")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Beneficial Effect of Genistein on Diabetes-Induced Brain Damage in the ob/ob Mouse Model
Authors Li R, Ding XW , Geetha T, Al-Nakkash L , Broderick TL, Babu JR
Received 13 February 2020
Accepted for publication 6 July 2020
Published 17 August 2020 Volume 2020:14 Pages 3325—3336
DOI https://doi.org/10.2147/DDDT.S249608
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Yan Zhu
Rong-zi Li,1 Xiao-Wen Ding,1 Thangiah Geetha,1 Layla Al-Nakkash,2 Tom L Broderick,2 Jeganathan Ramesh Babu1
1Department of Nutrition, Dietetics and Hospitality Management, Auburn University, Auburn, AL 36849, USA; 2Department of Physiology, Laboratory of Diabetes and Exercise Metabolism, College of Graduate Studies, Midwestern University, Glendale, AZ 85308, USA
Correspondence: Jeganathan Ramesh Babu
Department of Nutrition, Dietetics and Hospitality Management, Auburn University, Auburn, AL 36849, USA
Tel +1 334 844 3840 Fax +1 334 844 3268
Email [email protected]
Purpose: Diabetes mellitus (DM)-induced brain damage is characterized by cellular, molecular and functional changes. The mechanisms include oxidative stress, neuroinflammation, reduction of neurotrophic factors, insulin resistance, excessive amyloid beta (Aβ) deposition and Tau phosphorylation. Both antidiabetic and neuroprotective effects of the phytoestrogen genistein have been reported. However, the beneficial effect of genistein in brain of the ob/ob mouse model of severe obesity and diabetes remains to be determined.
Methods: In this study, female ob/ob mice and lean control mice were fed with either a standard diet or a diet containing genistein (600mg/kg) for a period of 4 weeks. Body weight was monitored weekly. Blood was collected for the measurement of glucose, insulin and common cytokines. Mice brains were isolated for Western immunoblotting analyses.
Results: Treatment with genistein reduced weight gain of ob/ob mice and decreased hyperglycemia compared to ob/ob mice fed the standard diet. The main findings show that genistein treatment increased insulin sensitivity and the expression levels of the neurotrophic factors nerve growth factor (NGF) and brain-derived neurotrophic factors (BDNF). In these mice, genistein also reduced Aβ deposition and the level of hyper-phosphorylated Tau protein.
Conclusion: The results of our study indicate the beneficial effects of genistein in the obese diabetic mouse brain, including improving brain insulin signaling, increasing neurotrophic support, and alleviating Alzheimer’s disease-related pathology.
Keywords: isoflavones, leptin-deficient mice, brain injury, insulin signaling, amyloid beta
Introduction
Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia. Type 1 DM (T1DM) is caused by impairment in insulin production from the pancreas. In contrast, type 2 DM (T2DM) is the result of peripheral insulin resistance in which the tissues fail to respond to insulin effectively. DM is associated with various complications, including retinopathy, nephropathy, and neuropathy.1 Several experimental and clinical studies have demonstrated brain damage in patients with DM. These individuals exhibit both cellular and functional changes, such as progressive cognitive deterioration and neurodegeneration.2–4 Systematic reviews and meta-analysis studies have reported that patients with T2DM have a 50–100% higher risk of developing dementia compared to the non-diabetic population.4 Results from longitudinal studies have shown that middle-aged to older adults with T2DM performed worse in memory, information-processing speed, attention tasks, and in overall cognitive function compare to non-diabetic controls over a follow-up of 3–6 years.5–7 In addition, T2DM patients are at increased risk of developing Alzheimer’s disease (AD).8 AD is the most common form of dementia, which is characterized by the accumulation of extracellular amyloid beta (Aβ) peptide formed senile plaques and intracellular hyperphosphorylated Tau-formed neurofibrillary tangles.9 The possible mechanisms of diabetes-induced brain damage include oxidative stress, neuroinflammation, neuronal apoptosis, reduction of neurotrophic factors, insulin receptor downregulation, insulin resistance, excessive Aβ deposition and Tau phosphorylation, mitochondria dysfunction.1,10
The consumption of plant-based foods rich in isoflavones is known to provide health benefits.11 Several studies have demonstrated beneficial effects of isoflavones, especially genistein, which is the most abundant isoflavone found in soy.12 Due to its similar structure with endogenous estrogen 17β-estradiol and to its affinity to the estrogen receptor, genistein displays estrogenic properties.13 Evidence from human and animal studies indicates that genistein could be a potential preventative and therapeutic treatment for DM.14,15 In experimental models of T1DM and T2DM, genistein treatment significantly decreased blood glucose, improved glucose tolerance and insulin sensitivity, possibly by modulating pancreatic beta cell function and alleviating inflammation and oxidative stress.13,16 Few studies, however, have reported the protective effects of genistein in the diabetic brain,17,18 especially in T2DM. Also, whether genistein exhibits beneficial effects within the brain of the genetic ob/ob model of severe obesity and insulin resistance is not well understood. The ob/ob mouse is an animal model of peripheral neuropathy of type 2 diabetes and obesity. Ob/ob mice possess a recessive mutation in the leptin, which is a hormone that helps to regulate energy expenditure and food intake. Mutation in the leptin gene results in hyperphagia, hyperglycemia, and subsequent development of insulin resistance, obesity and diabetes.19 Therefore, in this study, we used the naturally occurring soy isoflavone genistein to determine whether an improvement in insulin signaling is observed in the ob/ob mouse brain. We also examined the impacts of genistein on neurotrophic factors, Aβ deposition, and Tau metabolism. We hypothesize that genistein exerts beneficial effects by alleviating these neurodegenerative factors in this model of obesity and diabetes.
Materials and Methods
Animals
The study utilized female ob/ob mice (B6.Cg-Lepob/J) and lean control mice (C57BL/6J) purchased from Jackson Laboratory (Bar Harbor, ME, USA) at five weeks of age. Ob/ob mice display severe obesity and insulin resistance as a result of a spontaneous mutation of the leptin receptor gene. Although this mutation rarely causes diabetes,20 a feature of this model of leptin deficiency is the hyperphagia exhibited by mice and the gradual onset of diabetes, producing a phenotype similar to human patients with T2DM. Female mice were selected since AD is more prevalent in the female population.21 As a phytoestrogen that mimics estrogen, genistein may have more protective effects in the brain of female mice. All mice were housed in an animal facility maintained at a room temperature of 22°C with a 12-hour light/dark cycle and consumed food and water ad libitum. Body weight was measured weekly and the general health was monitored weekly during the study. This study was approved by the Institutional Animal Care and Use Committee at Midwestern University, and carefully followed the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals.
Study Design
After one week of acclimatization, mice were divided into four groups (n=8–13): (1) lean control mice fed a standard diet (SD), (2) lean mice fed a genistein diet, (3) ob/ob mice fed a SD, (4) ob/ob mice fed a genistein diet. The formulated genistein diet was prepared in powder form (Dyets Inc., Bethlehem, PA, USA) and contained 600 mg genistein/kg diet. The concentration of 600 mg/kg was chosen because it is effective in maintaining a significant improvement in plasma levels of genistein.22 Moreover, our previous studies have showed that 600 mg/kg induces significant physiological effects with four weeks of treatment in the mouse model.23–25 After four weeks of treatment, mice were sacrificed with CO2 asphyxiation, followed by bilateral pneumothorax. Brains were isolated and immediately snap-frozen in liquid nitrogen and stored at −80°C for Western immunoblotting analyses.
Plasma and Cytokine Assays
Cardiac blood was collected at the time of euthanasia and plasma maintained at −80°C until use. Plasma was utilized for the measurement of glucose using a colorimetric assay kit (Wako Chemicals USA, Richmond, VA, USA) and insulin (Alpco Research and Clinical Immunoassays, Salem, NH, USA) assayed according to manufacture specifications. In a separate series of age-matched female ob/ob treated with genistein, plasma was collected and assayed using for common cytokines using the Millipore Milliplex assay (Burlington, MA).
Antibodies
Antibodies against pAkt (Ser-473), pIR (Tyr-1150/1151) were purchased from Cell Signaling Technology (Danvers, MA). Antibodies against IR, Tau, PHF-6 (Tau phospho Thr 231), CDK-5 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Beta-actin, PSD-95 antibodies were obtained from Abcam (Cambridge, MA). 4G8 antibody was purchased from Covance (Princeton, NJ). Antibodies against pIRS-1(Ser307) and IRS-1 were purchased from Fisher Scientific (Hampton, NH). PHF-1 (Tau phospho Ser 396/Ser404) antibody was obtained from Alzforum. Antibody OC was obtained from Millipore (Burlington, MA). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Western Blot Analysis
Whole mouse brains were homogenized in Triton lysis buffer (50 mM Tris–HCl [pH 7.5], 150 mM NaCl, 10 mM NaF, 0.5% Triton X-100, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, and 2 μg/mL leupeptin, aprotinin and protease inhibitor cocktail). Samples were centrifuged at 12,000 rpm for 20 min at 4°C, and supernatants were collected. Protein concentrations of the brain lysates were measured with the Pierce 660-nm protein assay reagent (Thermo Scientific, Rockford, IL). The samples were separated on 8–15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gels and then transferred onto polyvinylidene difluoride membranes. Immunoblots were blocked for one hour in Tris-buffered saline Tween-20 (TBST) containing 5% dry milk. The blots were incubated with appropriate primary antibodies in TBST at 4°C overnight. After washing, the blots were then incubated with secondary antibodies for two hours at room temperature. The immunoreactive bands were visualized with Enhanced Chemiluminescence reagent.26
Statistics
The two-way analysis of variance (GraphPad Prism), followed by Tukey’s test was used to determine significant differences between different group means. All values are reported as mean ± SEM. P<0.05 were considered to be statistically significant.
Results
Genistein Reduces Weight Gain and Plasma Glucose of ob/ob Mice
At the beginning of treatment and at the time of sacrifice, ob/ob mice had higher body mass than lean mice. Ob/ob mice fed 600 mg genistein/kg diet for 4 weeks gained ~44% less weight compared to those SD-fed ob/ob mice, while no significant change of weight between SD-fed and genistein-fed lean control mice was observed (Figure 1). At the completion of the 4-week treatment, genistein-fed ob/ob mice showed a significant reduction (17%) in plasma glucose levels when compared with SD-fed ob/ob mice (Table 1). Typical for the ob/ob model of obesity and diabetes, serum insulin levels were significantly elevated in ob/ob mice compared to lean control mice. However, genistein diet had no significant effect on insulin levels in ob/ob mice. Interestingly, genistein diet significantly elevated serum insulin levels in lean control mice. This result is consistent with previous studies that genistein could improve pancreatic beta-cell function and stimulate insulin secretion.12,27 Cytokines measured in plasma of mice demonstrated the effects of obesity and diabetes. Plasma levels of LIX, IL-1a, CXCL1, IL-5, IL-6, and IL-13 were significantly increased in ob/ob mice compared to control mice. Treating ob/ob mice with genistein resulted in a significant reduction in plasma levels of LIX, IL-1a, and IL-13. All other cytokines measured using the Milliplex assay kit revealed either no effects of the obese diabetic state or treatment (Table 2).
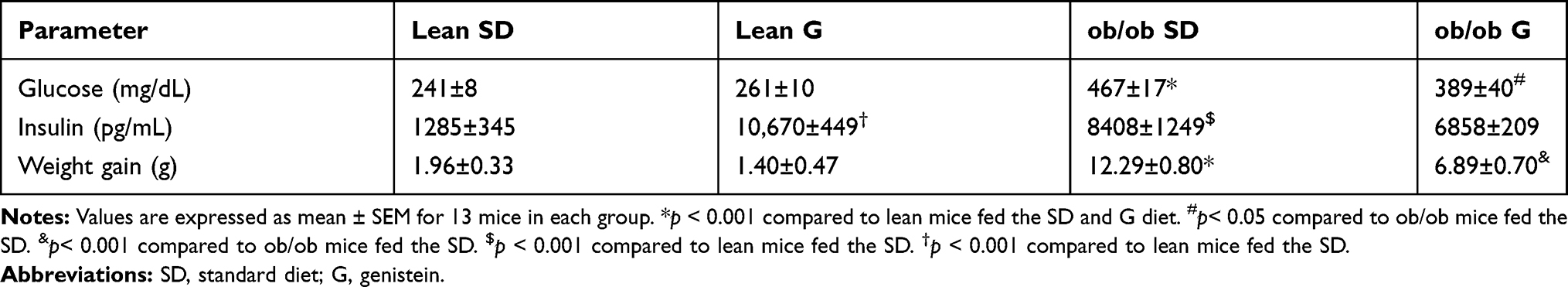 |
Table 1 Effect of Genistein on Blood Glucose Concentrations and Body Weight Gain of ob/ob Mice |
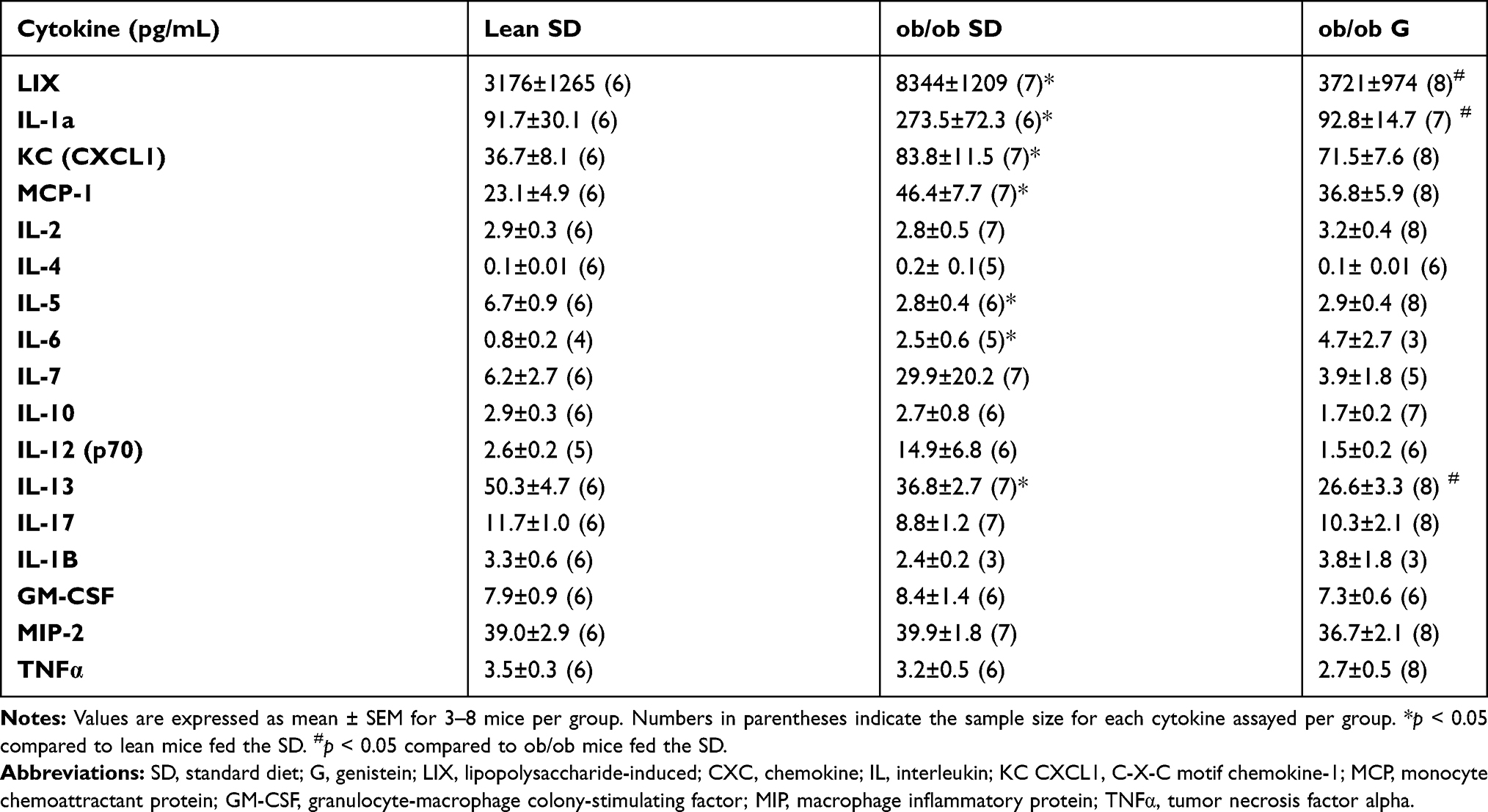 |
Table 2 Effect of Genistein on the Levels of Plasma Cytokines of ob/ob Mice |
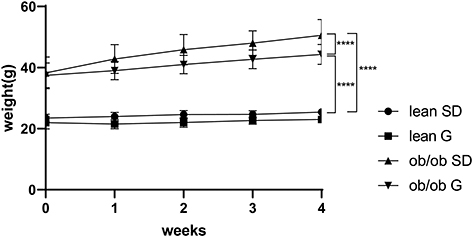 |
Figure 1 Weight change of mice during 4 weeks treatment. Lean mice and ob/ob mice were fed with a standard diet or genistein diet for 4 weeks. Body weight was monitored weekly. ****p ≤ 0.0001. Abbreviations: SD, standard diet; G, genistein. |
Genistein Mitigates Brain Insulin Resistance in ob/ob Mice
T2DM mice not only exhibit systemic insulin resistance (IR) but also brain IR. To evaluate insulin sensitivity in the brain of ob/ob mice and the effects of genistein, we analyzed the activation of insulin receptor and its downstream pathway. As shown in Figure 2, the levels of insulin receptor phosphorylation at Tyr-1150/1151 in SD-fed ob/ob mice were lower than SD-fed control mice (Figure 2A). Also, there was a significant increase in insulin receptor substrate-1 (IRS-1) phosphorylation at Ser307 in SD-fed ob/ob mice as compared to control lean mice (Figure 2B), which suggests the inactivation of IRS-1. Moreover, the activation or phosphorylation of Akt was inhibited in SD-fed ob/ob mice (Figure 2C). These data indicate increased insulin resistance in the brain of SD-fed ob/ob mice. However, genistein diet rescued the inactivation of the insulin receptor, IRS-1, and Akt in ob/ob mice, suggesting that short-term treatment with genistein improves brain insulin sensitivity in the ob/ob model of diabetic obesity. There were no significant differences in these markers of insulin sensitivity between SD-fed control mice and the genistein-fed control mice.
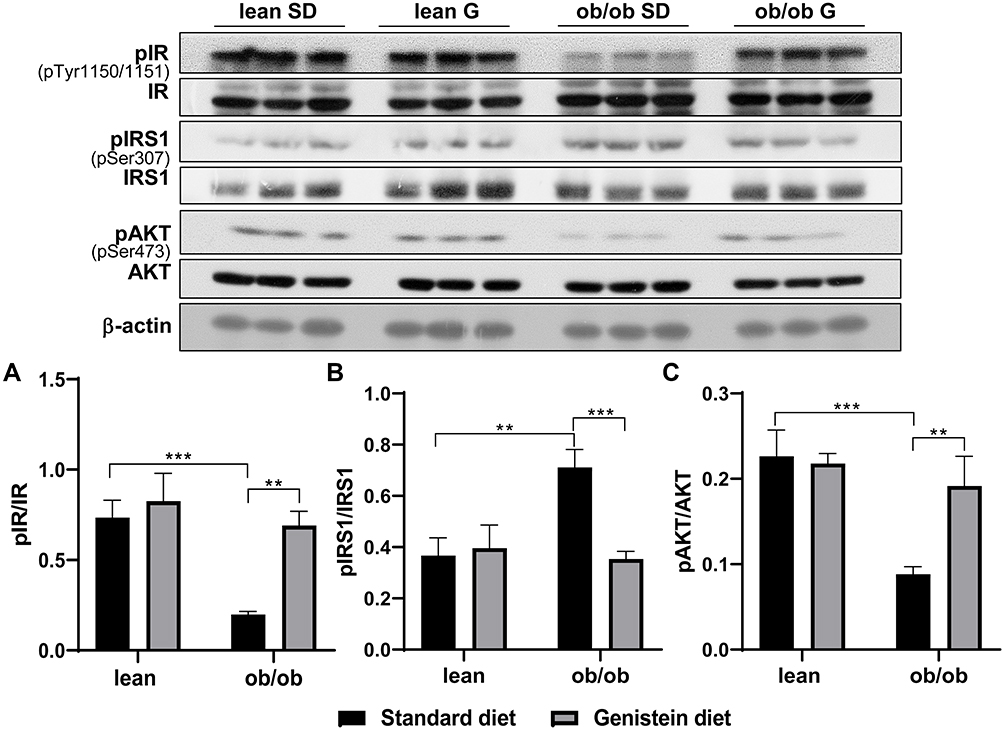 |
Figure 2 Effect of genistein on central insulin signaling in brain of ob/ob mice. (A) Representative blot images with corresponding densitometry measurements of the insulin receptor. (B) Representative blot images with the corresponding densitometry measurements of insulin receptor substrate-1. (C) Representative blot images with the corresponding densitometry measurements of Akt. pAkt, Akt phosphorylation at Ser473. Data are presented as mean ± SEM for 3 mice per group. **p ≤ 0.01, ***p ≤ 0.001. Abbreviations: IR, insulin receptor; pIR, IR phosphorylation at Tyr-1150/1151; IRS-1, insulin receptor substrate-1; pIRS-1, IRS-1 phosphorylation at Ser307; SD, standard diet; G, genistein. |
Genistein Improves the Level of Neurotrophic Factors in the Brain of ob/ob Mice
Neurotrophic factors are polypeptides known to regulate the proliferation, survival, differentiation, and migration of cells in the nervous system. Nerve growth factor (NGF) and brain-derived neurotrophic factors (BDNF) are the two main neurotrophic factors. Evidence indicates the role of neurotrophic factors in the pathogenesis of several neuronal and non-neuronal diseases, including AD and DM.28 As shown in Figure 3, SD-fed ob/ob mice exhibited diminished levels of NGF (Figure 3A) and BDNF (Figure 3B) compared to control lean mice. However, expression levels of NGF and BDNF were dramatically elevated by supplementation with genistein in the diet. In addition, it has been reported that compromised synaptic function could lead to various neurodegenerative disorders.29 In order to evaluate synaptic function in T2DM mice and the effect of genistein, the protein content of postsynaptic density protein (PSD95) was measured. As shown in Figure 3C, our results reveal a significant difference in PSD95 levels between SD-fed ob/ob mice and control lean mice. Genistein supplementation improved PSD95 levels in the brain of ob/ob mice, although this was not statistically different.
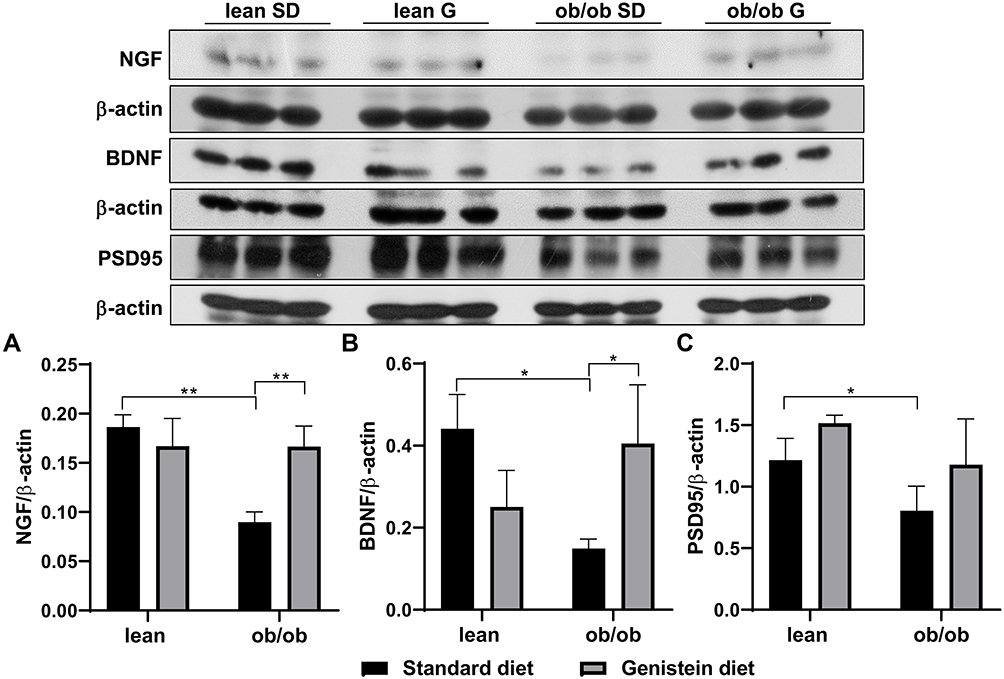 |
Figure 3 Effect of genistein on the levels of neurotrophins and synaptic marker in brain of ob/ob mice. (A) Representative blot images with the corresponding densitometry measurement of nerve growth factor (NGF). (B) Representative blot images with the corresponding densitometry measurements of brain-derived neurotropic factor (BDNF). (C) Representative blot images with the corresponding densitometry measurement of postsynaptic density 95 (PSD95). Data are presented as mean ± SEM for 3 mice per group. *p ≤ 0.05, **p ≤ 0.01. Abbreviations: SD, standard diet; G, genistein. |
Genistein Attenuates the Accumulation of Aβ and the Phosphorylation of Tau in the Brain of ob/ob Mice
Previous studies have demonstrated that diabetes-associated brain damage has similar pathology with AD. In addition to the decrease in neurotrophic and synaptic factors, we hypothesize that Aβ deposition is observed in ob/ob mice brain, and genistein is involved in the process of regulating Aβ deposition. Here, the antibody 4G8 was used to detect Aβ peptide and the antibody OC to recognize amyloid fibrils. As shown in Figure 4A, when compared with SD-fed lean mice, SD-fed ob/ob mice had significantly increased Aβ deposition, while treatment with genistein diet mitigated these changes. This benefit of genistein was also observed in OC-detected content (Figure 4B). In addition, we assessed the neurofibrillary tangle, through detecting the level of hyper-phosphorylated Tau protein. Antibody PHF-1 recognizes Tau protein phosphorylated at serine residues 396 and 404. The antibody PHF-6 recognizes Tau protein when phosphorylated at Thr231. As illustrated in Figure 5, brain from ob/ob mice possessed significantly higher levels of hyper-phosphorylated Tau than control mice. However, supplementation with genistein effectively reduced the phosphorylation level of Tau protein in those ob/ob mice. Furthermore, numerous serine/threonine kinases could phosphorylate Tau, such as cyclin dependent kinase 5 (CDK5). We observed enhanced expression of CDK5 in the brain of ob/ob mice compared to the control mice while treatment with genistein reduced levels of CDK5.
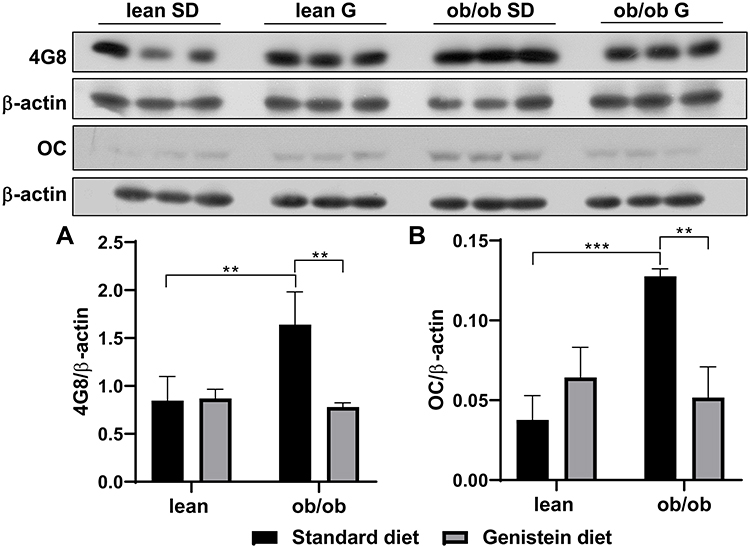 |
Figure 4 Effect of genistein on Aβ deposition in the brain of ob/ob mice. (A) Representative blot images with the corresponding densitometry measurement of 4G8. (B) Representative blot images with the corresponding densitometry measurement of OC. Data are presented as mean ± SEM for 3 mice per group. **p ≤ 0.01, ***p ≤ 0.001. Abbreviations: SD, standard diet; G, genistein. |
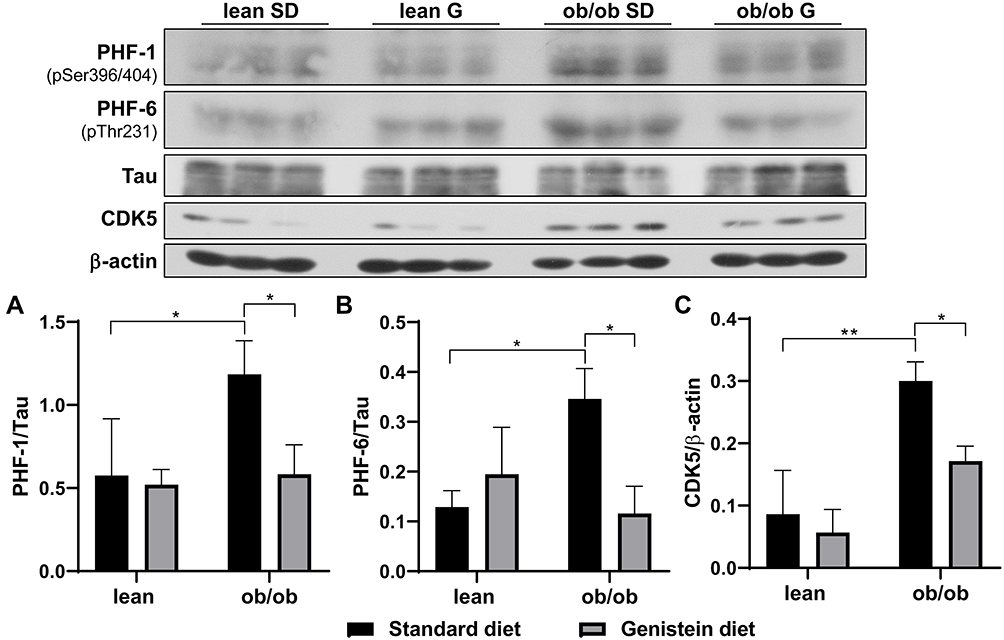 |
Figure 5 Effect of genistein on the level of hyper-phosphorylated Tau in the brain of ob/ob mice. (A) Representative blot images with the corresponding densitometry measurement of PHF-1. (B) Representative blot images with the corresponding densitometry measurement of PHF-6. (C) Representative blot images with the corresponding densitometry measurement of cyclin dependent kinase 5 (CDK5). Data are presented as mean ± SEM for 3 mice per group. *p ≤ 0.05, **p ≤ 0.01. Abbreviations: SD, standard diet; G, genistein. |
Discussion
In recent years, considerable attention has focused on diabetes pathology and the development of therapeutic or preventive strategies for the treatment of DM and its complications. Natural bioactive compounds attracted extensive interest due to abundant availability and few side effects.30 As an isoflavone found in a variety of plants, genistein exhibits anti-diabetic effects in both in vivo and in vitro studies due to its anti-antioxidant and estrogenic properties.12 Further, genistein treatment is reported to ameliorate diabetic peripheral neuropathy, inhibit oxidative stress and proinflammatory cytokine production, and restore NGF levels in brain of STZ-diabetic mice.18 In the high-fat diet (HFD)-fed ApoE knockout mice model, genistein supplementation not only reduced neuroinflammation and oxidative stress-mediated insulin resistance, but decreased the level of Aβ peptide and hyperphosphorylated Tau protein, two major hallmarks of AD pathology.17 Our results are consistent with these earlier reports, and we extend these observations by demonstrating beneficial effects on the brain afforded by genistein in the obese diabetic ob/ob mice.
Insulin receptors are expressed in the peripheral systems as well as in central nervous systems. Similar to systemic IR, brain IR is defined as reduced sensitivity in brain cells to the action of insulin.31 Evidence from experimental animal models suggests that systemic IR is linked with brain IR and both are observed in animal models of T2DM. Indeed, both HFD-induced diabetic mice and genetic diabetic mice (ob/ob and db/db mice) exhibit systemic and brain IR and abnormalities such as altered synaptic integrity and cognitive behaviors.32,33 The insulin receptor is a heterotetrameric transmembrane protein made up of two extracellular α-subunits and two transmembrane β-subunits.34 In the brain, binding of insulin to insulin receptors results in rapid autophosphorylation of the insulin receptor, which initiates a signal transduction cascade leading to tyrosine phosphorylation and activation of IRS-1. IRS proteins then activate phosphatidylinositol-3 kinase (PI3 kinases), followed by activation of downstream signaling cascade involving Akt and glycogen synthase kinase 3 (GSK3).9 Further, IRS-1 protein degradation could result from the serine phosphorylation of IRS-1. Our results demonstrate that ob/ob mice represent increased central IR, as evidenced by decreased insulin receptor phosphorylation, increased serine phosphorylation or inactivation of IRS-1 and inhibited Akt activation. However, feeding ob/ob mice with genistein diet improved insulin sensitivity. These results are consistent with previously published studies that genistein affords protection against central IR. In the cerebral cortex of aged female rats, acute treatment with genistein (40mg/kg body weight) reversed the deterioration in the interactions between PI3K and tyrosine phosphorylation of IRS-1, as well as stimulated Akt activation.35 Also, in HFD-fed ApoE knockout mice model, genistein supplementation (500mg/kg diet) was reported to lessen central IR by decreasing phosphorylated IRS-1 levels.17
Neurotrophic factors have multiple functions in the nervous system. In addition to their role in neuronal survival and growth, neurotrophic factors are capable of inducing synaptic plasticity, modulating the formation of long-term memories and maintaining neuron functions.36 Evidence for a role of NGF and BDNF in the pathogenesis of DM is supported by the observation that serum BDNF levels are lower in T2DM patients than non-diabetic subjects.37 Also, Passaro et al found that plasma BDNF levels are lower in patients with both T2DM and dementia than in non-diabetic patients with dementia.38 In the early stage of STZ-induced diabetes in mice, gene expression of BDNF and NGF is lower compared to non-diabetic mice.39 Furthermore, increasing evidence demonstrated a link between BDNF and steroid hormones such as estradiol. Harte-Hargrove et al reported that 17β-estradiol regulates hippocampal function by its impact on BDNF expression.40 Genistein has been suggested to improve spatial learning and memory by activating BDNF signaling.41 In the present study, our results show that genistein prevents a decrease in BDNF expression from occurring in the brain of ob/ob female mice. This effect is consistent with previous findings that estrogen plays a crucial role in BDNF expression and may improve the DM state. Furthermore, diminished levels of NGF were shown in the serum of both STZ-induced diabetic mice and diabetic patients.42 Tosaki et al indicated that high glucose condition induced the reduction of NGF secretion in Schwann cells, which could contribute to the disturbed axonal regeneration and may result in the development of diabetic neuropathy.43 Interestingly, repeated treatment with genistein improves NGF production in the sciatic nerve of STZ-induced diabetic mice, showing a neurotrophic activity.18 The results of our study further substantiate the effect of genistein on NGF normalization in the ob/ob genetic model of diabetes and obesity. Synaptic dysfunction contributes impairment of brain function and decline in learning and memory ability that occurs in the early stage of AD.44 In the present study, the synaptic protein PSD-95 was used to evaluate the synaptic function. Our data show that the expression of PSD-95 was significantly decreased in the brain of ob/ob female mice. Four-weeks treatment with genistein diet improved the level of PSD-95 by ~25% in ob/ob mice, although this was not significant. We hypothesize that increasing the duration of the treatment period beyond 4-weeks could result in significant changes in PSD-95 in the brain of ob/ob mice.
Previous studies have demonstrated that the pathology of diabetes-associated cognitive decline is similar to AD. The hallmark of AD, consisting of increased levels of aggregated Aβ protein, was observed in db/db mice and HFD-induced diabetic mice.4,33 Aβ peptides are formed from amyloid precursor protein through sequential proteolytic cleavage by two enzymes, β- and α-secretases. Aβ can oligomerize into amyloid fibrils, and excessive oligomerization creates senile plaques in neural tissues. Our results indicate that brain from ob/ob female mice exhibits both significantly enhanced Aβ and amyloid fibrils levels when compared to lean control mice. However, genistein supplementation significantly reduced Aβ deposition in these ob/ob mice. The inhibitory effect of genistein on Aβ aggregation has been proposed in a previous study: in an AD mouse model, treatment with genistein resulted in the rapid clearance of Aβ and the improvement of cognitive function.45 Genistein supplementation also alleviated Aβ deposition in HFD-fed Apo E knockout mice.17 Furthermore, several studies have reported a reduction in Aβ-induced neuronal apoptosis, astrogliosis, inflammation, and neurotoxicity following genistein treatment.46–49 In contrast, a recent study reported that genistein caused an accumulation in Aβ in human neuroblastoma cells.50 The reasons for these contrasting effects are not known, but may relate to the bioavailability of genistein in the plasma.51 Overall, the long-term impact of treatment with genistein cannot be overlooked, and further studies are needed to determine the effective concentration of genistein associated with neuroprotection.
Neurofibrillary tangles consist of hyperphosphorylated Tau protein. Under normal conditions, Tau modulates the stability of microtubes. In AD, Tau proteins undergo dynamic, site-specific phosphorylation by various kinases, such as CDK5, GSK3β.52 Inappropriate phosphorylation leads to dysfunction of Tau, decreased cell viability, and pathogenesis of the neurological disease.52 In this study, 4-weeks genistein diet significantly reduced the level of hyperphosphorylated Tau in ob/ob mice. The inhibition of CDK5 might partially contribute to this result. Previous studies indicate that genistein could modulate Tau hyperphosphorylation through regulating calcium/calmodulin-dependent protein kinase IV or GSK3β in AD mice or Apo E knockout mice.17,53 The autophagy-dependent mechanism may also be responsible for regulating the concentrations of hyperphosphorylated Tau by genistein in AD mice.54 In addition, a recent study demonstrated that the hyperphosphorylation of Tau in the brain of ob/ob mice might relate to hypothermia and the decreased thyroid hormone levels.55 Interestingly, treatment of ob/ob mice with genistein elevated thyroid hormone levels and induced a thermogenic change in these mice,25 suggesting that genistein might be a novel therapy in the elimination of hyper-phosphorylated Tau.
The diminished level of Aβ observed in the brain of genistein-fed ob/ob mice may be explained by improved insulin signaling. Brain IR has been suggested to accelerate AD pathology, including processing or aggregation of Aβ and hyperphosphorylation Tau. Diet-induced IR is suggested to promote Aβ generation in the brain of AD mice by enhancing γ- secretases activities and inhibiting insulin degrading enzyme (IDE) activities.56 The IR has also been implicated in Tau aggregation. The downstream signaling effect of PKB/Akt, GSK-3β functions as a kinase responsible for the phosphorylation of Tau.57 Also, administration of insulin can suppress protein phosphatase 2 activity, which is the primary Tau phosphatase contributes to Tau de-phosphorylation.58 Overall, brain IR may increase the tendency for Tau aggregation by improving Tau phosphorylation and inhibiting Tau de-phosphorylation.
In addition to its role in AD pathology, IR in brain has a direct effect on synaptic function and cognition. For instance, the downregulation of the hypothalamic insulin receptor in mice elicited a decreased expression of BDNF as well as an impaired long-term potentiation and spatial learning.59 The role of insulin as a synaptic modulator promoting neuroplasticity in the developing and adult brain has been proposed. Moreover, IR in T2DM impairs hippocampal synaptic plasticity.60,61 Our data indicate that ob/ob mice exhibit central IR accompanied with decreased synaptic plasticity. However, genistein supplementation improved brain insulin sensitivity and increased levels of the synaptic marker, PSD-95, by nearly 25%. Therefore, it is reasonable to hypothesize that genistein could ameliorate synaptic plasticity in the brain of diabetic mice by mitigating insulin resistance.
Conclusion
In summary, considering the high risk of deterioration in brain function induced by DM, the identification of preventive strategies and complementary therapies has been an important research goal. The results of this study indicate that genistein improved the obesity and diabetes-related neuronal damage in the brain of ob/ob mice. The beneficial effects of genistein are associated with increased insulin sensitivity and expression of neurotrophic factors as well as inhibiting Tau protein phosphorylation and Aβ deposition. As emerging evidence indicates that the putative mechanisms of genistein include estrogen receptor (ER) regulation,62 it is likely that genistein impacts survival and cell growth, synaptic plasticity, and cognitive function through the ER-mediated pathways.62,63 Therefore, further studies are needed to explore the role of ER-mediated or non-ER mediated mechanisms in neuroprotective effects of genistein in the DM animal model. Also, the present study only used female mice. The male mice model needs to be included in future studies to examine whether the effect of genistein is sex-dependent.
Acknowledgments
This work was supported by the Alabama Agricultural Experimental Station (AAES), Hatch/Multistate Funding Program and AAES Award for Interdisciplinary Research (AAES-AIR) to TG and JRB. Midwestern University Intramural funds (to LA) and Diabetes Action and Research Education Foundation (to LA).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hamed SA. Brain injury with diabetes mellitus: evidence, mechanisms and treatment implications. Expert Rev Clin Pharmacol. 2017;10(4):409–428. doi:10.1080/17512433.2017.1293521
2. den Heijer T, Vermeer SE, van Dijk EJ, et al. Type 2 diabetes and atrophy of medial temporal lobe structures on brain MRI. Diabetologia. 2003;46(12):1604–1610. doi:10.1007/s00125-003-1235-0
3. Sadeghi A, Hami J, Razavi S, Esfandiary E, Hejazi Z. The effect of diabetes mellitus on apoptosis in hippocampus: cellular and molecular aspects. Int J Prev Med. 2016;7:57. doi:10.4103/2008-7802.178531
4. Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol. 2006;5(1):64–74. doi:10.1016/S1474-4422(05)70284-2
5. Kanaya AM, Barrett-Connor E, Gildengorin G, Yaffe K. Change in cognitive function by glucose tolerance status in older adults: a 4-year prospective study of the Rancho Bernardo study cohort. Arch Intern Med. 2004;164(12):1327–1333. doi:10.1001/archinte.164.12.1327
6. Hassing LB, Grant MD, Hofer SM, et al. Type 2 diabetes mellitus contributes to cognitive decline in old age: a longitudinal population-based study. J Int Neuropsychol Soc. 2004;10(4):599–607. doi:10.1017/S1355617704104165
7. Gregg EW, Yaffe K, Cauley JA, et al. Is diabetes associated with cognitive impairment and cognitive decline among older women? Study of Osteoporotic Fractures Research Group. Arch Intern Med. 2000;160(2):174–180. doi:10.1001/archinte.160.2.174
8. Jayaraman A, Pike CJ. Alzheimer’s disease and type 2 diabetes: multiple mechanisms contribute to interactions. Curr Diab Rep. 2014;14(4):476. doi:10.1007/s11892-014-0476-2
9. Kothari V, Luo Y, Tornabene T, et al. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim Biophys Acta. 2017;1863(2):499–508. doi:10.1016/j.bbadis.2016.10.006
10. Wang T, Fu F, Han B, Zhang L, Zhang X. Danshensu ameliorates the cognitive decline in streptozotocin-induced diabetic mice by attenuating advanced glycation end product-mediated neuroinflammation. J Neuroimmunol. 2012;245(1–2):79–86. doi:10.1016/j.jneuroim.2012.02.008
11. Ososki AL, Kennelly EJ. Phytoestrogens: a review of the present state of research. Phytother Res PTR. 2003;17(8):845–869. doi:10.1002/ptr.1364
12. Gilbert ER, Liu D. Anti-diabetic functions of soy isoflavone genistein: mechanisms underlying its effects on pancreatic β-cell function. Food Funct. 2013;4(2):200–212. doi:10.1039/c2fo30199g
13. Behloul N, Wu G. Genistein: a promising therapeutic agent for obesity and diabetes treatment. Eur J Pharmacol. 2013;698(1–3):31–38. doi:10.1016/j.ejphar.2012.11.013
14. Fu Z, Zhang W, Zhen W, et al. Genistein induces pancreatic beta-cell proliferation through activation of multiple signaling pathways and prevents insulin-deficient diabetes in mice. Endocrinology. 2010;151(7):3026–3037. doi:10.1210/en.2009-1294
15. Elmarakby AA, Ibrahim AS, Faulkner J, Mozaffari MS, Liou GI, Abdelsayed R. Tyrosine kinase inhibitor, genistein, reduces renal inflammation and injury in streptozotocin-induced diabetic mice. Vascul Pharmacol. 2011;55(5–6):149–156. doi:10.1016/j.vph.2011.07.007
16. Fu Z, Gilbert ER, Pfeiffer L, Zhang Y, Fu Y, Liu D. Genistein ameliorates hyperglycemia in a mouse model of nongenetic type 2 diabetes. Appl Physiol Nutr Metab. 2012;37(3):480–488. doi:10.1139/h2012-005
17. Park Y-J, Ko JW, Jeon S, Kwon YH. Protective effect of genistein against neuronal degeneration in ApoE-/- mice fed a high-fat diet. Nutrients. 2016;8(11). doi:10.3390/nu8110692
18. Valsecchi AE, Franchi S, Panerai AE, Rossi A, Sacerdote P, Colleoni M. The soy isoflavone genistein reverses oxidative and inflammatory state, neuropathic pain, neurotrophic and vasculature deficits in diabetes mouse model. Eur J Pharmacol. 2011;650(2–3):694–702. doi:10.1016/j.ejphar.2010.10.060
19. Harris RBS, Zhou J, Redmann SM, et al. A Leptin dose-response study in obese (ob/ob) and lean (+/?) mice. Endocrinology. 1998;139(1):8–19. doi:10.1210/endo.139.1.5675
20. Roden M, Ludwig C, Nowotny P, et al. Relative hypoleptinemia in patients with type 1 and type 2 diabetes mellitus. Int J Obes. 2000;24(8):976–981. doi:10.1038/sj.ijo.0801266
21. Li R, Singh M. Sex differences in cognitive impairment and alzheimer’s disease. Front Neuroendocrinol. 2014;35(3):385–403. doi:10.1016/j.yfrne.2014.01.002
22. Al-Nakkash L, Clarke LL, Rottinghaus GE, Chen YJ, Cooper K, Rubin LJ. Dietary genistein stimulates anion secretion across female murine intestine. J Nutr. 2006;136(11):2785–2790. doi:10.1093/jn/136.11.2785
23. Michelin RM, Al-Nakkash L, Broderick TL, Plochocki JH. Genistein treatment increases bone mass in obese, hyperglycemic mice. Diabetes Metab Syndr Obes. 2016;9:63–70. doi:10.2147/DMSO.S97600
24. Al-Nakkash L, Schacht S, Dolan R, et al. Effect of genistein diet on jejunum contractility, motility and morphology in a mouse model of diabetic obesity. FASEB J. 2015;29(1_supplement):
25. Rockwood S, Broderick TL, Al-Nakkash L. Feeding obese diabetic mice a genistein diet induces thermogenic and metabolic change. J Med Food. 2018;21(4):332–339. doi:10.1089/jmf.2017.0084
26. Zheng C, Geetha T, Gearing M, Babu JR. Amyloid β-abrogated TrkA ubiquitination in PC12 cells analogous to alzheimer’s disease. J Neurochem. 2015;133(6):919–925. doi:10.1111/jnc.13076
27. Liu D, Zhen W, Yang Z, Carter JD, Si H, Reynolds KA. Genistein acutely stimulates insulin secretion in pancreatic beta-cells through a cAMP-dependent protein kinase pathway. Diabetes. 2006;55(4):1043–1050. doi:10.2337/diabetes.55.04.06.db05-1089
28. Yanev S, Aloe L, Fiore M, Chaldakov GN. Neurotrophic and metabotrophic potential of nerve growth factor and brain-derived neurotrophic factor: linking cardiometabolic and neuropsychiatric diseases. World J Pharmacol. 2013;2(4):92–99. doi:10.5497/wjp.v2.i4.92
29. Lepeta K, Lourenco MV, Schweitzer BC, et al. Synaptopathies: synaptic dysfunction in neurological disorders - A review from students to students. J Neurochem. 2016;138(6):785–805. doi:10.1111/jnc.13713
30. Li R, Zhang Y, Rasool S, Geetha T, Babu JR. Effects and underlying mechanisms of bioactive compounds on type 2 diabetes mellitus and alzheimer’s disease. Oxid Med Cell Longev. 2019;2019:8165707. doi:10.1155/2019/8165707
31. Arnold SE, Arvanitakis Z, Macauley-Rambach SL, et al. Brain insulin resistance in type 2 diabetes and alzheimer disease: concepts and conundrums. Nat Rev Neurol. 2018;14(3):168–181. doi:10.1038/nrneurol.2017.185
32. Arnold SE, Lucki I, Brookshire BR, et al. High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol Dis. 2014;67:79–87. doi:10.1016/j.nbd.2014.03.011
33. Sartorius T, Peter A, Schulz N, et al. Cinnamon extract improves insulin sensitivity in the brain and lowers liver fat in mouse models of obesity. PLoS One. 2014;9(3). doi:10.1371/journal.pone.0092358
34. Zeng Y, Zhang L, Hu Z. Cerebral insulin, insulin signaling pathway, and brain angiogenesis. Neurol Sci. 2016;37(1):9–16. doi:10.1007/s10072-015-2386-8
35. Morán J, Garrido P, Cabello E, Alonso A, González C. Effects of estradiol and genistein on the insulin signaling pathway in the cerebral cortex of aged female rats. Exp Gerontol. 2014;58:104–112. doi:10.1016/j.exger.2014.07.018
36. Tome D, Fonseca CP, Campos FL, Baltazar G. Role of neurotrophic factors in parkinson’s disease. Curr Pharm Des. 2017;23(5):809–838. doi:10.2174/1381612822666161208120422
37. Krabbe KS, Nielsen AR, Krogh-Madsen R, et al. Brain-derived neurotrophic factor (BDNF) and type 2 diabetes. Diabetologia. 2007;50(2):431–438. doi:10.1007/s00125-006-0537-4
38. Passaro A, Dalla Nora E, Morieri ML, et al. Brain-derived neurotrophic factor plasma levels: relationship with dementia and diabetes in the elderly population. J Gerontol a Biol Sci Med Sci. 2015;70(3):294–302. doi:10.1093/gerona/glu028
39. Matsui H, Musicki B, Sopko NA, et al. Early-stage type 2 diabetes mellitus impairs erectile function and neurite outgrowth from the major pelvic ganglion and downregulates the gene expression of neurotrophic factors. Urology. 2017;99:
40. Harte-Hargrove L, MacLusky NJ, Scharfman HE. BDNF-estrogen interactions in hippocampal mossy fiber pathway: implications for normal brain function and disease. Neuroscience. 2013;239:46–66. doi:10.1016/j.neuroscience.2012.12.029
41. Jiang T, Wang X-Q, Ding C, Du X-L. Genistein attenuates isoflurane-induced neurotoxicity and improves impaired spatial learning and memory by regulating cAMP/CREB and BDNF-TrkB-PI3K/Akt signaling. Korean J Physiol Pharmacol. 2017;21(6):579–589. doi:10.4196/kjpp.2017.21.6.579
42. Ordoñez G, Fernandez A, Perez R, Sotelo J. Low contents of nerve growth factor in serum and submaxillary gland of diabetic mice. A possible etiological element of diabetic neuropathy. J Neurol Sci. 1994;121(2):163–166. doi:10.1016/0022-510x(94)90346-8
43. Tosaki T, Kamiya H, Yasuda Y, et al. Reduced NGF secretion by schwann cells under the high glucose condition decreases neurite outgrowth of DRG neurons. Exp Neurol. 2008;213(2):381–387. doi:10.1016/j.expneurol.2008.06.017
44. Coleman P, Federoff H, Kurlan R. A focus on the synapse for neuroprotection in Alzheimer disease and other dementias. Neurology. 2004;63(7):1155–1162. doi:10.1212/01.wnl.0000140626.48118.0a
45. Valsecchi AE, Franchi S, Panerai AE, Sacerdote P, Trovato AE, Colleoni M. Genistein, a natural phytoestrogen from soy, relieves neuropathic pain following chronic constriction sciatic nerve injury in mice: anti-inflammatory and antioxidant activity. J Neurochem. 2008;107(1):230–240. doi:10.1111/j.1471-4159.2008.05614.x
46. Zeng H, Chen Q, Zhao B. Genistein ameliorates beta-amyloid peptide (25–35)-induced hippocampal neuronal apoptosis. Free Radic Biol Med. 2004;36(2):180–188. doi:10.1016/j.freeradbiomed.2003.10.018
47. Bagheri M, Rezakhani A, Nyström S, et al. Amyloid beta(1–40)-induced astrogliosis and the effect of genistein treatment in rat: a three-dimensional confocal morphometric and proteomic study. PLoS One. 2013;8(10):e76526. doi:10.1371/journal.pone.0076526
48. Bang OY, Hong HS, Kim DH, et al. Neuroprotective effect of genistein against beta amyloid-induced neurotoxicity. Neurobiol Dis. 2004;16(1):21–28. doi:10.1016/j.nbd.2003.12.017
49. Zhao X, Yuan L, Yu H, et al. Genistein inhibited amyloid-β induced inflammatory damage in C6 glial cells. Arch Med Res. 2014;45(2):152–157. doi:10.1016/j.arcmed.2013.12.008
50. Chatterjee G, Roy D, Khemka VK, Chattopadhyay M, Chakrabarti S. Genistein, the isoflavone in soybean, causes amyloid beta peptide accumulation in human neuroblastoma cell line: implications in alzheimer’s disease. Aging Dis. 2015;6(6):456–465. doi:10.14336/AD.2015.0327
51. Wiseman H, Casey K, Bowey EA, et al. Influence of 10 wk of soy consumption on plasma concentrations and excretion of isoflavonoids and on gut microflora metabolism in healthy adults. Am J Clin Nutr. 2004;80(3):692–699. doi:10.1093/ajcn/80.3.692
52. Johnson GVW, Stoothoff WH. Tau phosphorylation in neuronal cell function and dysfunction. J Cell Sci. 2004;117(Pt 24):5721–5729. doi:10.1242/jcs.01558
53. Ye S, Wang -T-T, Cai B, et al. Genistein protects hippocampal neurons against injury by regulating calcium/calmodulin dependent protein kinase IV protein levels in alzheimer’s disease model rats. Neural Regen Res. 2017;12(9):1479–1484. doi:10.4103/1673-5374.215260
54. Pierzynowska K, Podlacha M, Gaffke L, et al. Autophagy-dependent mechanism of genistein-mediated elimination of behavioral and biochemical defects in the rat model of sporadic alzheimer’s disease. Neuropharmacology. 2019;148:332–346. doi:10.1016/j.neuropharm.2019.01.030
55. Gratuze M, El Khoury NB, Turgeon A, et al. Tau hyperphosphorylation in the brain of ob/ob mice is due to hypothermia: importance of thermoregulation in linking diabetes and alzheimer’s disease. Neurobiol Dis. 2017;98:1–8. doi:10.1016/j.nbd.2016.10.004
56. Ho L, Qin W, Pompl PN, et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of alzheimer’s disease. FASEB J. 2004;18(7):902–904. doi:10.1096/fj.03-0978fje
57. Hong M, Lee VM. Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J Biol Chem. 1997;272(31):19547–19553. doi:10.1074/jbc.272.31.19547
58. Kins S, Crameri A, Evans DR, Hemmings BA, Nitsch RM, Gotz J. Reduced protein phosphatase 2A activity induces hyperphosphorylation and altered compartmentalization of tau in transgenic mice. J Biol Chem. 2001;276(41):38193–38200. doi:10.1074/jbc.M102621200
59. Grillo CA, Piroli GG, Kaigler KF, Wilson SP, Wilson MA, Reagan LP. Downregulation of hypothalamic insulin receptor expression elicits depressive-like behaviors in rats. Behav Brain Res. 2011;222(1):230–235. doi:10.1016/j.bbr.2011.03.052
60. Spinelli M, Fusco S, Mainardi M, et al. Brain insulin resistance impairs hippocampal synaptic plasticity and memory by increasing GluA1 palmitoylation through FoxO3a. Nat Commun. 2017;8(1):2009. doi:10.1038/s41467-017-02221-9
61. Ferrario CR, Reagan LP. Insulin-mediated synaptic plasticity in the CNS: anatomical, functional and temporal contexts. Neuropharmacology. 2018;136(Pt B):182–191. doi:10.1016/j.neuropharm.2017.12.001
62. Lee Y-B, Lee HJ, Sohn HS. Soy isoflavones and cognitive function. J Nutr Biochem. 2005;16(11):641–649. doi:10.1016/j.jnutbio.2005.06.010
63. Crider A, Pillai A. Estrogen signaling as a therapeutic target in neurodevelopmental disorders. J Pharmacol Exp Ther. 2017;360(1):48–58. doi:10.1124/jpet.116.237412
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.