")
Back to Journals » Infection and Drug Resistance » Volume 11
Baseline hepatitis C virus resistance-associated substitutions present at frequencies lower than 15% may be clinically significant
Authors Perales C, Chen Q, Soria ME, Gregori J , Garcia-Cehic D, Nieto-Aponte L, Castells L, Imaz A, Llorens-Revull M , Domingo E, Buti M, Esteban JI, Rodriguez-Frias F , Quer J
Received 25 April 2018
Accepted for publication 10 August 2018
Published 8 November 2018 Volume 2018:11 Pages 2207—2210
DOI https://doi.org/10.2147/IDR.S172226
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Eric Nulens
Celia Perales,1,2,* Qian Chen,1,2,* Maria Eugenia Soria,1 Josep Gregori,1–3 Damir Garcia-Cehic,1,2 Leonardo Nieto-Aponte,4 Lluis Castells,1,2 Arkaitz Imaz,5 Meritxell Llorens-Revull,1 Esteban Domingo,2,6 Maria Buti,1,2,7 Juan Ignacio Esteban,1,2,7 Francisco Rodriguez-Frias,2,4,7 Josep Quer1,2,7
1Liver Unit, Liver Disease Laboratory-Viral Hepatitis, Internal Medicine Department, Vall d’Hebron Institut of Research (VHIR)- Vall d’Hebron University Hospital(HUVH), 08035, Barcelona, Spain; 2Centro de Investigación Biomédica en Red (CIBER) de Enfermedades Hepáticas y Digestivas (CIBERehd) del Instituto de Salud Carlos III, 28029, Madrid, Spain; 3Roche Diagnostics SL, Sant Cugat del Vallès, 08174, Barcelona, Spain; 4Liver Pathology Unit, Department of Biochemistry and Microbiology, HUVH, 08035, Barcelona, Spain; 5HIV and STD Unit, Infectious Diseases Department, Bellvitge University Hospital-Bellvitge Biomedical Research Institut (IDIBELL), Hospitalet de Llobregat, 08907, Barcelona, Spain; 6Virology and Microbiology Department, Centro de Biología Molecular “Severo Ochoa” (CSIC-UAM), Campus de Cantoblanco, 28049, Madrid, Spain; 7Medicine Department. Autonomous University of Barcelona, Bellaterra, 08193, Barcelona, Spain
*These authors contributed equally to this work
Background: Controversy is ongoing about whether a minority mutant present at frequencies below 15% may be clinically relevant and should be considered to guide treatment.
Methods: Resistance-associated substitution (RAS) studies were performed in patients before and at failure of antiviral treatments using Next-generation hepatitis C virus (HCV) sequencing (NGS).
Results: We have found two patients with genotype 1a infection having RAS in 3.5%–7.1% of the viral population at baseline that were selected during ledipasvir + sofosbuvir treatment. Co-selection of RAS located in a region not directly affected by the antiviral treatment also occurred. This observation calls into question, the recommendations to guide RAS-based direct-acting antiviral (DAA) treatment only when RAS are present in >15% of the sequences generated.
Conclusion: Our results suggests that RAS study should include all three HCV DAA target proteins and minority mutants should be considered as clinically relevant.
Keywords: HCV, minority mutants, NGS, antiviral resistance
Introduction
Although new-generation direct-acting antivirals (DAAs) are effective for treating hepatitis C virus (HCV) infection, 5%–10% of patients fail the treatment because of resistance-associated substitutions (RAS).1 Controversy is ongoing about whether anti-HCV treatment should be guided by RAS detection (reviewed in the study by Pérez et al2). International guidelines recommend that physicians with access to reliable tests (population or deep sequencing) assessing HCV resistance to NS5A inhibitors should use this information to guide treatment. However, only RAS present in >15% of sequences are considered clinically significant.3,4 In our center, RAS study based on massively parallel, next-generation sequencing (NGS) using the MiSeq platform is included in routine HCV management with DAA-based treatment. We systematically study the three HCV target genes – NS3, NS5A, and NS5B. Here, we report on two HCV G1a-infected patients failing treatment with Ledipasvir[NS5Ai]+ Sofosbuvir[NS5Bi]) associated with mutants present at much less than 15% of the viral quasispecies at baseline.
Patients and methods
Patients
This study includes two patients both infected with G1a subtype: patient 1, a treatment-naïve man – with HCV–HIV coinfection (HIV–RNA <40 copies/mL at initiation of anti-HCV therapy), fibrosis F2 (7.8 kpa) measured by noninvasive liver elastography (Fibroscan), and viral load 2.19×106 IU/mL – was treated with LDV + SOF for 8 weeks. Adherence, estimated by patient’s report and pill count at the hospital pharmacy together with viral decline at weeks 4 and 8, was good. HCV–RNA was undetectable at the end of treatment. During follow-up, a viral load of 5.89×103 IU/mL was detected 19 weeks after stopping LDV + SOF. Patient 2, a treatment-naïve man – with chronic HCV infection, fibrosis F3 (10.5 kpa) measured by Fibroscan, and viral load 1.04×107 IU/mL– was treated with LDV + SOF for 12 weeks. Adherence to treatment was good, and HCV DNA was undetectable at 8 weeks and at treatment completion. During follow-up, a viral load of 1.30×107 IU/mL was detected 12 weeks after stopping LDV + SOF treatment.
The risk factor for HCV acquisition for patient 1 was injecting drug use in the 1980. Since then, the patient has not taken any drugs. For patient 2, after continuous deep interviewing, we were unable to identify any risk factor for HCV infection.
Two serum samples of each patient, at baseline and during treatment failure time, were analyzed. Briefly, HCV–RNA was extracted either manually from 140 µL of serum using the QIAamp Viral RNA Mini Kit (Qiagen NV, Venlo, the Netherlands) or automatically using a total nucleic acid isolation kit in a COBAS/AmpliPrep system (Hoffman-La Roche Ltd., Basel, Switzerland) following the manufacturers’ instructions. Standard measures to avoid contamination were strictly enforced.5 A high-resolution HCV subtyping method was used with samples before and after treatment failure to conclusively identify HCV subtype and to determine whether there was more than one subtype at relapse.6 Besides, a phylogenetic analysis at the NS5B region was performed to discard reinfection. The three DAA–HCV targeted regions (NS3, NS5A, and NS5B) were amplified by RT-PCR-nested and deep sequenced using MiSeq (Illumina, San Diego, CA, USA) platform. The raw data generated by MiSeq in 2×300 mode were analyzed using in-house R scripts7 to obtain the final RAS reports.
RT-PCR-nested HCV amplification
Reverse transcription and PCR (RT-PCR) were performed using the OneStep RT-PCR Transcriptor kit (Roche Applied Science, Basel, Switzerland). Subtype-specific primers were used to amplify the NS3, NS5A, and NS5B regions (primer sequences are not provided, as they are under patent study). A second internal or nested PCR amplifying a shorter fragment was performed to obtain sufficient DNA product for sequencing. This nested PCR used the FastStart High Fidelity PCR system, dNTPack (Roche Applied Science), with a previously described protocol.56
Amplification products were analyzed by electrophoresis on 2% agarose gel. Negative controls (amplification in the absence of RNA) were included in all amplification runs to ensure absence of contamination. Size-specific expected bands were purified from agarose gel (Agarose MP, Roche Indianapolis, IN, USA) using the QIAquick Gel Extraction Kit (Qiagen, Valencia, CA, USA) and quantified using Nanodrop (Thermo Fisher Scientific, Waltham, MA, USA).
Deep sequencing
Nested-PCR products were purified from agarose gel bands (QIAquick Gel Extraction Kit, Qiagen), quantified (QubitTM dsDNA BR Assay Kit, Thermo Fisher Scientific), and tested for quality (Bioanalyzer DNA 1000, Agilent Technologies, Santa Clara, CA, USA). PCR products from a single patient were pooled at a normalized concentration and purified (Agentcourt® AM0050ure® XP, Beckman Coulter, Brea, CA, USA). Another normalization was performed to 1.5 ng/µL, followed by the library preparation protocol using the Kapa Hyper Prep Kit (Roche Applied Science, Pleasanton, CA, USA) and the SeqCap Adapter Kit A/B (24 Index Nimblegen, Roche Pleasanton, CA, USA) for the pool indexing. A second cleanup was performed (Kapa Pure Beads, KapaBiosystems, Roche, Pleasanton, CA, USA) and a quality control assay was done using the Bioanalyzer. Each pool was normalized to 4 nM, and appropriated volumes of each pool were added to the final library, which was quantified by LightCycler480 (Kapa Library Quantification Kit, KapaBiosystems). Last dilution of the library was prepared and mixed with an internal DNA control (PhiX control V3, Illumina) before being sequenced using the MiSeq platform with MiSeq Reagent Kit V3 (Illumina).
Ultra deep sequencing data management
We have developed a bioinformatic haplotype-centric procedure to exclude full reads that do not meet minimum quality requirements. Once acquiring raw data from MiSeq (fastq), the first step included the overlap of paired-end reads using FLASH. The FLASH parameters were established as a minimum overlap between R1 and R2 of 20 bp with a maximum of 10% differences. Reads not fulfilling this requirement were discarded. The yield of this process ranged from 60% to 80% in all our experiments. The quality profile of the overlapped reads was substantially better than the original reads, with a 5% lower quantile consistently above Q30, and just slightly below Q30 for the ~50 bp in the center. A filter step on the FLASH reads was added to improve both sensitivity and specificity, the threshold was established by experiments with spikes and controls, finally excluding all reads with more than 5% bp with Phred score below Q30. The yield of this filter is very sensitive to the general quality of the sequencing run and has been found to be in the range of 70%–90%. The third step was demultiplexing by specific primers to obtain a fasta file by region. Reads were then collapsed into haplotypes with corresponding frequencies. Haplotypes were aligned with the reference sequence, and haplotypes containing more than two indeterminations, three gaps, or 99 differences were also discarded. Accepted indeterminations and gaps were repaired as per the contents of the dominant haplotype. The yield of this process was above 90% in all cases. Then, reads were translated to amino acids, and the intersection between forward and reverse haplotypes with abundances not below 0.2% was performed. The yield of this process has been found in the range 45%–60%. Based on the controls, all amino acid variants by site or haplotype at 1% or above are reported. The global yield is 15%–30% of raw reads.
Results and discussion
Between 21,000 and 114,000 reads/sample were obtained (Table 1). At baseline in patient 1, the S122G substitution in NS3 and E62D in NS5A was present in 7.1% and 3.7% of the viral population, respectively. At failure, the two substitutions were present in 100% of the sequences. According to International guidelines,4 E62D substitution confers resistance to NS5A inhibitors in G1b patients. Although this effect has not been reported for G1a, this substitution may explain the treatment failure.
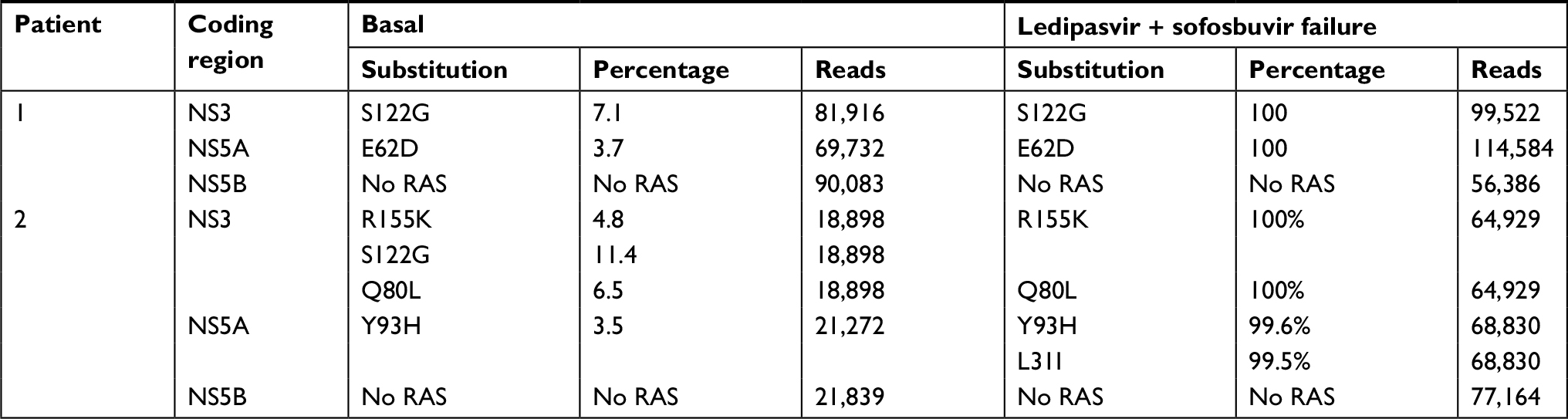 | Table 1 Frequency of RAS detected at baseline and at failure after LDV + SOF treatment. Reads refer to the number of sequences analyzed by next-generation sequencing (MiSeq platform) Abbreviations: LDV, Ledipasvir; RAS, resistance-associated substitutions; SOF, sofosbuvir. |
At baseline in patient 2, the Q80L and R155K substitution in NS3 and Y93H in NS5A were present at 6.5%, 4.8%, and 3.5%, respectively. At failure, NS3 and NS5A were observed in 100% of the sequences obtained from this sample. Moreover, the Y93H NS5A substitution was found in combination with the L31I RAS (not detected at baseline) in 99.5% of genomes. Both Y93H and L31I confer resistance to ledipasvir in HCV G1a patients. NS5B mutations were not observed in either patient (Table 1).
These observations suggest that minority RAS present at much lower than 15%, which may go unnoticed in population sequencing, may be relevant for treatment planning. Clinically relevant mutants have long been known to be present as minority components of replicating viral populations.8 Implication of preexisting minority viral variants and its implication on drug treatment failure needs further studies.9–13 Specifically, our results support reports that baseline RAS using a 1% cutoff resulted in lower SVR rates than the rates in patients showing no RAS.1 Actually, there is no theoretical or experimental rationale other than the limit of detection of population sequencing to use the 15% cutoff.
Of note, NS3 RAS were also observed at failure, even though antiviral treatment was directed against NS5A and NS5B. NS3 RAS could not have been specifically selected by the drug regimen. In this scenario, three alternative or concomitant explanations are possible. First of all, the phenomenon can be caused by a random event in which the NS3 RAS mutation appeared and gave advantage to the viral genome (improve fitness). Second, because of a mutation in NS5A is compensated with a mutation in NS3, both proteins interact with the replication complex. Third, by the phenomenon of hitchhiking, in which the genome carrying the resistance mutation (RAS to NS5A) is selected and dominates the viral population. This selection sweep can produce the hitchhiking of mutations, such as resistance mutations to other antiviral targets such as NS3 that accompany the selected mutation in the same genome. Anyway, this result suggests that contrary to international guidelines recommendation RAS study should always include the three target regions (NS3, NS5A, and NS5B) independently of the treatment and consider RAS mutations at frequencies of around 1%, not being so useful population sequencing.
In contrast to recommendations in international guidelines, our results suggest that RAS mutations at <15% may be clinically relevant and should be taken into consideration in treatment decisions. Detailed information on substitutions in all three DAA target regions (NS3, NS5A, and NS5B) should be provided by NGS, regardless of the failed treatment, in order to design the most suitable salvage therapy.
Acknowledgments
This study has been supported by Ministerio de Sanidad, Servicios Sociales e Igualdad grant name “Plan Estratégico Nacional contra la Hepatitis C”; Instituto de Salud Carlos III cofinanced by the European Regional Development Fund (ERDF) grant number PI16/00337; and Centro para el Desarrollo Tecnológico Industrial-CDTI from the Spanish Ministry of Economics and Competitiveness (MINECO) grant number IDI-20151125. Work at CSIC-UAM supported by grants SAF2014-52400-R, SAF2017-87846-R from MINECO, S2013/ABI-2906 (PLATESA from Comunidad Autónoma de Madrid/FEDER), and institutional grants from Fundación Ramón Areces and Banco Santander. CP is supported by the Miguel Servet program of the Instituto de Salud Carlos III, grant CP14/00121, cofinanced by the ERDF. The authors thank Celine Cavallo for English language support and helpful editing suggestions.
Author contributions
MLR, QC, MES and DGC performed the RAS studies, LNA performed the subtyping, LC, AI, and MB collected patients samples and followed patients, JIE and ED revised the manuscript, JG developed the software tools, CP, FRF, and JQ directed the work, wrote the manuscript, analyzed data, and validated RAS reports. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Sarrazin C, Dvory-Sobol H, Svarovskaia ES, et al. Prevalence of resistance-associated substitutions in HCV NS5A, NS5B, or NS3 and outcomes of treatment with Ledipasvir and Sofosbuvir. Gastroenterology. 2016;151(3):501–512.e1. | ||
Pérez AB, Chueca N, García F. Resistance testing for the treatment of chronic hepatitis C with direct acting antivirals: when and for how long? Germs. 2017;7(1):40–44. | ||
Hepatitis C Guidance 2018 Update: AASLD-IDSA Recommendations for Testing, Managing, and Treating Hepatitis C Virus Infection. AASLD-IDSA HCV Guidance Panel. Clin Infect Dis. 2018 Sep 12. [Epub ahead of print]. | ||
European Association for the Study of the Liver. EASL recommendations on treatment of hepatitis C 2016. J Hepatol. 2017;66(1):153–194. | ||
Kwok S, Higuchi R. Avoiding false positives with PCR. Nature. 1989;339(6221):237–238. | ||
Quer J, Gregori J, Rodríguez-Frias F, et al. High-resolution hepatitis C virus subtyping using NS5B deep sequencing and phylogeny, an alternative to current methods. J Clin Microbiol. 2015;53(1):219–226. | ||
R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2016. | ||
Briones C, Domingo E. Minority report: hidden memory genomes in HIV-1 quasispecies and possible clinical implications. AIDS Rev. 2008;10(2):93–109. | ||
Verbinnen T, Jacobs T, Vijgen L, et al. Replication capacity of minority variants in viral populations can affect the assessment of resistance in HCV chimeric replicon phenotyping assays. J Antimicrob Chemother. 2012;67(10):2327–2337. | ||
Zeuzem S, Mizokami M, Pianko S, et al. NS5A resistance-associated substitutions in patients with genotype 1 hepatitis C virus: prevalence and effect on treatment outcome. J Hepatol. 2017;66(5):910–918. | ||
Jacobson IM, Lawitz E, Kwo PY, et al. Safety and efficacy of elbasvir/grazoprevir in patients with hepatitis C virus infection and compensated cirrhosis: an integrated analysis. Gastroenterology. 2017;152(6):1372–1382.e2. | ||
Kai Y, Hikita H, Morishita N, et al. Baseline quasispecies selection and novel mutations contribute to emerging resistance-associated substitutions in hepatitis C virus after direct-acting antiviral treatment. Sci Rep. 2017;7:41660. | ||
Komatsu TE, Boyd S, Sherwat A, et al. Regulatory analysis of effects of hepatitis C virus NS5A polymorphisms on efficacy of elbasvir and grazoprevir. Gastroenterology. 2017;152(3):586–597. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.