respectively. At 6 months, mean 6-minute walk distance was 388.7±59.2 versus 368.6±61.1 m, and 403.7±53.8 versus 350.0±82.9 m at 12 months in Group 1 and Group 2, respectively, without statistical significance (Table 2).
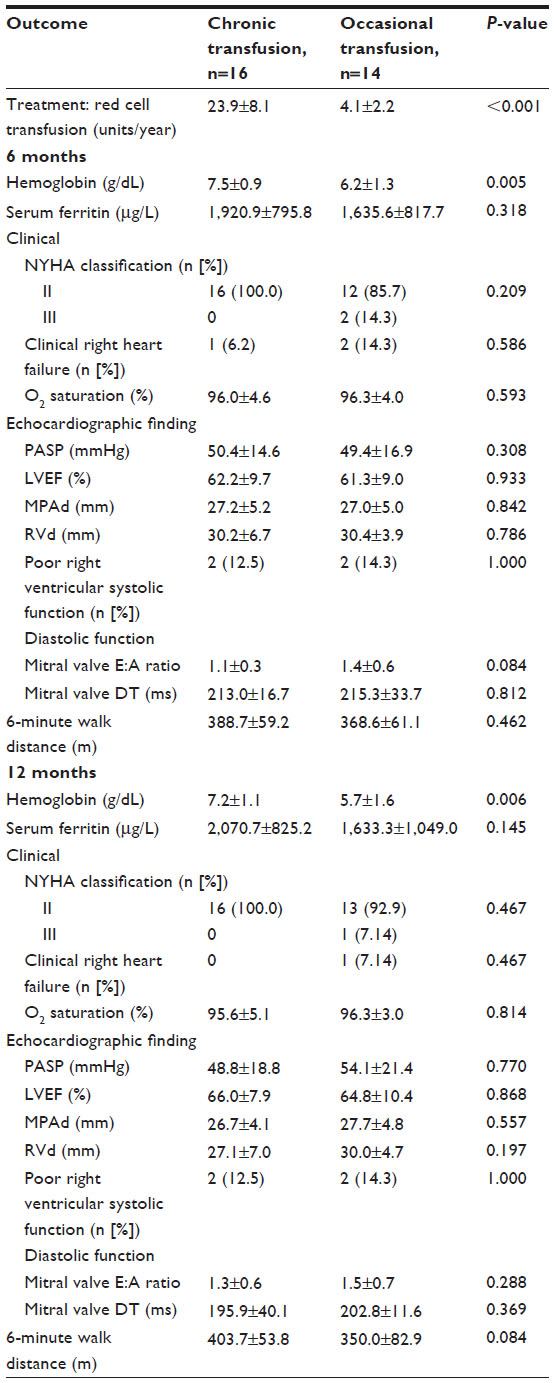 | Table 2 Comparison of clinical outcomes, echocardiographic findings, and 6-minute walk distance at 6 and 12 monthsa |
After adjusting for propensity score (age, splenectomy, acetyl salicylic acid, NYHA class, clinical right heart failure, O2 saturation, baseline PASP, left ventricular ejection fraction, diastolic function, hemoglobin level, platelet count, nucleated red cell, baseline serum ferritin level, and baseline 6-minute walk distance) and right ventricular systolic function, after 12 months of treatment, Group 1 had a mean PASP reduction greater than that of Group 2 (adjusted mean difference, −16.83; 95% confidence interval [CI], −26.35 to −7.32; P=0.001). The mean 6-minute walk distance at 12 months in Group 1 was greater than that in Group 2 (adjusted mean difference, 46.55; 95% CI, 18.08 to 75.02; P=0.001; Table 3). Chronic blood transfusions improved functional class status and clinical outcome of clinical right heart failure at 6 and 12 months after treatment. None of the patients had clinical evidence of thromboembolism (deep-vein thrombosis, pulmonary embolism, stroke). No patients were prematurely terminated from the study.
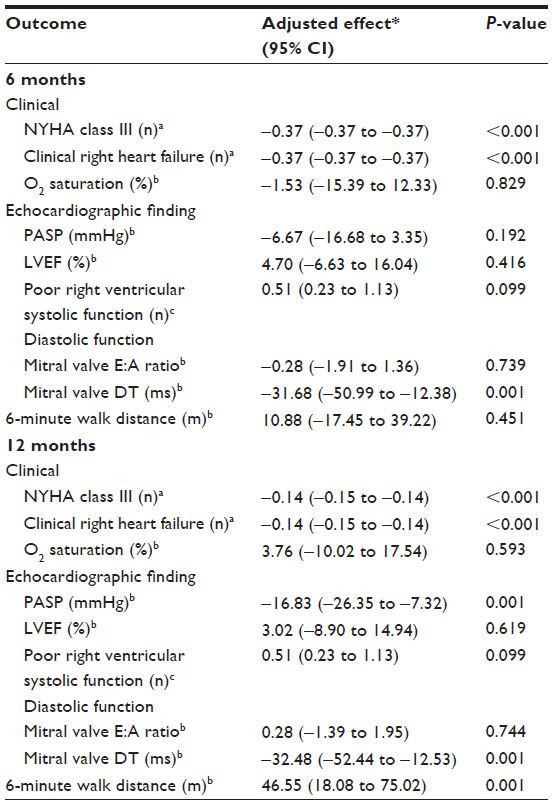 | Table 3 Effect of chronic blood transfusion on clinical outcomes, echocardiographic findings and 6-minute walk distance |
Discussion
In the study reported here, chronic blood transfusions had a significant effect on PASP and mean 6-minute walk distance after 12 months. The results of this study support the treatment of thalassemic patients with PAH with chronic blood transfusions, according to a previous study that showed that correction of hypercoagulability and amelioration of pulmonary arterial hypertension by chronic blood transfusions was seen in an asplenic E/β-Thal patient.18
A previous study showed that PAH is typical in TI patients who do not receive transfusions.11 PAH in thalassemia has also been associated with platelet activation and a hypercoagulable state.4 Diverse factors contributing to the hypercoagulable state in patients with thalassemia have been identified.19 Hypercoagulability is a well-described comorbid state in β-thalassemics who do not receive transfusions.8 Plasma heparin cofactor II levels were found to be significantly lower in TI patients who did not receive transfusions than in thalassemia major patients who received transfusions.20
Management of PAH is symptomatic and supportive to improve right ventricular function. Acetyl salicylic acid21,22 and an anticoagulant8,23 are also prescribed by some. Response to treatment is poor and the survival of patients of NYHA functional class III to IV is about 1 to 2 years.
The marked reduction in PASP achieved in our study suggests an improvement in thrombotic pulmonary arteriopathy as a result of chronic blood transfusions.
The preference 14 patients had for occasional transfusions was due to the amount of time required to travel to and from hospital; consequently, they could not manage visiting more than once per month.
The present study may have some limitations as it was not a randomized controlled trial due to ethics considerations, but the propensity score adjustment was applied to cope with such limitations.
Conclusion
The findings of the study presented here suggest that chronic blood transfusions might have beneficial effects on PAH in thalassemia patients over 1 year.
Acknowledgments
The authors wish to thank the medical staff members of Chiang Rai Hospital for their assistance and Chiang Mai University for its financial support.
Disclosure
The research was supported by a grant from the Faculty of Medicine, Chiang Mai University, Thailand. The authors declare no other conflicts of interest in this work.
References
Aessopos A, Farmakis D, Karagiorga M, et al. Cardiac involvement in thalassemia intermedia: a multicenter study. Blood. 2001;97(11):3411–3416. | |
Phrommintikul A, Sukonthasarn A, Kanjanavanit R, Nawarawong W. Splenectomy: a strong risk factor for pulmonary hypertension in patients with thalassaemia. Heart. 2006;92(10):1467–1472. | |
Sonakul D, Pacharee P, Laohapand T, Fucharoen S, Wasi P. Pulmonary artery obstruction in thalassaemia. Southeast Asian J Trop Med Public Health. 1980;11(4):516–523. | |
Singer ST, Kuypers FA, Styles L, Vichinsky EP, Foote D, Rosenfeld H. Pulmonary hypertension in thalassemia: association with platelet activation and hypercoagulable state. Am J Hematol. 2006;81(9):670–675. | |
Atichartakarn V, Likittanasombat K, Chuncharunee S, et al. Pulmonary arterial hypertension in previously splenectomized patients with beta-thalassemic disorders. Int J Hematol. 2003;78(2):139–145. | |
Ruf A, Pick M, Deutsch V, et al. In-vivo platelet activation correlates with red cell anionic phospholipid exposure in patients with beta-thalassaemia major. Br J Haematol. 1997;98(1):51–56. | |
Cappellini MD, Robbiolo L, Bottasso BM, Coppola R, Fiorelli G, Mannucci AP. Venous thromboembolism and hypercoagulability in splenectomized patients with thalassaemia intermedia. Br J Haematol. 2000;111(2):467–473. | |
Eldor A, Rachmilewitz EA. The hypercoagulable state in thalassemia. Blood. 2002;99(1):36–43. | |
Eldor A, Lellouche F, Goldfarb A, Rachmilewitz EA, Maclouf J. In vivo platelet activation in beta-thalassemia major reflected by increased platelet-thromboxane urinary metabolites. Blood. 1991;77(8):1749–1753. | |
Opartkiattikul N, Funahara Y, Fucharoen S, Talalak P. Increase in spontaneous platelet aggregation in beta-thalassemia/hemoglobin E disease: a consequence of splenectomy. Southeast Asian J Trop Med Public Health. 1993;23 Suppl 2:S36–S41. | |
Aessopos A, Farmakis D, Deftereos S, et al. Thalassemia heart disease: a comparative evaluation of thalassemia major and thalassemia intermedia. Chest. 2005;127(5):1523–1530. | |
Chiewsilp P, editor. The Appropriate Use of Blood and Blood Components Physician Handbook. 1st ed. Bangkok: Thai Red Cross Society’s National Blood Center; 2011. | |
ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med. 2002;166(1):111–117. | |
Rubin LJ; American College of Chest Physicians. Diagnosis and management of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126(Suppl 1):7S–10S. | |
Yock PG, Popp RL. Noninvasive estimation of right ventricular systolic pressure by Doppler ultrasound in patients with tricuspid regurgitation. Circulation. 1984;70(4):657–662. | |
Otto CM. Echocardiographic evaluation of left and right ventricular systolic function. In: Otto CM, editor. Textbook of Clinical Echocardiography. 2nd ed. Philadelphia, PA: WB Saunders; 2000:100–128. | |
Barst RJ, McGoon M, Torbicki A, et al. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):40S–47S. | |
Atichartakarn V, Chuncharunee S, Chandanamattha P, Likittanasombat K, Aryurachai K. Correction of hypercoagulability and amelioration of pulmonary arterial hypertension by chronic blood transfusion in an asplenic hemoglobin E/beta-thalassemia patient. Blood. 2004;103(7):2844–2846. | |
Taher AT, Otrock ZK, Uthman I, Cappellini MD. Thalassemia and hypercoagulability. Blood Rev. 2008;22(5):283–292. | |
O’Driscoll A, Mackie IJ, Porter JB, Machin SJ. Low plasma heparin cofactor II levels in thalassaemia syndromes are corrected by chronic blood transfusion. Br J Haematol. 1995;90(1):65–70. | |
Fucharoen S, Youngchaiyud P, Wasi P. Hypoxaemia and the effect of aspirin in thalassaemia. Southeast Asian J Trop Med Public Health. 1981;12(1):90–93. | |
Robbins IM, Kawut SM, Yung D, et al. A study of aspirin and clopidogrel in idiopathic pulmonary arterial hypertension. Eur Respir J. 2006;27(3):578–584. | |
Atichartakarn V, Angchaisuksiri P, Aryurachai K, et al. Relationship between hypercoagulable state and erythrocyte phosphatidylserine exposure in splenectomized haemoglobin E/beta-thalassaemic patients. Br J Haematol. 2002;118(3):893–898. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.