")
Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 11
BCL2 Family Inhibitors in the Biology and Treatment of Multiple Myeloma
Authors Gupta VA , Ackley J, Kaufman JL, Boise LH
Received 5 January 2021
Accepted for publication 26 February 2021
Published 12 March 2021 Volume 2021:11 Pages 11—24
DOI https://doi.org/10.2147/BLCTT.S245191
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Wilson Gonsalves
Vikas A Gupta,1 James Ackley,2 Jonathan L Kaufman,1 Lawrence H Boise1
1Department of Hematology and Medical Oncology, Winship Cancer Institute of Emory University, Emory University School of Medicine, Atlanta, GA, USA; 2Cancer Biology Graduate Program, Winship Cancer Institute of Emory University, Emory University School of Medicine, Atlanta, GA, USA
Correspondence: Lawrence H Boise
Department of Hematology and Medical Oncology, Winship Cancer Institute of Emory University, Emory University School of Medicine, 1365 Clifton Road NE, Rm C4078, Atlanta, GA, 30322, USA
Tel +1 404-778-4724
Fax +1 404-778-5530
Email [email protected]
Vikas A Gupta
Department of Hematology and Medical Oncology, Winship Cancer Institute of Emory University, Emory University School of Medicine, 1365 Clifton Road NE, Rm C4090, Atlanta, GA, 30322, USA
Tel +1 404-778-6485
Fax +1 404-778-3260
Email [email protected]
Abstract: Although much progress has been made in the treatment of multiple myeloma, the majority of patients fail to be cured and require numerous lines of therapy. Inhibitors of the BCL2 family represent an exciting new class of drugs with a novel mechanism of action that are likely to have activity as single agents and in combination with existing myeloma therapies. The BCL2 proteins are oncogenes that promote cell survival and are frequently upregulated in multiple myeloma, making them attractive targets. Venetoclax, a BCL2 specific inhibitor, is furthest along in development and has shown promising results in a subset of myeloma characterized by the t(11;14) translocation. Combining venetoclax with proteasome inhibitors and monoclonal antibodies has improved responses in a broader group of patients, but has come at the expense of a toxicity safety signal that requires additional follow-up. MCL1 inhibitors are likely to be effective in a broader range of patients and are currently in early clinical trials. This review will cover much of what is known about the biology of these drugs, biomarkers that predict response, mechanisms of resistance, and unanswered questions as they pertain to multiple myeloma.
Keywords: multiple myeloma, BCL2 family inhibitors, venetoclax
Introduction
Resistance to apoptosis is a hallmark of cancer biology.1 Cell survival is a delicate balance of pro-survival and pro-death signals that in turn feed into pro-apoptotic and anti-apoptotic proteins of the BCL2 family. In order to survive the numerous genetic insults and pro-apoptotic changes in cell biology such as growth factor withdrawal, loss of adhesion, and hypoxia, malignant cells must upregulate the anti-apoptotic BCL2 proteins, and as a result, become highly dependent on the sustained activity of these proteins.2 This dependence therefore makes for an attractive therapeutic target. Within the last several years, multiple highly selective inhibitors of the BCL2 family have been developed leading to the approval of the first drug in this class, venetoclax, for the treatment of chronic lymphocytic leukemia (CLL) and acute myeloma leukemia (AML) in combination with chemotherapy or hypomethylating agents. This review will discuss the pre-clinical and clinical work examining the role of BCL2 inhibitors specifically in the plasma cell malignancy multiple myeloma.
Multiple Myeloma
Multiple myeloma is the result of clonal proliferation of malignant plasma cells.3 These plasma cells retain much of their normal biology, including bone marrow residence and secretion of large quantities of monoclonal antibody.4 Myeloma is also highly heterogeneous both clinically and genetically. As such, therapeutically targeting its plasma cell biology rather than its tumor biology has proven to be far more effective clinically.4 The use of proteasome inhibitors, immunomodulatory agents, monoclonal antibodies, and routine high dose melphalan with autologous stem cell rescue has dramatically improved the outcomes in multiple myeloma patients. Long-term follow-up for induction therapy with the triplet lenalidomide, bortezomib, and dexamethasone and autologous stem cell transplant has demonstrated a 98.5% response rate, median progression-free survival of 65 months, and overall survival of 126 months.5 The addition of daratumumab is likely to further improve those outcomes as well as the key goal of achieving negative measurable residual disease.6–9 However, despite these excellent results, the majority of myeloma patients ultimately relapse and require additional lines of therapy. Identifying drugs with novel mechanisms of action to avoid cross-resistance is therefore of critical importance. Inhibitors of the BCL2 family are a promising new class of drugs that represent decades of research into the mechanisms of apoptosis as well as a tour de force of structural and chemical biology during drug development.
BCL2 Family
The BCL2 family consists of numerous pro- and anti-apoptotic proteins (Figure 1).10 The anti-apoptotic proteins include BCL2, BCLXL, MCL, BCLW, and BFL1. These proteins contain 4 BCL2 homology (BH) domains and a transmembrane domain that allows them to insert into the endoplasmic reticulum or mitochondrial membrane. Their expression varies from cell type to cell type, and one cell type can express multiple proteins. Cancer cells often upregulate expression of one or more anti-apoptotic proteins in order to survive.11 The pro-apoptotic proteins can be subdivided into two groups, the BH3 only proteins and the effector proteins.10 Like their name suggests, the BH3 only proteins contain a BH3 domain, but lack the BH1, 2, and 4 domains found in anti-apoptotic proteins. Proteins in this group include BIM, BID, PUMA, BAD, NOXA, BIK, BMF, and HRK. BAK and BAX are effector proteins containing the BH1-3 domains and promote apoptosis by homo- or hetero-oligomerizing in the mitochondrial outer membrane upon activation, forming a pore that allows release of cytochrome C from the mitochondria. Cytosolic cytochrome C provides the seed which triggers the formation of the apoptosome, a large macromolecular complex that activates caspases, the final step in execution of apoptosis.12 The BH3 only proteins are typically induced by various cellular stresses such as DNA damage or growth factor withdrawal. BIM, BID, and PUMA are considered activator BH3 only proteins because of their ability to directly activate BAK and BAX, whereas the remaining BH3 only proteins are sensitizers. The sensitizers bind to the anti-apoptotic proteins, but do not directly activate BAK and BAX.
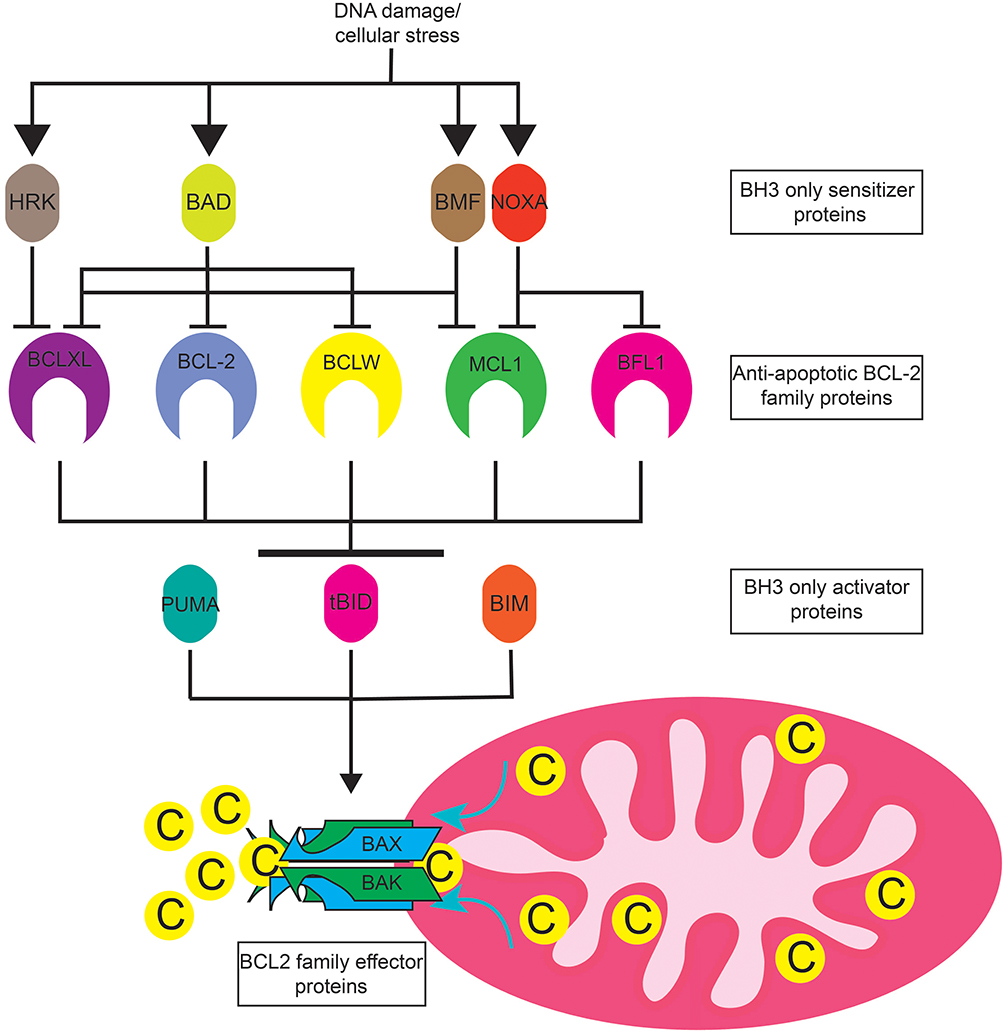 |
Figure 1 BCL-2 family protein interactions. DNA damage and cellular stress such as that caused by DNA damaging agents or proteosome inhibitors induce the expression of apoptotic sensitizers HRK, BAD, NOXA, BMF, and BIK (not pictured). These proteins selectively bind to and inhibit the anti-apoptotic BCL2 family members BCLXL BCL2, BCLW, MCL1, and BFL1. Anti-apoptotic BCL2 family proteins sequester apoptotic activator proteins PUMA, tBID, and BIM. Free apoptotic activators induce the oligomerization of apoptotic effectors BAK and BAX at the mitochondrial membrane. BAK and BAX oligomerization causes mitochondrial outer membrane permeabilization and the release of cytochrome C into the cytosol resulting in apoptosome formation and caspase activation. |
Together, the pro- and anti-apoptotic proteins bind to one another in a network of interactions that ultimately determine whether a cell lives or dies. Specifically, the pro-apoptotic proteins bind via their BH3 domain to a hydrophobic groove in the anti-apoptotic proteins. BCL2, BCLXL, MCL1, BCLW, and BFL1 bind to and neutralize the activator BH3 only proteins BIM, BID, and PUMA, thus preventing them from activating BAK and BAX.11 BIM, BID, and PUMA are capable of binding to all of the anti-apoptotic proteins. Upon induction, the sensitizer proteins function by binding to the anti-apoptotic proteins and displacing BIM, BID, and PUMA, thus allowing these activator proteins to activate BAK and BAX. In contrast to activator BH3 only proteins, the sensitizers have a more restrictive pattern of binding to the anti-apoptotic proteins. For example, NOXA binds only to MCL1 and BFL1, while BAD binds to BCL2, BCLXL, and BCLW. In addition to neutralizing BIM, BID, and PUMA, the anti-apoptotic proteins can directly bind to activated BAK and BAX, preventing their oligomerization until being released by the BH3 only proteins.
BCL2 Family Dependence in Multiple Myeloma
The effectiveness of a BCL2 family inhibitor in a particular cell type is based on the dependence of that cell type for that BCL2 family member or members, which in turn is dictated by the expression pattern and interactions of the BCL2 proteins within that cell. Normal plasma cells from which multiple myeloma arises are primarily MCL1 dependent. The use of ABT-737, an inhibitor of BCL2 and BCLXL, resulted in the loss of newly formed plasma cells, but had no effect on the number of long-lived plasma cells in mice.13 Similarly, ABT-737 or RNA interference knockdown of BCLXL induced apoptosis of in vitro differentiating plasma cells, consistent with a BCLXL dependent stage in early plasma cell development.14 In contrast, MCL1 appears to be crucial for long-lived plasma cells.15 Conditional knockout of MCL1 in plasma cells reduced their numbers significantly. However, reflecting the overall heterogeneity of multiple myeloma, the BCL2 family dependence in malignant plasma cells is also much more heterogeneous. A number of studies have examined BCL2 family expression in multiple myeloma with varying results. BCL2, BCLXL, and MCL1 have all been reported to be expressed in multiple myeloma.16–18 The expression of MCL1 and possibly BCLXL can be induced by IL-6, a key plasma cell survival cytokine, though this induction is cell line dependent rather than universal.19–25 Increased expression of BCLXL and MCL1 has also been associated with worse patient outcome.16,18 Furthermore, MCL1 expression is increased with disease progression.18 The importance of MCL1 for myeloma cell survival was first established by the use of antisense oligonucleotides (ASO) to knockdown MCL1. Only MCL1 ASO and not BCL2 or BCLXL ASO resulted in myeloma cell death.26,27 Knockdown of MCL1 by siRNA or CRISPR Cas9 mediated knockout confirmed that the majority of myeloma cell lines are MCL1 dependent.28,29
Early Putative BCL2 Inhibitors
Given the understanding of BCL2 family proteins and their regulation of apoptosis, inhibitors of BCL2, BCLXL, and MCL1 should have a common mechanism of action.30 Numerous compounds including obatoclax, subatoclax, maritoclax, gossypol, apogossypol, TW-37, UMI-77, and BDA-366, have been developed as specific or pan-BCL2 inhibitors, some of which have entered clinical trials. However, a number of recent studies using sophisticated tools have demonstrated that these drugs have off-target and BCL2 independent effects that contribute to cell death.31–33 Although these drugs bind their proposed targets, if they induce cytochrome C release, they do so indirectly through other mitochondrial and endoplasmic reticulum stress that results in transcriptional induction of BH3 sensitizers such as NOXA.31,33 Most importantly, many of these drugs are capable of killing BAK/BAX deficient cells, thus distinguishing them from true BH3 mimetics.32–34 The inhibitors discussed below not only bind to their targets with much higher affinity than the putative BCL2 inhibitors above, they induce apoptosis through activation of BAK and BAX and mitochondrial cytochrome C release as expected for true direct inhibitors of the BCL2, BCLXL, and MCL1.32
Pre-Clinical Studies with BCL2 Inhibitors in Myeloma
Although the data described above point to MCL1 as a natural target for anti-myeloma therapy, the first specific small molecule inhibitor of the BCL2 family, ABT-737, targeted BCL2, BCLXL, and BCLW (Figure 2). ABT-737 was developed using nuclear magnetic resonance (NMR) to identify a small molecule capable of binding the same hydrophobic groove of anti-apoptotic proteins responsible for binding the BH3 domain of anti-apoptotic proteins, thus disrupting their interaction.35 ABT-737 demonstrates nanomolar affinity for the BH3 binding site of BCL2, BCLXL, and BCLW and is capable of inducing apoptosis in a range of hematologic and solid tumor cell lines.35 ABT-737 was modified to make it orally bioavailable for clinical use, resulting in ABT-263 or navitoclax. Unfortunately, navitoclax’s clinical utility was limited by significant thrombocytopenia as a result of BCLXL dependent platelet apoptosis.36 ABT-263 was therefore modified to make it BCL2 specific resulting in ABT-199 or venetoclax.37 By inhibiting only BCL2, venetoclax does not cause platelet apoptosis in vitro or thrombocytopenia in vivo and therefore expanded into clinical trials for various hematologic malignancies.37 Venetoclax has subnanomolar affinity for BCL2 while binding BCLXL and BCLW 1000-fold less effectively and was able to induce cell death more efficiently than ABT-737, particularly in cells with high BCL2 expression. It also disrupted binding of BIM and PUMA to BCL2, but not BCLXL.38,39 Importantly, both ABT-737 and venetoclax act through BAK and BAX, indicating that they are indeed inducing apoptosis through the mitochondrial pathway.34,37 In myeloma, treatment of both cell lines and patient samples with ABT-737 was able to induce cell death and synergized with other myeloma therapies such as dexamethasone, bortezomib, and melphalan.40–42 Cells sensitive to ABT-737 tend to express more BCL2 and BCLXL than resistant cells, which express more MCL1. Higher MCL1 expression may serve as a source of resistance to venetoclax by acting as a sink to bind and neutralize BIM displaced from BCL2 by venetoclax. Indeed, eliminating that sink with short interfering RNA knockdown of MCL1 sensitized cells to venetoclax.43 However, given the complex interactions between pro- and anti-apoptotic proteins, expression alone cannot fully account for response. Co-immunoprecipitation experiments demonstrate that ABT-737 sensitive cells also have more of the pro-apoptotic protein BIM bound to BCL2 and BCLXL.19,29 A number of factors both intrinsic and extrinsic are likely to influence the distribution of BIM binding to anti-apoptotic proteins. In addition to increasing MCL1 expression, IL-6 induces BIM phosphorylation through the mitogen-activated protein kinase pathway (MAPK), which correlates with increased BIM association with MCL1 and resistance to ABT-737 and venetoclax.19 In addition, dexamethasone has been shown to increase BIM expression as well as its binding to BCL2, thus sensitizing cells to BCL2 inhibition.38 Glutamine withdrawal and inhibition of glutamine metabolism had a similar effect.44 Other aspects of cellular metabolism such as the oxygen consumption rate and electron transport chain activity are decreased in venetoclax sensitive myeloma, suggesting that they may contribute to BCL2 dependence as well.45
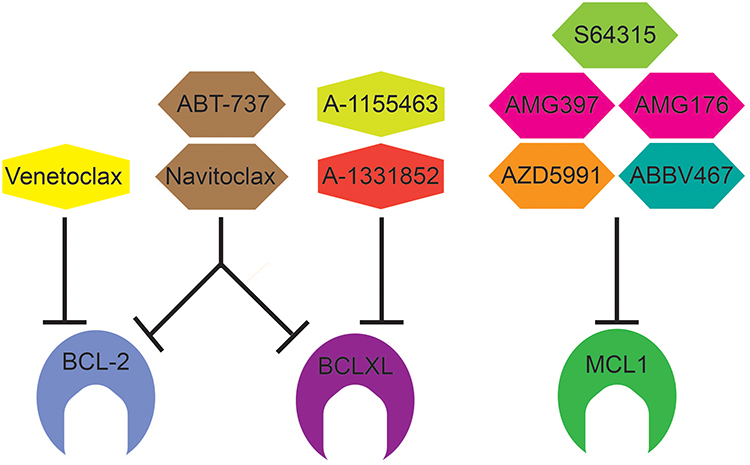 |
Figure 2 BCL-2 Family inhibitors and their targets. Venetoclax (ABT-199) is a BCL2 selective inhibitor and approved for use in CLL, SLL, and AML under the trade name VENCLEXTA®. ABT-737 inhibits BCL2, BCLXL, and BCLW (not shown). Similar to ABT-737, Navitoclax inhibits BCL-2, BCLXL, and BCLW but is orally bioavailable. A-1155463 and A-1331852 are selective BCLXL inhibitors. There are five selective MCL1 inhibitors in early clinical trials: S64315 (MIK665), AZD5991, AMG176, AMG379, and ABBV467. |
Further testing of myeloma cell lines revealed that a subset of myeloma characterized by the t(11;14) translocation is particularly sensitive to ABT-737 and venetoclax.28,39,46–49 This translocation juxtaposes the cyclin D1 gene with the immunoglobulin heavy chain enhancer, resulting in cyclin D1 overexpression, and is one of the first biomarkers in multiple myeloma to predict response to therapy.
Clinical Studies of Venetoclax in Multiple Myeloma
The promising pre-clinical results with venetoclax led to a Phase I study in relapsed/refractory multiple myeloma.50 This study enrolled 66 patients, 30 of whom were positive for t(11;14), a fraction far higher than the 15–20% prevalence of t(11;14) in the general myeloma population. The increased enrollment of t(11;14) patients is likely a reflection of the pre-clinical data demonstrating increased response in the t(11;14) subset. The patients enrolled represented a heavily pre-treated group with a median of 5 lines of prior therapy (range 1–15). The majority of patients were refractory to bortezomib (70%), lenalidomide (77%), and pomalidomide (53%) and had undergone autologous stem cell transplant (76%). Venetoclax was well tolerated with hematologic toxicities being the most common grade 3 or 4 adverse events (AE). Thrombocytopenia occurred in 26%, neutropenia in 21%, anemia in 14%, and leukopenia in 14% (Table 1). Gastrointestinal toxicities including nausea (47%), diarrhea (36%), and vomiting (21%) were the most common AEs. Importantly, and in contrast to CLL, no events of tumor lysis syndrome occurred.51 The maximum tolerated dose was not reached, and thus the maximum planned dose of 1200 mg daily was used for the expansion cohort. In the total population, the overall response rate was 21%, with 15% achieving a very good partial response (VGPR) or better (Table 2). In the t(11;14) subpopulation, the overall response rate (ORR) was 40% with 27% achieving a VGPR or better. Twelve of the 14 responders were positive for t(11;14), confirming the value of t(11;14) as a biomarker for venetoclax response, but also highlighting that responses occur in non-t(11;14) patients as well. In the t(11;14) group, the median duration of response (DOR) was 9.7 months.
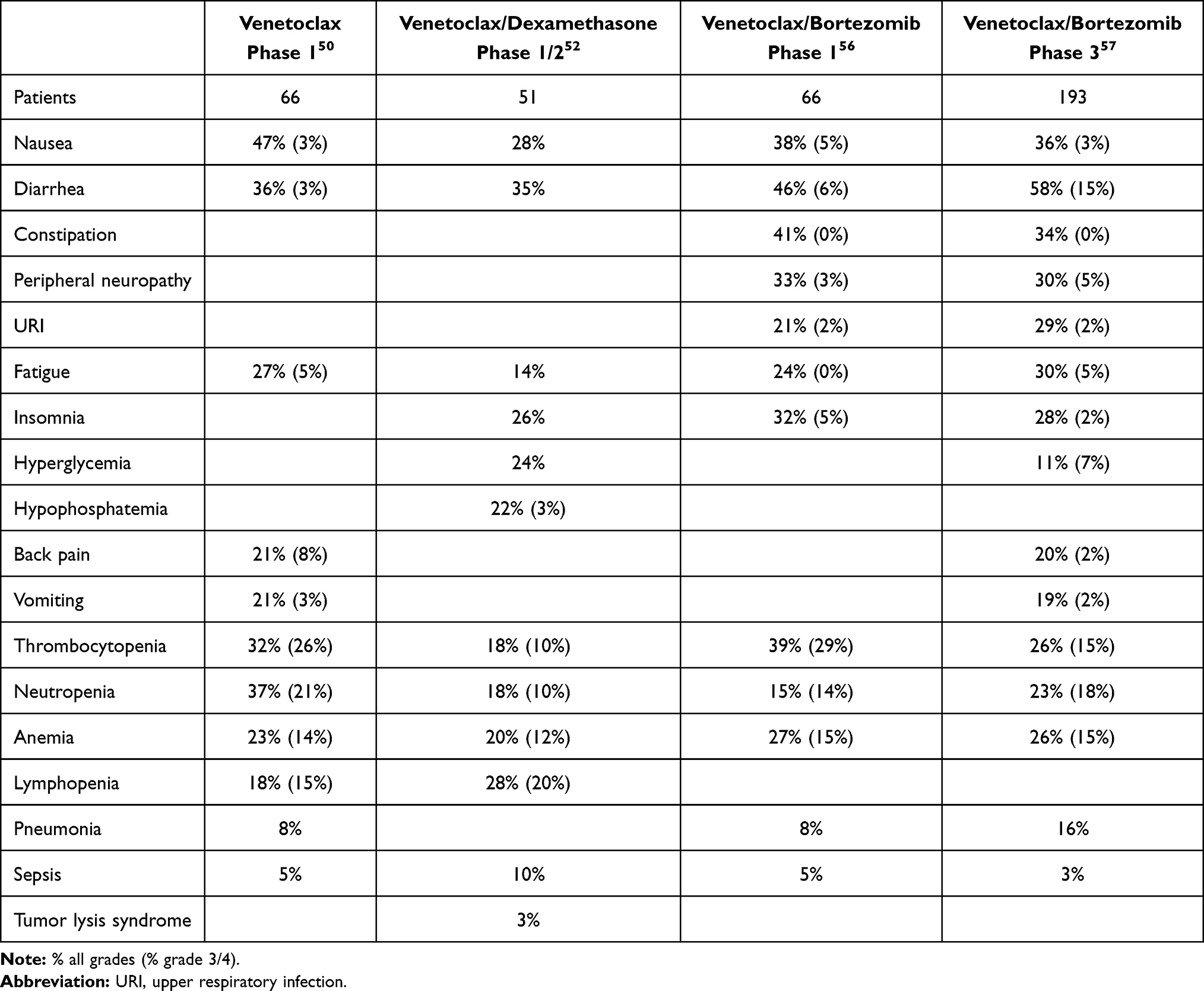 |
Table 1 Venetoclax Toxicities |
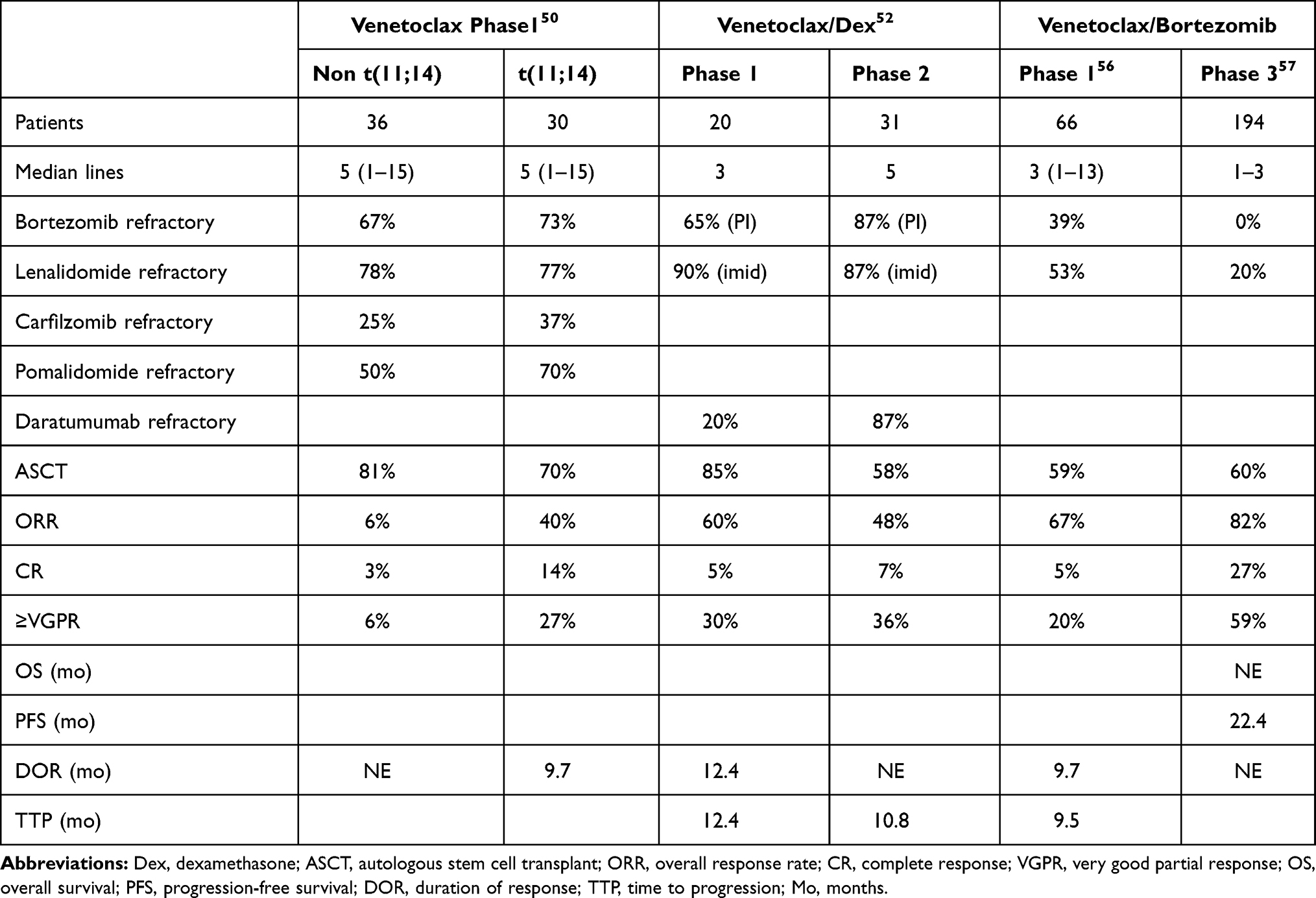 |
Table 2 Previous Therapy and Responses in Venetoclax Trials |
Based on the response rate in t(11;14) and the pre-clinical data demonstrating synergy with venetoclax and dexamethasone, a phase I/II study of this combination specifically in t(11;14) relapse/refractory myeloma was initiated.52 All patients received venetoclax 800 mg daily and dexamethasone 40 mg weekly. The phase I portion of the trial enrolled 20 patients with a median of 3 prior therapies. Treatment-emergent adverse events were similar to the venetoclax monotherapy phase I study, with the addition of insomnia (45%), hypophosphatemia (40%), and hyperglycemia (35%), likely due to the addition of dexamethasone (Table 1). Tumor lysis syndrome was seen in 10% of patients with no deaths. The ORR was 60% with 30% achieving a VGPR or better (Table 2). The median duration of response was 12.4 months. Thirty-one patients with a median of 5 prior lines of therapy participated in the Phase II portion. Adverse events were similar, though 11 deaths occurred, 8 due to progressive disease and 2 due to an adverse event. The ORR was 48% with 36% achieving a VGPR or better. Median DOR and overall survival (OS) had not been reached at the time of publication. Although the lower ORR in the phase II group is likely due to heavier pre-treatment in this population, it remains remarkable for a single agent plus dexamethasone and compares favorably to response rates for other agents. Furthermore, in a single-institution retrospective analysis of 68 patients selected for venetoclax treatment based on the presence of t(11;14), the ORR was 71% (48.5% VGPR or better) with a median progression-free survival (PFS) of 14.1 months.53 For patients with less than 3 prior lines of therapy, the PFS was 23.2 months. These promising results have led to an ongoing Phase III study randomizing patients with t(11;14) myeloma to either venetoclax and dexamethasone or pomalidomide and dexamethasone.54 The primary endpoint will be PFS, with safety, tolerability, ORR, rate of VGPR or better, DOR, and OS being assessed as well.
In addition to having single-agent activity, venetoclax may serve to decrease the apoptotic threshold of myeloma cells, sensitizing them to other active agents. Thus, a number of studies are investigating additional venetoclax combinations. Proteasome inhibitors are of particular interest based on their ability to upregulate the BH3 only protein NOXA.55 NOXA binds specifically to MCL1, thus inhibiting a potential source of resistance to venetoclax. This finding provided the rationale for a study of bortezomib plus venetoclax and dexamethasone which enrolled 66 patients.56 All cytogenetic subtypes were allowed, thus only 14% of patients had t(11;14). Patients had received a median of 3 prior lines of therapy. Importantly 80% were bortezomib exposed and 39% were bortezomib refractory. An additional 53% were lenalidomide refractory and 59% had undergone autologous stem cell transplant. In terms of safety, the most common AEs were gastrointestinal, including diarrhea (46%), constipation (41%), and nausea (38%, Table 1). Cytopenias were the most common grade 3 or 4 AEs and included thrombocytopenia (29%), anemia (15%), and neutropenia (14%). Thrombocytopenia is a known side effect of bortezomib, and patients who remained on venetoclax monotherapy saw their platelet counts stabilize. Serious AEs included pneumonia in 8% of patients. No tumor lysis syndrome was reported. In the total population, the ORR was 67% with 42% achieving a VGPR or better (Table 2). The median DOR was 9.7 months. The response rate was higher in patients not refractory to bortezomib (90%) compared to those who were refractory (31%). Of the bortezomib refractory responders, 38% were t(11;14), suggesting they may have responded to venetoclax alone. The number of prior therapies influences response as well. In patients exposed to 1–3 prior lines, the response rate was 89% compared to 50% for those with 4–6 prior lines and only 11% for those with greater than 6.
These results led to the design of the phase III BELLINI study comparing venetoclax and placebo in combination with bortezomib and dexamethasone.57 Patients with 1–3 prior lines of therapy, but not proteasome inhibitor refractory disease, were included. Prior proteasome exposure was allowed. Randomization was 2:1 to receive venetoclax (194 patients) or placebo (97 patients). Previous proteasome inhibitor exposure occurred in 70% of patients, while 30% were PI naïve. Patients with t(11;14) made up 10% of the venetoclax arm and 15% of the placebo arm. The PFS in the venetoclax arm was significantly longer than that of the placebo arm, 22.4 months vs 11.5 months (Table 2), with the patients deriving the greatest benefit having either t(11;14) or high BCL2 expression. The overall response rate in the venetoclax arm was 82% (59% VGPR or better) compared to 68% (36% VGPR or better) in the placebo arm. However, a surprising and concerning increase in death was observed in the venetoclax arm, 21% vs 11% in the placebo group. These deaths appear to be limited to the non-t(11;14), BCL2 low patients and were primarily related to infection, particularly at the time of disease progression. Although the overall rate of infections was similar in the two groups, 8 fatal infections occurred in the venetoclax arm compared to none in the placebo arm. Some of these infections occurred in the setting of early disease progression, raising the question of whether venetoclax may antagonize bortezomib in a small subset of patients. Alternatively, patients who progress on or do not respond to venetoclax may have shorter OS due to failure to respond to subsequent therapies. However, in a retrospective analysis of 38 patients who progressed on venetoclax, 84% went on to receive an additional line of therapy and experienced a median OS of 31.4 months from the time of venetoclax refractoriness, arguing against any inferior outcomes post-venetoclax.53 The safety concerns from this trial have increased scrutiny of other venetoclax combination trials in myeloma and led to adjustments such as routine antimicrobial prophylaxis and closer infectious monitoring.
Venetoclax is also being combined with the monoclonal antibody daratumumab with or without bortezomib (NCT03314181, Table 3). Part 1 of this trial is a dose escalation of venetoclax in combination with daratumumab and dexamethasone for patients with t(11;14). Part 2 involves a dose escalation of venetoclax with bortezomib, daratumumab and dexamethasone for relapsed/refractory patients. Preliminary results from this trial were reported in May 2020.58 Twenty-four patients with t(11;14) had been enrolled in part 1 and another 24 patients in part 2, 6 with t(11;14). Adverse events were similar to previous venetoclax trials with common side effects including fatigue, diarrhea, and nausea. Nine patients had infection-related AEs of grade 3 or worse. Given the results of the BELLINI study, an additional non-venetoclax containing arm consistent of bortezomib, daratumumab, and dexamethasone has been added as a comparator to monitor for toxicity related to venetoclax. The ORR for both parts was greater than 90%. Another active combination trial with venetoclax in relapsed/refractory myeloma includes the addition of carfilzomib to venetoclax and dexamethasone (NCT02899052). Carfilzomib is a second-generation proteasome inhibitor and thus has a similar rationale for combination with venetoclax as bortezomib. This ongoing phase II trial has enrolled 49 patients with 1–3 prior lines of therapy.59 The authors did not report any new safety signals. The maximum tolerated dose had not been reached, and venetoclax 800 mg with carfilzomib 70 mg/m2 was chosen for expansion. In t(11;14) patients with a median of 2 lines of prior therapy, the ORR was 92% (85% VGPR or better). Together these data suggest venetoclax combinations are highly effective, particularly in t(11;14) patients, but additional follow-up and monitoring for infectious complications is necessary.
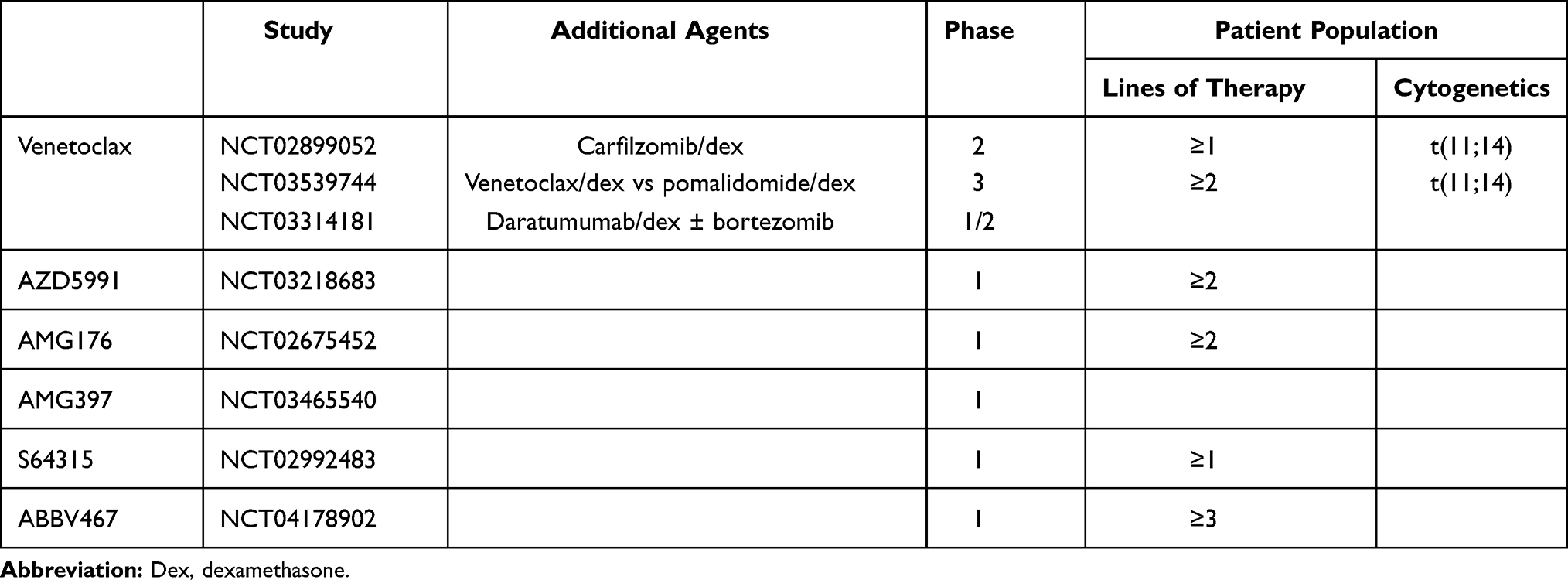 |
Table 3 Currently Open Clinical Trials Investigating BCL2 Family Inhibitors |
Predicting Response to Venetoclax
Another approach to reducing the risk of venetoclax combinations is to use biomarkers to select the patients most likely to respond. Although t(11;14) enriches for venetoclax sensitive patients, not all t(11;14) patients responds and there are non-t(11;14) patients that do, making a more sensitive and specific marker a potentially valuable tool. Knowing that BCL2 is the target of venetoclax and that BCLXL and MCL1 may contribute to resistance, expression of these BCL2 family members has been examined in a number of studies. In cell lines, BCL2 expression was positively correlated with ABT-737 and venetoclax response, while BCLXL and MCL1 expression was negative correlated.39,46,47 The BCL2/MCL1 and BCL2/BCLXL ratios were also significantly higher in sensitive cell lines and patient samples.39,49 This approach has also been examined in the clinical trials of venetoclax. In the phase I trial of venetoclax monotherapy, 44 patients were evaluated for BCL2, BCLXL, and MCL1 gene expression.60 Both the BCL2/BCLXL and BCL2/MCL1 ratios were higher in patients achieving a PR or better. A high BCL2/BCLXL ratio (log2≥2.3) was selected to maximize the likelihood of a patient response in a post hoc analysis using bootstrapping and aggregating of thresholds from trees (BATT).61 A high ratio was present in 23% of patients; and the ORR in this group was 80%, while it was only 9% in the remaining patients with a low ratio. In the Phase 1/2 study of venetoclax and dexamethasone, only the BCL2 expression was statistically higher in the responsive group, while the BCLXL expression and BCL2/BCLXL ratio were unchanged.52 Similarly, in the phase I study of venetoclax plus bortezomib, only BCL2 expression was significantly higher in the responding group.56 Of the 40% of patients with high BCL2, 94% achieved a PR or better compared to 59% in the BCL2 low group. The BELLINI trial used both immunohistochemistry (IHC) and gene expression to assess BCL2 levels. IHC classified 79% of patients as BCL2 high based on 50% or more of tumor cells having a cytoplasmic intensity score of 2 or more on a 0–3 scale.57 The hazard ratio for PFS was 0.5 in the BCL2 high group and 1.39 in the BCL2 low group. High BCL2 gene expression was again determined in a post hoc analysis using a sequential BATT process. The PFS in the BCL2 high group 22.4 months compared to 9.9 months in the BCL2 low group. Although t(11;14) appears to enrich for increased BCL2 expression it remains unclear whether t(11;14) is directly responsible for the higher expression or the two are just correlated. While BCL2 expression and ratios may be more selective than t(11;14), their general use may be challenging. In all of the phase I trials where individual patient data are reported, there is a significant overlap among the sensitive and resistant patients in terms of expression and ratios, and all three studies required a post hoc analysis to identify the optimal cutoff. Furthermore, gene expression fails to account for the post-translational regulation of some BCL2 family members as well as the complex interactions of pro- and anti-apoptotic proteins that contribute to BCL2 dependence.
An alternative to gene expression analysis is ex vivo testing of patient samples using either peptides from the BH3 only proteins, also referred to as BH3 profiling, or the therapeutic drugs themselves. This type of testing captures all of the complexities of the BCL2 family and directly assesses BCL2 dependence. BH3 profiling takes advantage of the specific binding patterns of the pro-apoptotic BH3 only proteins.62 Cells are gently permeabilized with a detergent that allows small peptides to pass through the cell membrane without affecting the mitochondria. The cells are then treated with a small peptide from the BH3 only proteins and the apoptotic response of the cell is measured by mitochondrial cytochrome C release to determine BCL2 dependence. For example, the BH3 peptide from BAD only binds to BCL2 and BCLXL. Therefore, a cell that is BCL2 or BCLXL dependent will undergo cytochrome C release upon treatment with the BAD peptide but would not respond to a peptide from NOXA which binds MCL1. Ex vivo drug testing involves using inhibitors such as venetoclax specific for different BCL2 family members. Both of these techniques have been used on cell lines and patient samples with very good correlation.28,47–49 Ex vivo testing also correlates with clinical response.63 When applied to 16 bone marrow aspirates from patients who went on to receive venetoclax, 10 patients were predicted to be sensitive, 9 of whom achieved a partial response (PR) or better. Of the 6 patients predicted to be resistant, only 1 had a partial response. Despite the appeal of ex vivo testing for selecting venetoclax sensitive patients, it requires a highly specialized laboratory to be performed and thus has its own limitations.
Acquired Venetoclax Resistance
Like other therapies, most myeloma patients who initially respond to venetoclax will ultimately relapse. Much of this acquired resistance may occur through shifts in the dependence of the cell from BCL2 to BCLXL or MCL1. Ex vivo testing with venetoclax prior to clinical treatment and again upon relapse post-venetoclax in three patients confirmed a decrease in BCL2 dependence.63 This shift in BCL2 dependence can occur through copy number gains of either MCL1 and BCLXL.64 Ex vivo testing in a patient who had expansion of a clone with gain of MCL1 post-venetoclax again demonstrated a greater than 10 fold decrease in BCL2 dependence compared to testing performed pre-venetoclax and simultaneously demonstrated marked MCL1 dependence. In studies of CLL patients progressing on venetoclax, sequencing of the BCL2 gene has also revealed recurrent acquired mutations in the BH3 binding domain that disrupt the ability of venetoclax to bind to BCL2, the most common being G101V.65–67 A de novo D111A mutation in BCL2 has also been reported in a myeloma patient progressing on venetoclax.64 This mutation conferred resistance to venetoclax when overexpressed in the venetoclax sensitive t(11;14) cell line KMS12PE and also prevented release of BIM from BCL2 upon venetoclax treatment.
MCL1 Inhibitors
As noted above, the majority of multiple myeloma is hypothesized to be MCL1 dependent, and therefore the development of MCL1 specific inhibitors has been eagerly anticipated. There are currently 5 MCL1 inhibitors in early clinical trials, S64315 (MIK665), AZD5991, AMG176, AMG397, and ABBV467 (Table 3). S63845 (a pre-clinical variant of S64315), AZD5991, AMG176, and AMG397 all have picomolar affinity for MCL1 with little to no binding of BCL2 or BCLXL.68–72 They are capable of disrupting MCL1 binding to BIM and, as expected, require BAK and BAX for activity. All four are effective against a range of cell lines representing various hematologic malignancies, both in vitro and in vivo. Not surprisingly, multiple myeloma was among the most sensitive cell types to these MCL1 inhibitors. Unlike venetoclax, responses were seen beyond t(11;14) myeloma. Of 48 myeloma patient samples treated ex vivo, 71% were sensitive to AZD5991, a fraction greater than would be expected from venetoclax alone. In vivo testing was done with mouse xenografts. However, due to differences between human and mouse MCL1, these MCL1 inhibitors are not effective against mouse MCL1, making toxicity assessments difficult. To overcome this limitation, AMG176 and S63845 were tested in a “humanized” mouse in which the mouse MCL1 was replaced with human MCL1.68,73 In these mice MCL1 inhibition caused dose-dependent decreases in B cells, monocytes, and neutrophils. No other systemic toxicity was reported, however, conditional knockout of MCL1 in mice has revealed an important role for MCL1 in cardiac myocytes.74,75 Mice in which MCL1 was genetically deleted from the heart rapidly developed cardiomyopathy and died. Interestingly, the defects in MCL1 deficient cardiac myocytes are only partly related to apoptosis, but also mitochondrial function, suggesting MCL1 participates in more than just cell survival. The risk of cardiac toxicity has already been confirmed in the phase I trial of AMG397, leading to a temporary hold in the trials of both AMG397 and AMG176. The remaining MCL1 inhibitor trials have not reported any significant cardiac adverse events, which could be influenced by dose and timing of drug administration, and thus the therapeutic window for each drug may be slightly different. Identification of a biomarker for MCL1 inhibitor sensitivity would allow for more selective treatment of the patients most likely to respond, thus improving the risk/benefit ratio. The MCL1 gene is located on chromosome 1q, which is frequently amplified in multiple myeloma and results in increased MCL1 expression, potentially increasing MCL1 dependency in these cells. In one study of 44 patient samples treated ex vivo with S63845, 75% of the 12 samples with 1q gain were highly sensitive compared to 19% of the 1q wildtype samples.76 However, a second study of 27 patient samples did not report any difference in sensitivity between the two groups.77
Like venetoclax, resistance to MCL1 inhibitors can be mediated by the presence of other anti-apoptotic proteins. The effectiveness of both S63485 and AMG176 was inversely correlated with BCL2 and BCLXL expression in myeloma, AML, and CML.68,69,76,77 Furthermore, the BIM released from MCL1 by MCL1 inhibitors has been observed to shift to BCL2 or BCLXL.78,79 Given the potential for other BCL2 proteins to contribute to MCL1 inhibitor resistance, a number of studies have examined combinations of different BCL2 family inhibitors. Both AMG176 and AZD5991 in combination venetoclax was more effective than any of the drugs alone in a panel of AML cell lines, and a phase I trial of venetoclax plus S64315 is currently enrolling (NCT03672695).68,70 Such combinations produce similar results in multiple myeloma and are more effective against cell lines such as U266 that are resistant to MCL1 or BCL2 inhibition alone.76–80 However, targeting multiple anti-apoptotic proteins simultaneously increases the risk of inducing apoptosis in normal cells, particularly lymphocytes, and must be approached cautiously. The bone marrow microenvironment is likely to play a role in response to MCL1 inhibitors as well. Cell-cell contact with bone marrow stromal cells protects myeloma cells from MCL1 inhibitor-induced death.78 In contrast to venetoclax, the increased expression of MCL1 by IL-6 should have no effect on MCL1 inhibitors, though other secreted factors may affect response.78 To improve the efficacy of MCL1 inhibitors, combinations with venetoclax and other active myeloma agents will likely be investigated once the safety of these MCL1 inhibitors as single agents is established.
BCLXL Inhibitors
Despite the risk of thrombocytopenia due to platelet dependence on BCLXL, BCLXL inhibitors continue to be developed. Two BCLXL specific inhibitors have been described, A-1155463 and A-1331852.81,82 Both have picomolar affinity for BCLXL with 1000 fold less binding to BCL2 and MCL1.83 A-1155463 disrupts the binding of BIM to BCLXL, but not BCL2, and effectively kills BCLXL dependent cell lines. A-1331852 is 10 fold more potent than A-1155463 and is orally bioavailable as well. Although A-1331852 did cause expected thrombocytopenia in rats, it did not affect neutrophil counts, which can be reduced with BCL2 inhibition.83 Neither of these compounds have entered clinical development yet. However, two dual BCL2/BCLXL inhibitors are being studied. ABT-263 is an orally bioavailable version of ABT-737 that is being studied in solid tumors where normal marrow function may mitigate the degree of thrombocytopenia,84 while AZD4320 is being studied in hematologic malignancies including multiple myeloma (NCT04214093).85 Unlike ABT-263, which is given orally daily, AZD4320 is given as an intravenous infusion once a week and has a shorter half-life.85 These properties allow platelets to recover between doses, while still having meaningful anti-tumor effects, and may therefore help AZD4320 avoid the dose-limiting thrombocytopenia seen with ABT-263. Another interesting approach to targeting BCLXL without inducing thrombocytopenia was demonstrated with the use of a PROTAC.86 These bifunctional molecules bind to a target protein and recruit them to an E3 ubiquitin ligase for proteasomal degradation. ABT-263 served as the basis for the PROTAC DT2216, which targets BCLXL to the Von Hippel Lindau (VHL) E3 ligase. VHL was specifically chosen because it is not expressed in platelets, thus sparing the BCLXL in platelets and avoiding thrombocytopenia.
Conclusions and Future Directions
BCL2 inhibitors have the potential to make a significant impact in the treatment of multiple myeloma. Like other malignancies, myeloma becomes increasingly dependent on the BCL2 family for survival during tumorigenesis. While most myeloma is expected to be MCL1 dependent, there is already evidence of heterogeneity that is influenced by the genetics and course of the disease. The 15–20% of myeloma this is positive for t(11;14) has shown a response rate of 40–60% with venetoclax and dexamethasone, demonstrating that a subset of myeloma is BCL2 dependent. Many of these responses are both deep and durable, a remarkable result for a single agent that is orally available and thus convenient for patients. Combinations with venetoclax may expand the group of patients that stand to benefit; however, the increased death observed in the BELLINI trial of venetoclax plus bortezomib is cause for concern. Although the safety signal appears to be limited to the non-t(11;14) population, the FDA has slowed the progress of venetoclax trials to allow for more time to understand these observations. Multiple MCL1 inhibitors are currently being investigated in clinical trials and data on tolerability and response rates are eagerly awaited. Based on the broader MCL1 dependence in multiple myeloma, one or more of these drugs could benefit a significant number of patients. Even BCLXL inhibitors may find a place in the treatment of myeloma, but they will certainly serve as useful tools in the laboratory.
Of particular importance for all of these drugs is an understanding of the biology driving the heterogeneity in myeloma BCL2 dependence, which is likely influenced by both intrinsic factors such as tumor-specific genetics and metabolism, and extrinsic factors such as the bone marrow microenvironment. As more patients are treated and relapse on these inhibitors, studying the patterns of acquired resistance will also be essential. Together, this knowledge will aid in the development of biomarkers to predict BCL2 dependence, thus allowing for the selection of patients most likely to respond to particular inhibitors, as well as rationally designed combination therapy to safely improve responses and target resistance mechanisms.
Author Contributions
V.A.G. and J.A. prepared the manuscript. V.A.G., J.A., J.L.K., and L.H.B. assisted with the review and editing of the manuscript. All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
J.L.K. receives honoraria from Tecnofarma; research support from BMS, Janssen, Bluebird, Sutro Biopharma, Amgen, Abbvie, Fortis Therapeutics, GSK, Genentech, and Celgene; reports consultancy for BMS, Janssen, Genentech, and Celgene; and data safety monitoring board for TG therapeutics. L.H.B. performs consultancy for Genentech and Abbvie: and reports research funding, consultancy, and honoraria from AstraZeneca. The authors report no other potential conflicts of interest for this work.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674.
2. Letai AG. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat Rev Cancer. 2008;8(2):121–132. doi:10.1038/nrc2297
3. Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364(11):1046–1060. doi:10.1056/NEJMra1011442
4. Boise LH, Kaufman JL, Bahlis NJ, Lonial S, Lee KP. The tao of myeloma. Blood. 2014;124(12):1873–1879. doi:10.1182/blood-2014-05-578732
5. Joseph NS, Kaufman JL, Dhodapkar MV, et al. Long-term follow-up results of lenalidomide, bortezomib, and dexamethasone induction therapy and risk-adapted maintenance approach in newly diagnosed multiple myeloma. J Clin Oncol. 2020;38(17):1928–1937. doi:10.1200/JCO.19.02515
6. Mateos MV, Dimopoulos MA, Cavo M, et al. Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N Engl J Med. 2018;378(6):518–528. doi:10.1056/NEJMoa1714678
7. Facon T, Kumar S, Plesner T, et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med. 2019;380(22):2104–2115. doi:10.1056/NEJMoa1817249
8. Moreau P, Attal M, Hulin C, et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open-label, Phase 3 study. Lancet. 2019;394(10192):29–38. doi:10.1016/S0140-6736(19)31240-1
9. Voorhees PM, Kaufman JL, Laubach J, et al. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplant-eligible newly diagnosed multiple myeloma: the GRIFFIN trial. Blood. 2020;136(8):936–945. doi:10.1182/blood.2020005288
10. Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 Family reunion. Mol Cell. 2010;37(3):299–310. doi:10.1016/j.molcel.2010.01.025
11. Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011;30(18):3667–3683. doi:10.1038/emboj.2011.307
12. Kalkavan H, Green DR. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018;25(1):46–55. doi:10.1038/cdd.2017.179
13. Carrington EM, Vikstrom IB, Light A, et al. BH3 mimetics antagonizing restricted prosurvival Bcl-2 proteins represent another class of selective immune modulatory drugs. Proc Natl Acad Sci U S A. 2010;107(24):10967–10971. doi:10.1073/pnas.1005256107
14. Gaudette BT, Iwakoshi NN, Boise LH. Bcl-xL protein protects from C/EBP homologous protein (CHOP)-dependent apoptosis during plasma cell differentiation. J Biol Chem. 2014;289(34):23629–23640. doi:10.1074/jbc.M114.569376
15. Peperzak V, Vikstrom I, Walker J, et al. Mcl-1 is essential for the survival of plasma cells. Nat Immunol. 2013;14(3):290–297. doi:10.1038/ni.2527
16. Tu Y, Renner S, Xu F, et al. BCL-X expression in multiple myeloma: possible indicator of chemoresistance. Cancer Res. 1998;58(2):256–262.
17. Puthier D, Pellat-Deceunynck C, Barillé S, et al. Differential expression of Bcl-2 in human plasma cell disorders according to proliferation status and malignancy. Leukemia. 1999;13(2):289–294. doi:10.1038/sj.leu.2401302
18. Wuillème-Toumi S, Robillard N, Gomez P, et al. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia. 2005;19(7):1248–1252. doi:10.1038/sj.leu.2403784
19. Gupta VA, Matulis SM, Conage-Pough JE, et al. Bone marrow microenvironment-derived signals induce Mcl-1 dependence in multiple myeloma. Blood. 2017;129(14):1969–1979. doi:10.1182/blood-2016-10-745059
20. Puthier D, Bataille R, Amiot M. IL-6 up-regulates mcl-1 in human myeloma cells through JAK/STAT rather than ras/MAP kinase pathway. Eur J Immunol. 1999;29(12):3945–3950. doi:10.1002/(SICI)1521-4141(199912)29:12<3945::AID-IMMU3945>3.0.CO;2-O
21. Puthier D, Derenne S, Barille S, et al. Mcl-1 and Bcl-xL are co-regulated by IL-6 in human myeloma cells. Br J Haematol. 1999;107(2):392–395. doi:10.1046/j.1365-2141.1999.01705.x
22. Jourdan M, De Vos J, Mechti N, Klein B. Regulation of Bcl-2-family proteins in myeloma cells by three myeloma survival factors: interleukin-6, interferon-alpha and insulin-like growth factor 1. Cell Death Differ. 2000;7(12):1244–1252. doi:10.1038/sj.cdd.4400758
23. Jourdan M, Veyrune JL, De Vos J, Redal N, Couderc G, Klein B. A major role for Mcl-1 antiapoptotic protein in the IL-6-induced survival of human myeloma cells. Oncogene. 2003;22(19):2950–2959. doi:10.1038/sj.onc.1206423
24. Catlett-Falcone R, Landowski TH, Oshiro MM, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10(1):105–115. doi:10.1016/S1074-7613(00)80011-4
25. Kawano M, Hirano T, Matsuda T, et al. Autocrine generation and requirement of BSF-2/IL-6 for human multiple myelomas. Nature. 1988;332(6159):83–85. doi:10.1038/332083a0
26. Derenne S, Monia B, Dean NM, et al. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood. 2002;100(1):194–199. doi:10.1182/blood.V100.1.194
27. Zhang B, Gojo I, Fenton RG. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood. 2002;99(6):1885–1893. doi:10.1182/blood.V99.6.1885
28. Gong J-N, Khong T, Segal D, et al. Hierarchy for targeting prosurvival BCL2 family proteins in multiple myeloma: pivotal role of MCL1. Blood. 2016;128(14):1834. doi:10.1182/blood-2016-03-704908
29. Morales AA, Kurtoglu M, Matulis SM, et al. Distribution of Bim determines Mcl-1 dependence or codependence with Bcl-xL/Bcl-2 in Mcl-1-expressing myeloma cells. Blood. 2011;118(5):1329–1339. doi:10.1182/blood-2011-01-327197
30. Soderquist S, Eastman A. BCL2 inhibitors as anticancer drugs: a plethora of misleading BH3 mimetics. Mol Cancer Ther. 2016;15(9):2011. doi:10.1158/1535-7163.MCT-16-0031
31. Albershardt TC, Salerni BL, Soderquist RS, et al. Multiple BH3 mimetics antagonize antiapoptotic MCL1 protein by inducing the endoplasmic reticulum stress response and up-regulating BH3-only protein NOXA. J Biol Chem. 2011;286(28):24882–24895. doi:10.1074/jbc.M111.255828
32. Villalobos-Ortiz M, Ryan J, Mashaka TN, Opferman JT, Letai A. BH3 profiling discriminates on-target small molecule BH3 mimetics from putative mimetics. Cell Death Differ. 2020;27(3):999–1007. doi:10.1038/s41418-019-0391-9
33. Vogler M, Weber K, Dinsdale D, et al. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ. 2009;16(7):1030–1039. doi:10.1038/cdd.2009.48
34. van Delft MF, Wei AH, Mason KD, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10(5):389–399. doi:10.1016/j.ccr.2006.08.027
35. Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681. doi:10.1038/nature03579
36. Roberts AW, Seymour JF, Brown JR, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30(5):488–496. doi:10.1200/JCO.2011.34.7898
37. Souers AJ, Leverson JD, Boghaert ER, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19(2):202–208. doi:10.1038/nm.3048
38. Matulis SM, Gupta VA, Nooka AK, et al. Dexamethasone treatment promotes Bcl-2 dependence in multiple myeloma resulting in sensitivity to venetoclax. Leukemia. 2016;30(5):1086–1093. doi:10.1038/leu.2015.350
39. Touzeau C, Dousset C, Le Gouill S, et al. The Bcl-2 specific BH3 mimetic ABT-199: a promising targeted therapy for t(11;14) multiple myeloma. Leukemia. 2014;28(1):210–212. doi:10.1038/leu.2013.216
40. Chauhan D, Velankar M, Brahmandam M, et al. A novel Bcl-2/Bcl-X(L)/Bcl-w inhibitor ABT-737 as therapy in multiple myeloma. Oncogene. 2007;26(16):2374–2380. doi:10.1038/sj.onc.1210028
41. Kline MP, Rajkumar SV, Timm MM, et al. ABT-737, an inhibitor of Bcl-2 family proteins, is a potent inducer of apoptosis in multiple myeloma cells. Leukemia. 2007;21(7):1549–1560. doi:10.1038/sj.leu.2404719
42. Trudel S, Stewart AK, Li Z, et al. The Bcl-2 family protein inhibitor, ABT-737, has substantial antimyeloma activity and shows synergistic effect with dexamethasone and melphalan. Clin Cancer Res. 2007;13(2 Pt 1):621–629. doi:10.1158/1078-0432.CCR-06-1526
43. Dousset C, Maiga S, Gomez-Bougie P, et al. BH3 profiling as a tool to identify acquired resistance to venetoclax in multiple myeloma. Br J Haematol. 2017;179(4):684–688.
44. Bajpai R, Matulis SM, Wei C, et al. Targeting glutamine metabolism in multiple myeloma enhances BIM binding to BCL-2 eliciting synthetic lethality to venetoclax. Oncogene. 2016;35(30):3955–3964. doi:10.1038/onc.2015.464
45. Bajpai R, Sharma A, Achreja A, et al. Electron transport chain activity is a predictor and target for venetoclax sensitivity in multiple myeloma. Nat Commun. 2020;11(1):1228. doi:10.1038/s41467-020-15051-z
46. Bodet L, Gomez-Bougie P, Touzeau C, et al. ABT-737 is highly effective against molecular subgroups of multiple myeloma. Blood. 2011;118(14):3901–3910. doi:10.1182/blood-2010-11-317438
47. Punnoose EA, Leverson JD, Peale F, et al. Expression profile of BCL-2, BCL-XL, and MCL-1 predicts pharmacological response to the BCL-2 selective antagonist venetoclax in multiple myeloma models. Mol Cancer Ther. 2016;15(5):1132–1144. doi:10.1158/1535-7163.MCT-15-0730
48. Touzeau C, Ryan J, Guerriero J, et al. BH3 profiling identifies heterogeneous dependency on Bcl-2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia. 2016;30(3):761–764. doi:10.1038/leu.2015.184
49. Gomez-Bougie P, Maiga S, Tessoulin B, et al. BH3-mimetic toolkit guides the respective use of BCL2 and MCL1 BH3-mimetics in myeloma treatment. Blood. 2018;132(25):2656–2669. doi:10.1182/blood-2018-03-836718
50. Kumar S, Kaufman JL, Gasparetto C, et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood. 2017;130(22):2401–2409. doi:10.1182/blood-2017-06-788786
51. Roberts AW, Davids MS, Pagel JM, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):311–322. doi:10.1056/NEJMoa1513257
52. Kaufman JL, Gasparetto C, Schjesvold FH, et al. Targeting BCL-2 with venetoclax and dexamethasone in patients with relapsed/refractory t(11;14) multiple myeloma. Am J Hematol. 2020;In press.
53. Maples KT, Nooka AK, Gupta V, et al. Natural history of multiple myeloma patients refractory to venetoclax: a single center experience. Am J Hematol. 2020;96(3):E68–E71. doi:10.1002/ajh.26064
54. Mateos M-V, Moreau P, Dimopoulos MA, et al. A Phase III, randomized, multicenter, open-label study of venetoclax or pomalidomide in combination with dexamethasone in patients with t(11;14)-positive relapsed/refractory multiple myeloma. J Clin Oncol. 2020;38(15_suppl):TPS8554–TPS8554. doi:10.1200/JCO.2020.38.15_suppl.TPS8554
55. Qin JZ, Ziffra J, Stennett L, et al. Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res. 2005;65(14):6282–6293. doi:10.1158/0008-5472.CAN-05-0676
56. Moreau P, Chanan-Khan A, Roberts AW, et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood. 2017;130(22):2392–2400. doi:10.1182/blood-2017-06-788323
57. Kumar SK, Harrison SJ, Cavo M, et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2020;21(12):1630–1642. doi:10.1016/S1470-2045(20)30525-8
58. Kaufman JL, Baz RC, Harrison SJ, et al. Updated analysis of a phase I/II study of venetoclax in combination with daratumumab and dexamethasone, ± bortezomib, in patients with relapsed/refractory multiple myeloma. J Clin Oncol. 2020;38(15_suppl):8511. doi:10.1200/JCO.2020.38.15_suppl.8511
59. Costa LJ, Burwick N, Jakubowiak A, et al. Assessment of minimal residual disease by next-generation sequencing and fluorodeoxyglucose-positron emission tomography in patients with relapsed/refractory multiple myeloma treated with venetoclax in combination with carfilzomib and dexamethasone. Blood. 2020;136(Supplement 1):4–5. doi:10.1182/blood-2020-134737
60. Kumar SK, Bensinger WI, Zimmerman TM, et al. Phase 1 study of weekly dosing with the investigational oral proteasome inhibitor ixazomib in relapsed/refractory multiple myeloma. Blood. 2014;124(7):1047–1055. doi:10.1182/blood-2014-01-548941
61. Huang X, Sun Y, Trow P, et al. Patient subgroup identification for clinical drug development. Stat Med. 2017;36(9):1414–1428. doi:10.1002/sim.7236
62. Certo M, Moore VDG, Nishino M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9(5):351–365. doi:10.1016/j.ccr.2006.03.027
63. Matulis SM, Gupta VA, Neri P, et al. Functional profiling of venetoclax sensitivity can predict clinical response in multiple myeloma. Leukemia. 2019;33(5):1291–1296. doi:10.1038/s41375-018-0374-8
64. Neri P, Maity R, Alberge J-B, et al. Mutations and copy number gains of the BCL2 family members mediate resistance to venetoclax in multiple myeloma (MM) patients. Blood. 2019;134(Supplement_1):572. doi:10.1182/blood-2019-127593
65. Birkinshaw RW, Gong JN, Luo CS, et al. Structures of BCL-2 in complex with venetoclax reveal the molecular basis of resistance mutations. Nat Commun. 2019;10(1):2385. doi:10.1038/s41467-019-10363-1
66. Tausch E, Close W, Dolnik A, et al. Venetoclax resistance and acquired BCL2 mutations in chronic lymphocytic leukemia. Haematologica. 2019;104(9):e434–e437. doi:10.3324/haematol.2019.222588
67. Blombery P, Thompson ER, Nguyen T, et al. Multiple BCL2 mutations cooccurring with Gly101Val emerge in chronic lymphocytic leukemia progression on venetoclax. Blood. 2020;135(10):773–777. doi:10.1182/blood.2019004205
68. Caenepeel S, Brown SP, Belmontes B, et al. AMG 176, a selective MCL1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer Discov. 2018;8(12):1582–1597. doi:10.1158/2159-8290.CD-18-0387
69. Kotschy A, Szlavik Z, Murray J, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538(7626):477–482. doi:10.1038/nature19830
70. Tron AE, Belmonte MA, Adam A, et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun. 2018;9(1):5341. doi:10.1038/s41467-018-07551-w
71. Szlavik Z, Csekei M, Paczal A, et al. Discovery of S64315, a potent and selective Mcl-1 inhibitor. J Med Chem. 2020;63(22):13762–13795. doi:10.1021/acs.jmedchem.0c01234
72. Caenepeel S, Karen R, Belmontes B, et al. Abstract 6218: discovery and preclinical evaluation of AMG 397, a potent, selective and orally bioavailable MCL1 inhibitor. Cancer Res. 2020;80(16 Supplement):6218.
73. Brennan MS, Chang C, Tai L, et al. Humanized Mcl-1 mice enable accurate preclinical evaluation of MCL-1 inhibitors destined for clinical use. Blood. 2018;132(15):1573–1583. doi:10.1182/blood-2018-06-859405
74. Thomas RL, Roberts DJ, Kubli DA, et al. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 2013;27(12):1365–1377.
75. Wang X, Bathina M, Lynch J, et al. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 2013;27(12):1351–1364. doi:10.1101/gad.215855.113
76. Slomp A, Moesbergen LM, Gong JN, et al. Multiple myeloma with 1q21 amplification is highly sensitive to MCL-1 targeting. Blood Adv. 2019;3(24):4202–4214. doi:10.1182/bloodadvances.2019000702
77. Seiller C, Maiga S, Touzeau C, et al. Dual targeting of BCL2 and MCL1 rescues myeloma cells resistant to BCL2 and MCL1 inhibitors associated with the formation of BAX/BAK hetero-complexes. Cell Death Dis. 2020;11(5):316. doi:10.1038/s41419-020-2505-1
78. Siu KT, Huang C, Panaroni C, et al. BCL2 blockade overcomes MCL1 resistance in multiple myeloma. Leukemia. 2019;33(8):2098–2102. doi:10.1038/s41375-019-0421-0
79. Algarín EM, Díaz-Tejedor A, Mogollón P, et al. Preclinical evaluation of the simultaneous inhibition of MCL-1 and BCL-2 with the combination of S63845 and venetoclax in multiple myeloma. Haematologica. 2020;105(3):e116–e120. doi:10.3324/haematol.2018.212308
80. Wong KY, Chim CS. Venetoclax, bortezomib and S63845, an MCL1 inhibitor, in multiple myeloma. J Pharm Pharmacol. 2020;72(5):728–737. doi:10.1111/jphp.13240
81. Tao ZF, Hasvold L, Wang L, et al. Discovery of a potent and selective BCL-XL inhibitor with in vivo activity. ACS Med Chem Lett. 2014;5(10):1088–1093. doi:10.1021/ml5001867
82. Wang L, Doherty GA, Judd AS, et al. Discovery of A-1331852, a first-in-class, potent, and orally-bioavailable BCL-X(L) inhibitor. ACS Med Chem Lett. 2020;11(10):1829–1836. doi:10.1021/acsmedchemlett.9b00568
83. Leverson JD, Phillips DC, Mitten MJ, et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci Transl Med. 2015;7(279):279ra240. doi:10.1126/scitranslmed.aaa4642
84. Tse C, Shoemaker AR, Adickes J, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68(9):3421–3428. doi:10.1158/0008-5472.CAN-07-5836
85. Balachander SB, Criscione SW, Byth KF, et al. AZD4320, a dual inhibitor of Bcl-2 and Bcl-x(L), induces tumor regression in hematologic cancer models without dose-limiting thrombocytopenia. Clin Cancer Res. 2020;26(24):6535–6549. doi:10.1158/1078-0432.CCR-20-0863
86. Khan S, Zhang X, Lv D, et al. A selective BCL-X(L) PROTAC degrader achieves safe and potent antitumor activity. Nat Med. 2019;25(12):1938–1947. doi:10.1038/s41591-019-0668-z
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.