")
Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 11
Bartter syndrome: causes, diagnosis, and treatment
Authors Cunha TDS , Heilberg IP
Received 20 June 2018
Accepted for publication 5 September 2018
Published 9 November 2018 Volume 2018:11 Pages 291—301
DOI https://doi.org/10.2147/IJNRD.S155397
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 7
Editor who approved publication: Professor Pravin Singhal
Tamara da Silva Cunha, Ita Pfeferman Heilberg
Nephrology Division, Universidade Federal de São Paulo (UNIFESP), Escola Paulista de Medicina, São Paulo, Brazil
Abstract: Bartter syndrome is an inherited renal tubular disorder caused by a defective salt reabsorption in the thick ascending limb of loop of Henle, resulting in salt wasting, hypokalemia, and metabolic alkalosis. Mutations of several genes encoding the transporters and channels involved in salt reabsorption in the thick ascending limb cause different types of Bartter syndrome. A poor phenotype–genotype relationship due to the interaction with other cotransporters and different degrees of compensation through alternative pathways is currently reported. However, phenotypic identification still remains the first step to guide the suspicion of Bartter syndrome. Given the rarity of the syndrome, and the lack of genetic characterization in most cases, limited clinical evidence for treatment is available and the therapy is based mainly on the comprehension of renal physiology and relies on the physician’s personal experiences. A better understanding of the mutated channels and transporters could possibly generate targets for specific treatment in the future, also encompassing drugs aiming to correct deficiencies in folding or plasma membrane expression of the mutated proteins.
Keywords: Bartter syndrome, metabolic alkalosis, hypokalemia, Gitelman syndrome, tubulopathy
Introduction
In 1962, Bartter et al identified a new syndrome characterized by hypokalemia and metabolic alkalosis with hyperaldosteronism and hyperplasia of the juxtaglomerular apparatus (JGA).1 These patients were different from the typical patients with hyperaldosteronism because they were younger, had normal blood pressure, and also had growth retardation. Bartter syndrome (BS) is currently recognized as a rare inherited renal tubular disorder that affects around 1 in 1,000,000 of the population, caused by a defective salt reabsorption in the thick ascending limb (TAL) of loop of Henle, resulting in salt wasting, hypokalemia, and metabolic alkalosis with relatively low levels of serum chloride.2 In addition, patients with BS present hyperreninemic hyperaldosteronism with normal/low blood pressure, reduced peripheral resistance, and hyporesponsiveness to antihypertensives.3 This syndrome is associated with an increased antenatal and neonatal mortality because many patients fail to thrive.
How can we suspect BS?
Children and young patients with symptoms of hypokalemia associated with metabolic alkalosis should have BS and Gitelman syndrome (GS) included in their clinical and laboratorial investigation aiming to start therapy promptly. In adults, the presence of BS or GS can be suspected after more common causes such as diuretic abuse and surreptitious vomiting have been excluded.
In recent years, the genetic characterization and detection of different ion channels and their regulations involved in this disease helped physicians to better understand the underlying mechanisms. First, different genotypes were discovered and thought to represent groups sharing the same phenotypes of BS (Table 1). However, important clinical and laboratorial differences among patients in these groups have been progressively disclosed resulting in different presentations of the same entity.4
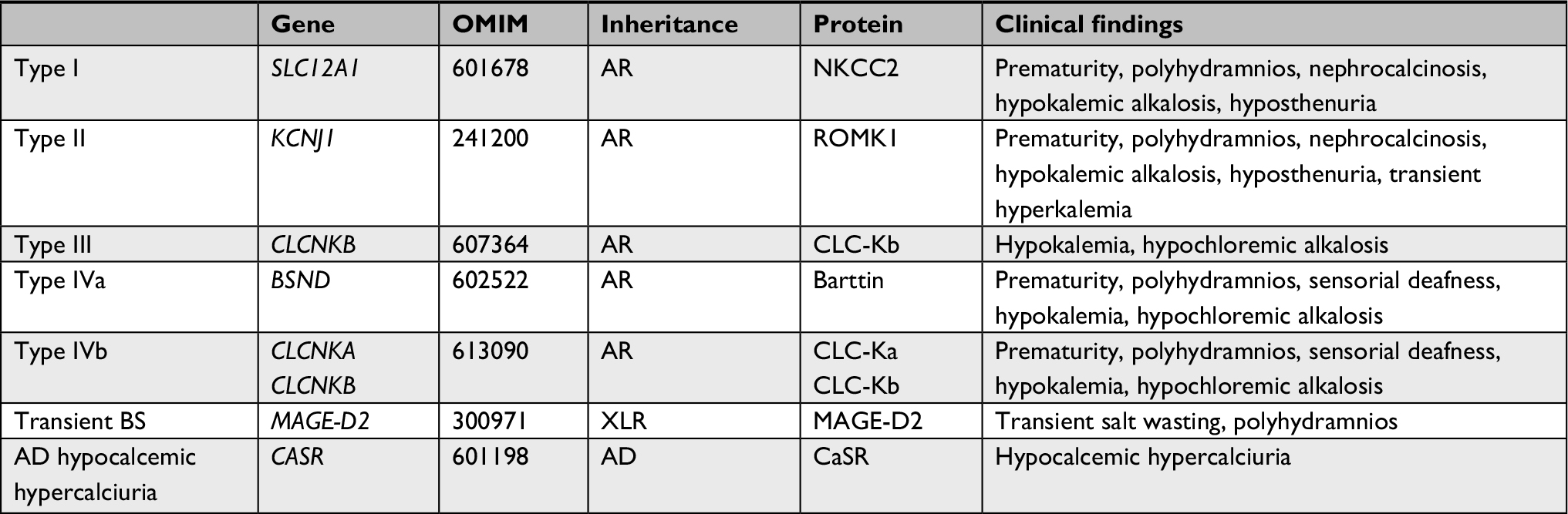 | Table 1 Genetics of Bartter syndrome and typical clinical findings Abbreviations: AD, autosomal dominant; AR, autosomal recessive; XLR, X-linked recessive. |
TAL function
The TAL reabsorbs ~25% filtered load of sodium and plays an important role in extracellular volume and electrolyte balance, as shown in Figure 1.5 The main ion channel at the luminal membrane is NKCC2 (furosemide-sensitive sodium–potassium–chloride cotransporter), which belongs to a family of sodium-coupled electrolyte transporters, encoded by the solute carrier family of SLC12A genes. NKCC2 is responsible for carrying sodium from the tubular lumen into the cell coupled with potassium and chloride. This passive electroneutral transport is driven by the electrochemical gradient established by the ATP-dependent basolateral sodium-potassium adenosine-triphosphatase (Na+K+ATPase) pump.
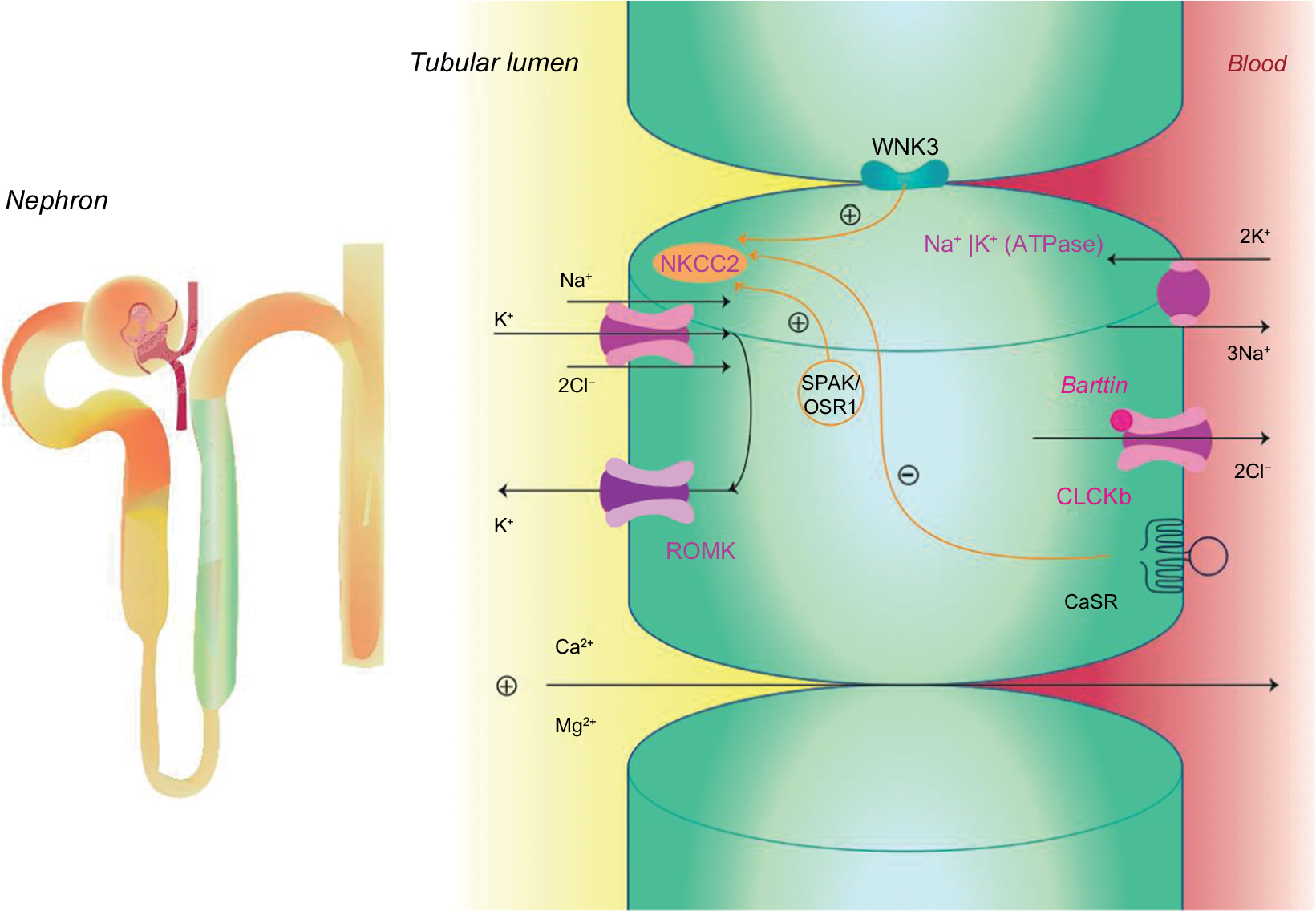 | Figure 1 Sodium reabsorption in the thick ascending limb. Abbreviations: CaSR, calcium-sensing receptor; CLC-Kb, chloride channel; NKCC2, furosemide-sensitive sodium–potassium–chloride cotransporter; OSR1, oxidative stress-responsive kinase 1; ROMK, renal outer medullary potassium channel; SPAK, sterile 20/SPS-1-related proline alanine-rich kinase; WNK3, with-no-lysine [K] family of kinases. |
NKCC2 is a product of the SLC12A1 gene located in chromosome 15q21.1 and is regulated by kinases and the ubiquitination pathway.6 WNK3 kinase (with-no-lysine [K] family of kinases) can modulate the activity of NKCC2 by altering its expression on the plasma membrane. Sterile 20/SPS-1-related proline alanine-rich kinase and oxidative stress-responsive kinase 1 increase the phosphorylation of NKCC2, which increase the cotransporter activity in the TAL. On the contrary, activation of the calcium-sensing receptor (CaSR) inhibits the activity of NKCC2, and thus, the paracellular permeability decreases in response to a reduction of the lumen-positive voltage.7–9
The movement of sodium (Na+), potassium (K+), and chloride (Cl−) ions inside the cell alters the intracellular ion balance, and thus, they must be relocated either to the interstitium or to the tubular lumen. Therefore, Na+ is actively pumped out of TAL cell by Na+K+ATPase and K+ is recycled to the tubular lumen by the renal outer medullary K+ (ROMK) channel, also known as inwardly rectifying Kir1.1, encoded by KCNJ1 gene and which is located at the luminal surface of the epithelial cell.10 Cl− can exit the cell via at least two specific basolateral chloride channels namely ClC-Ka and ClC-Kb (encoded by the CLCNKA and CLCNKB genes). The presence of a subunit barttin, encoded by the BSND gene, is required by both these chloride channels for normal function because it is important to channel integrity, efficient membrane transport, and channel gating. The ClC-Ka is predominantly expressed in the thin limb of the Henle’s loop and the ClC-Kb in the TAL, distal convolute tubule (DCT), and in collecting duct-intercalated cells.11,12 These two proteins with the barttin subunit are also expressed in the inner ear and contribute to the production of endolymph.
Transepithelial salt movement into the medullary interstitium, together with the low permeability of the luminal membrane of the TAL to water, is important to allow the kidneys to develop interstitial hypertonicity and support the urinary concentrating ability. Henle’s loop has another important role in the reabsorption of divalent cations such as calcium (Ca2+) and magnesium (Mg2+), which are reabsorbed by a passive paracellular mechanism secondary to the driving force produced by active salt reabsorption.13
Another essential portion of TAL is the macula densa, a region with an interface with afferent glomerular arterioles, namely JGA, which mediates the tubuloglomerular feedback (TGF). Hypertrophy of the JGA is part of the original description of BS and is considered a hallmark of the disease.
In physiological conditions, the reduction of intracellular chloride concentration at the level of macula densa cells at JGA indicates diminished filtration, resulting in activation of the TGF hence stimulating renin release and afferent arteriolar dilatation with hyperfiltration.14 In patients with BS, reduced reabsorption of chloride occurs due to the genetic defects, and an increase in chloride delivery to the macula densa with an abnormal volume sensing ensues. Therefore, the control of filtration becomes uncoupled from volume status.15
This impaired entry of sodium and chloride into the macula densa increases the expression of cyclooxygenase-2 (COX2) stimulating the renal production of prostaglandin E2 (PGE2), also resulting in afferent arteriolar dilatation and activation of renin release by JGA.16,17
Therefore, the use of prostaglandin synthesis inhibitors (eg, NSAIDs) may be considered for the management of patients with BS. However, careful monitoring is required because significant adverse effects including renal and gastrointestinal toxicity can occur.
Genotype classification of BS
Mutations of several genes encoding the transporters involved in salt reabsorption in the TAL cause different types of BS, which is currently classified into different subtypes according to the gene mutations involved: type I BS caused by mutations in NKCC2 (SLC12A1); type II BS by mutations in ROMK (KCNJ1)18; type III BS by mutations in CLC-Kb (CLCNKB)19; type IVa BS by mutations in barttin (BSND)20; and type IVb BS by mutations in CLC-Ka and CLC-Kb (CLCNKA and CLCNKB).21,22 All these four types are recessive disorders. An additional distinct subtype of BS, considered as type V BS by many investigators, is ascribed to gain-of-function mutations of CASR and is characterized by an autosomal dominant hypocalcemic hypercalciuria.23 More recently, mutations in melanoma-associated antigen-D2 (MAGE-D2) have been implicated in a transient form of antenatal BS, also referred to as type V BS according to some reports.24 This newly recognized form of BS is characterized in most cases by a very early onset of severe polyhydramnios and complete resolution of symptoms after birth.
To avoid misunderstanding, for the purpose of this review we have classified BS into four types (types I–IV) and described the CASR and MAGE-D2 mutations separately.
Previously, another terminology had been proposed to separate BS into “antenatal BS” (types I, II, and IV BS), associated to a more severe presentation, from “classic” BS (type III BS) with a later presentation in childhood. However, the latest findings show a wide spectrum of severity in all forms of BS: some patients with types I, II, or IV BS presenting with late-onset forms, whereas some patients with type III BS may appear with a severe antenatal presentation.
A complete diagram showing the pathophysiology of gene defects in BS is presented in Figure 2.
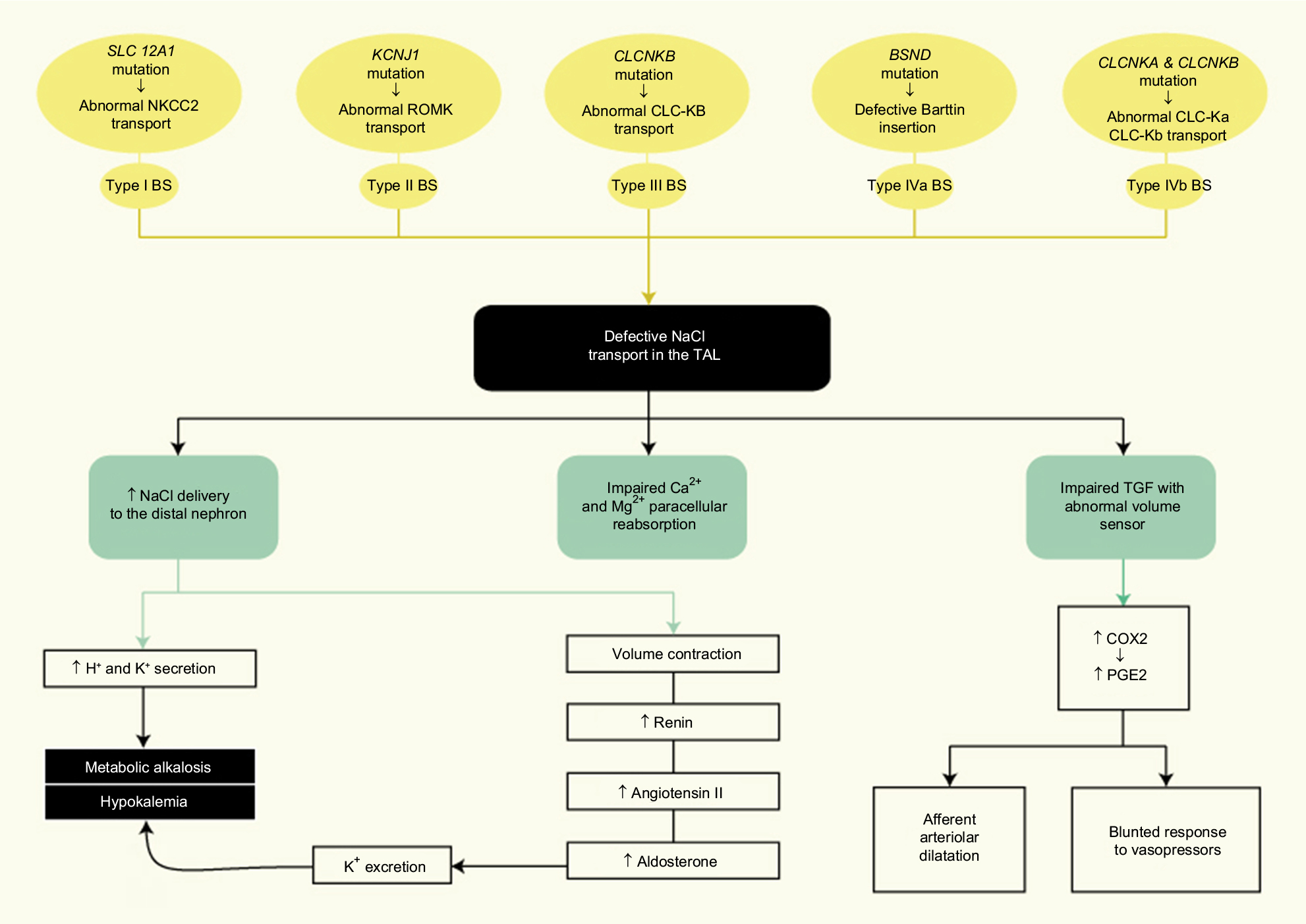 | Figure 2 Gene defects pathophysiology in Bartter syndrome. Abbreviations: COX2, cycloxygenase-2; NaCl, sodium chloride; PGE2, prostaglandin E2; TAL, thick ascending limb; TGF, tubuloglomerular feedback. |
Type I BS
Type I BS is genetically heterogeneous and >63 mutations have been reported, including missense/nonsense mutations, splicing mutations, small insertions, and small deletions.25 In previously reported cases of type I BS, most of patients exhibited homozygous mutations or the presence of a mutation in one allele and a large deletion in the other allele.26
In type I BS, symptoms are usually present at birth with severe salt wasting, hyposthenuria, increased production of PGE2, and failure to thrive. Some of these symptoms already occur in utero and can cause polyhydramnios and premature delivery. A common feature is marked hypercalciuria, which may lead to nephrocalcinosis and/or osteopenia.27 Types I and II BS were considered antenatal variants, also termed “hyperprostaglandin E syndrome,” but recent findings have shown variable phenotypic expression and severity.28
Recently, a novel loss-of-function SLC12A1 gene mutation associated with hypokalemic metabolic alkalosis, hyperparathyroidism, hypercalcemia, nephrogenic diabetes insipidus, and nephrocalcinosis has been described.29 The relation between calcium and parathyroid hormone abnormalities in this different group with SLC12A1 mutations is still an unsolved issue.
Early diagnosis of type I BS is mandatory because the disorder is associated with significant morbidity and mortality if not treated appropriately. Prenatal diagnosis could be useful to start an appropriate management but studies on amniotic fluid to establish the diagnosis had conflicting results.30 Amniotic fluid aldosterone did not show consistent diagnostic information, whereas high chloride levels in amniotic fluid were found in some cases with no other electrolyte abnormalities.31One previous study showed that amniotic fluid chloride concentration ranged from 114 to 123 mEq/L in antenatal BS patients, which is high compared with normal amniotic fluid chloride concentration (around 107–109 mEq/L between gestational age of 25 and 37 weeks).32 The use of an amniotic fluid index measuring the total protein and alpha-fetoprotein in the amniotic fluid has also been proposed and may be helpful.33
Type II BS
Type II BS is also called antenatal variant of BS because it occurs more frequently with severe symptoms in the neonatal period. However, late onset of type II BS (equivalent to type I BS) is also reported due to the broad phenotype of the disease.34,35
More than 40 KCNJ1 mutations, causing loss of function of ROMK channel, have been described and most of these are missense/nonsense mutations in the exon 2, which encode an important domain of ROMK. As a consequence, a production of truncated proteins with loss-of-function occurs, which reduces or eliminates ROMK surface expression due to misfolding or mistrafficking. The paradox of potassium wasting in this setting may be explained by the compensatory overexpression of big potassium (BK) channels permitting potassium excretion in the high-flow setting of BS.36
Hypokalemia is usually milder in this Bartter type, and a transient hyperkalemia in the first days of life may be observed probably due to Na+K+ATPase pump immaturity and also due to the expression of ROMK channel in the cortical collecting duct where it functions in K+ secretion.37,38
More recently, experimental data have shown that some disease-causing mutations in ROMK trap the protein in a folding-deficient conformation, which targets the protein for endoplasmic reticulum (ER)–associated degradation pathway. These interesting findings suggest that future therapeutic strategies should focus on correcting deficiencies in ROMK folding.39
Interestingly, individuals heterozygous for a type II Bartter mutant allele exhibit resistance to hypertension, most likely because of decreased NKCC2-mediated reabsorption of sodium.40
Type III BS
The first identified patients with these mutations showed symptoms during infancy or early childhood with hypokalemia, salt wasting, polyuria, polydipsia, and growth retardation. Hypochloremia is usually more severe in patients with type III BS compared with patients with types I and II BS, probably due to the expression of the CLC-Kb in other parts of the nephron, contributing to chloride depletion. Nephrocalcinosis may be present in type III BS but occurs less frequently than in types I and II BS. In a large Spanish cohort, urinary calcium was variable with hypercalciuria occurring in 31% of patients and hypocalciuria in 23%. In the same cohort, nephrocalcinosis was diagnosed in 20% of type III BS patients.41
Type III BS was thought to be “classic BS” but considerable phenotypic variability has been described in this group of patients, including neonatal BS, antenatal BS, and Gitelman-like phenotype.42 In a large type III BS French cohort, the phenotypic variation consisted of around 30% of antenatal/neonatal BS, 45% of classic BS, and 25% of Gitelman-like phenotype.43 The types of CLCNKB gene mutations in patients with type III BS include missense mutations in most patients, frameshift, nonsense, splicing, and large deletion mutations. This can lead to full impairment, reduced expression at the membrane, and/or reduced chloride channel proper function.44 More than 75 mutations in the CLCNKB gene have been described, and the effect of the kind of mutation in the phenotype is not clearly related.45 However, large deletions were observed to be more frequent in patients with early-onset and severe phenotypes. Some authors defend the hypothesis that phenotypic heterogeneity could be due to various degrees of compensation through alternative basolateral chloride efflux.46
Type IVa and IVb BS
Mutations in the BSND gene cause type IVa BS and result in impairment of barttin insertion in the plasma membrane of CLC-Kb and CLC-Ka channels in the Henle’s loop as well as in the inner ear, which interfere with epithelial salt transport.
On the contrary, type IVb is a digenic disorder with mutations in both CLCNKA and CLCNKB genes, which can lead to impairment in the functioning of two chloride channels and as a result, severe salt wasting and deafness.
Polyhydramnios and preterm delivery are typical characteristics of patients with either BSND or CLCNKA and CLCNKB mutations. Patients with BSND mutations typically have an important defect in urinary concentrating ability. However, the latter is not significantly impaired in cases with CLCNKA and CLCNKB mutations. The presence of chronic hypokalemia could exacerbate the defect in urinary concentration ability because the former may induce a nephrogenic diabetes insipidus (NDI). Nevertheless, some BS cases with NDI but no hypokalemia have been reported, suggesting the presence of an independent pathway as well.47
BS IVa and BS IVb patients typically show severe phenotypes with growth retardation, poor response to NSAIDs, and sometimes, early onset of end-stage renal disease (ESRD). In some patients, a fall in glomerular filtration rate begins at a young age.
Six mutations, which can cause barttin dysfunctions (type IVa BS), have been previously studied (R8L, R8W, G10S, Q32X, G47R, and E88X). The chloride channels were nonfunctional when barttin carried the mutations R8L, R8W, G10S, and Q32X. Furthermore, the distribution of the chloride channels was totally changed when E88X mutation was present leading to severe symptoms in both cases. Conversely, the mutation G47R, which does not prevent chloride channel insertion into the surface membrane, may occur in patients with a late onset of the disease and preserved kidney function.48 Rare unrelated cases due to this G47R mutation, responsible for such forms of late onset of type IV, have been described in Brazil, Portugal, Japan, and Spain.49–52
The sensory function of the inner ear becomes impaired in type IVa and IVb BS. In type IVa, barttin mutations impair potassium secretion in the stria vascularis and the vestibular labyrinth, whereas in type IVb mutations they occur in both chloride channels impairing their normal function in the inner ear.53
A recent experimental study by Nomura et al evidenced that the retention of mutant forms of barttin in the ER is one of the underlying mechanisms responsible for the development of type IVa BS. These investigators reported that the treatment of MDCK cells with 17-allylamino-17-demethoxygeldanamycin (17-AAG), an Hsp90 inhibitor, enhanced the plasma membrane expression of mutant barttin (R8L and G47R), ameliorating hypokalemia, metabolic alkalosis, and hearing loss.54
Transient antenatal BS
Loss of function mutations in MAGE-D2 causes X-linked polyhydramnios with prematurity and a severe but transient form of antenatal BS. MAGE-D2 encodes melanoma-associated antigen D2, which interacts with NKCC2 and increases their cell-surface expression and activity. Mutations in MAGE-D2 reduce this expression of NKCC2 in the TAL and in the NCCT (thiazide-sensitive sodium chloride cotransporter), which is expressed in the DCT.
This phenotype is characterized by the spontaneous resolution of polyuria, a decrease in the concentrations of renin and aldosterone, as well as a decrease in urinary PGE2 levels. The explanation for this transient nature of the phenotype is unclear. One hypothesis is that MAGE-D2 is required in the hypoxic fetal renal medulla to allow proper expression of the cotransporters. After birth, the increasing tissue oxygen levels in the renal medulla and the increasing sensitivity of adenylate cyclase activity to vasopressin result in a reduced dependence on MAGE-D2.55
In a French cohort, MAGE-D2 mutations explained 9% of antenatal BS cases.56 In the same cohort, severe polyhydramnios occurred in all pregnancies and most patients had a weight and/or length above the 90th percentile. Plasma chloride is higher in patients with MAGE-D2 mutations compared with other antenatal BS and, together with birth weight percentile, contribute to differentiate the transient form. Genetic screening for MAGE-D2 mutations should be routinely performed in patients with suspected antenatal BS.
Autosomal dominant hypocalcemia with BS
Gain-of-function mutations of CASR cause hypocalcemia, hypercalciuria, and salt wasting. Hypokalemia, metabolic alkalosis with hyperreninemia, as well as hyperaldosteronism are all common findings.
In the kidney, CaSR is expressed on the basolateral cell surface of the TAL. Several mechanisms have been proposed to explain the inhibitory effect of the active CaSR on paracellular divalent cation transport, which is probably related to the inhibition of NKCC2 phosphorylation and activation. Impairment on divalent calcium reabsorption with consequent renal loss of Ca2+ and Mg2+ is observed.57
Several mutations have been identified in the CASR gene, and the phenotype differs according to its type. In physiological conditions, activation of CaSR by increases in serum Ca2+ and Mg2+ concentrations inhibits divalent calcium reabsorption. The extracellular Ca2+ level at which normal CaSR exhibits half of its maximum activity (EC50) is 3.4 mmol/L (13.6 mg/dL). In CaSR with K47N or P221L mutations, the EC50 is around 2.0–2.2 mmol/L (8.0–8.8 mg/dL) and 0.5 mmol/L (2.0 mg/dL) for CASR with A843E or C131W mutations. A lower level of EC50 can induce a higher activity of CaSR and a faster onset of the phenotype. This can explain why patients with A843E or C131W mutations have the most severe symptoms of BS (Table 1).
BS vs GS
GS is also a rare recessive salt-losing tubulopathy with impaired salt reabsorption in the DCT, which affects 1 in 25,000 of the population and probably is the most common renal salt-wasting disorder. The typical clinical findings include hypokalemia, metabolic alkalosis, hypocalciuria, and hypomagnesemia.58
In the past, BS used to be distinguished to GS by some clinical findings like early age of onset, severity, presence of hypercalciuria, polyhydramnios, or growth retardation. However, advances in medical knowledge showed that patients with BS could have a late presentation, and some findings like hypercalciuria in BS are not present in all cases.
In general, most cases of BS present in neonates and subsequent difficulties can include failure to thrive and growth retardation. Maternal polyhydramnios with premature labor is a common finding of BS. Vomiting, diarrhea, and fever have also been reported. Some infants can have an appearance typical of BS, including a prominent forehead, triangular face with drooping mouth, large eyes, and pinnae. Nevertheless, the diagnostic approach should include genetic evaluation because some phenotypes can be similar.59
A flowchart providing a clinical approach for diagnosis and differential diagnosis of BS or GS in adult patient is presented in Figure 3.
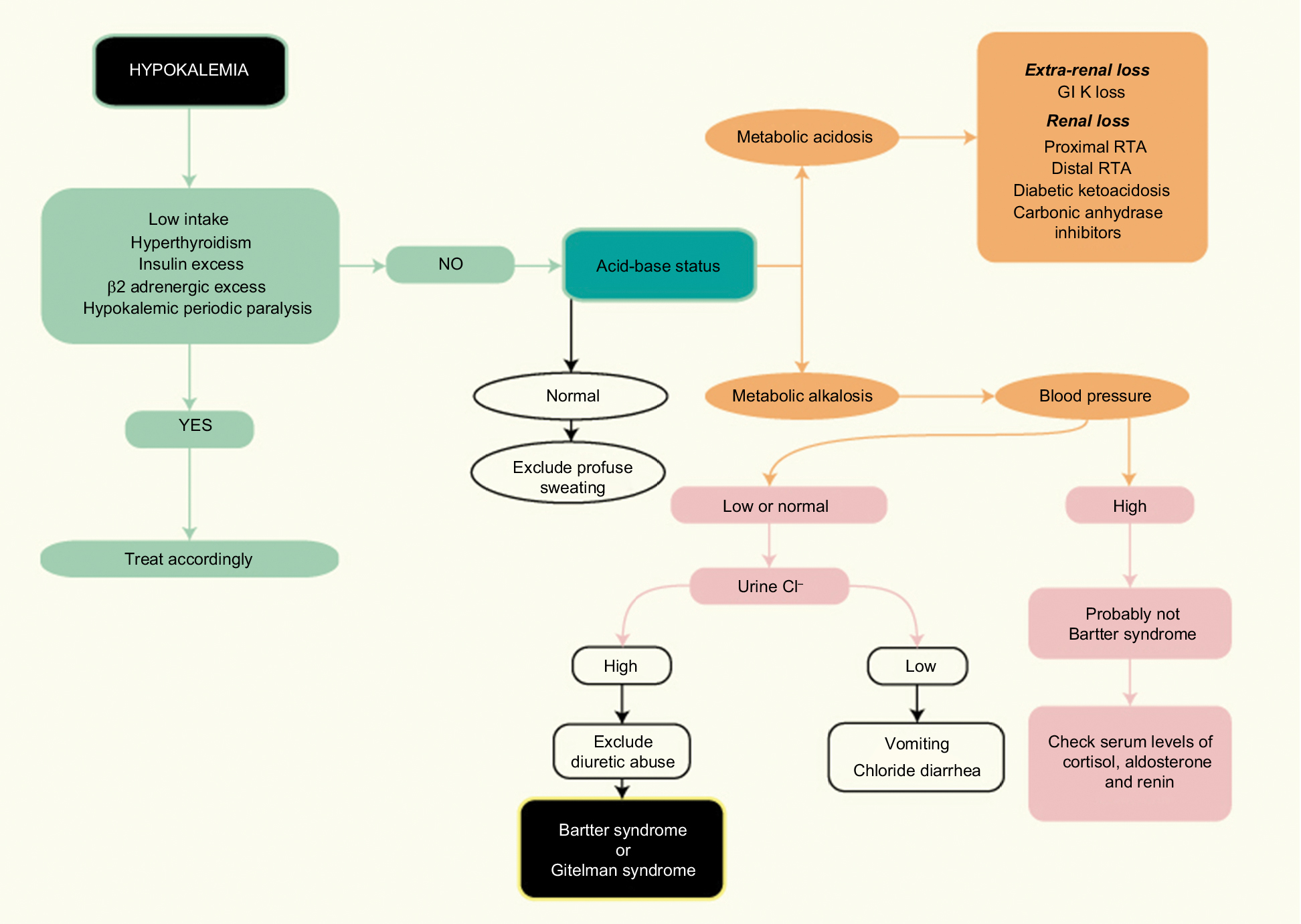 | Figure 3 Flowchart for the diagnosis of Bartter syndrome in adult patients. Abbreviations: GI, gastrointestinal; RTA, renal tubular acidosis. |
A thiazide test has been proposed to help distinguish between BS and GS and involves oral administration of hydrochlorothiazide (1 mg/kg up to 50 mg), after a 7-day “wash-out” period during which all medications except potassium and magnesium supplements are paused. In patients with GS, the fractional excretion of chloride shows very little changes (<2.3%) because the defect is in the NCCT. In patients with BS, this limited response is not seen and instead a more pronounced excretion is observed. However, due to a higher risk of volume depletion, the test is not routinely recommended.60
Bartter-like syndrome
Nephrotoxic agents (eg, aminoglycosides, amphotericin B, and heavy metal intoxication) have also been reported to be associated with a phenotype of BS.61
Extrarenal loss of sodium chloride via gastrointestinal tract (congenital chloride diarrhea) or by the skin (cystic fibrosis) may also be associated with Bartter phenotype, whereas with a low urinary excretion fraction of sodium, as there is normal tubular function.62,63
Diuretic abuse and bulimia also have to be ruled out in all suspected patients of BS.64,65 The urine chloride concentration is low in patients with surreptitious vomiting and bulimia. On the contrary, repeated measurements of urine chloride at different times may help to identify diuretic abuse. The urine chloride concentration is highly changeable in diuretic abusers and persistently elevated in BS patients.
Impact and limitations of treatment
Limited clinical evidence regarding treatment exists due to the rarity of the syndrome. Treatment can vary widely among physicians and clinics and is based mainly on the understanding of renal physiology, reported clinical observations, and individual experiences. The classical pharmacological therapy includes potassium chloride supplementation, prostaglandin inhibitor (indomethacin), and aldosterone antagonist (spironolactone).66
Despite the treatment, hypokalemia usually persists, but the low levels of serum K+ will be less pronounced than at the time of diagnosis.
In addition to potassium supplementation, which is often not well tolerated in higher doses, the use of potassium-sparing diuretics, such as spironolactone, eplerenone, or amiloride, should help to raise serum potassium and reverse metabolic alkalosis. Spironolactone and eplerenone block aldosterone receptor but because hypokalemia may induce lower aldosterone levels, some investigators have hypothesized that amiloride (a direct ENaC channel inhibitor) could possibly work better. Besides, gynecomastia, an undesirable effect, which may limit spironolactone use, is not observed with amiloride.67
Angiotensin-converting enzyme inhibitors can be used either when proteinuria is present or to help correction of low K+ levels.68,69 Because polyuria and dehydration are common symptoms, caution is necessary due to the potential risk of acute kidney injury in this high-risk group of patients. Some authors suggest starting therapy using doses below the minimum one and always providing adequate fluid intake.70 Angiotensin receptor blockers probably have similar performance, but there are lack of data in this group of patients with BS.
Implementation of therapy has led to a great improvement in growth velocity and a stability of metabolic and electrolyte levels by some authors.71
Gastrointestinal side effects can be frequent and even severe, which suggest the necessity of routine endoscopic evaluation of these patients under a long-term use of prostaglandin inhibitors. The use of COX2 selective inhibitor as rofecoxib has suppressed hyperreninemia to an equal level as indomethacin and probably with less gastric side effects.72 However, recent evidence about a high risk of cardiovascular events in patients receiving COX2 selective inhibitors has limited this use.73 Therefore, whether or not COX inhibitors provide best efficacy with less side effects and if it should be removed gradually during school age or maintained lifelong remain unsolved questions.74
Maternal treatment with Nonsteroidal anti-inflammatory drugs (NSAIDs) in antenatal presentations is controversial because this could affect the maturation of the kidneys and the total number of nephrons. In a recent report, prenatal BS with severe polyhydramnios was diagnosed and patients were given NSAIDs until gestational week 31 and avoided later because they may cause premature closure of the ductus arteriosus.75 The risk of premature closure of the patent ductus, which could result in cardiovascular problems, should be considered in this decision.
A summary of treatment options and clinical controversies in BS are presented in Table 2.
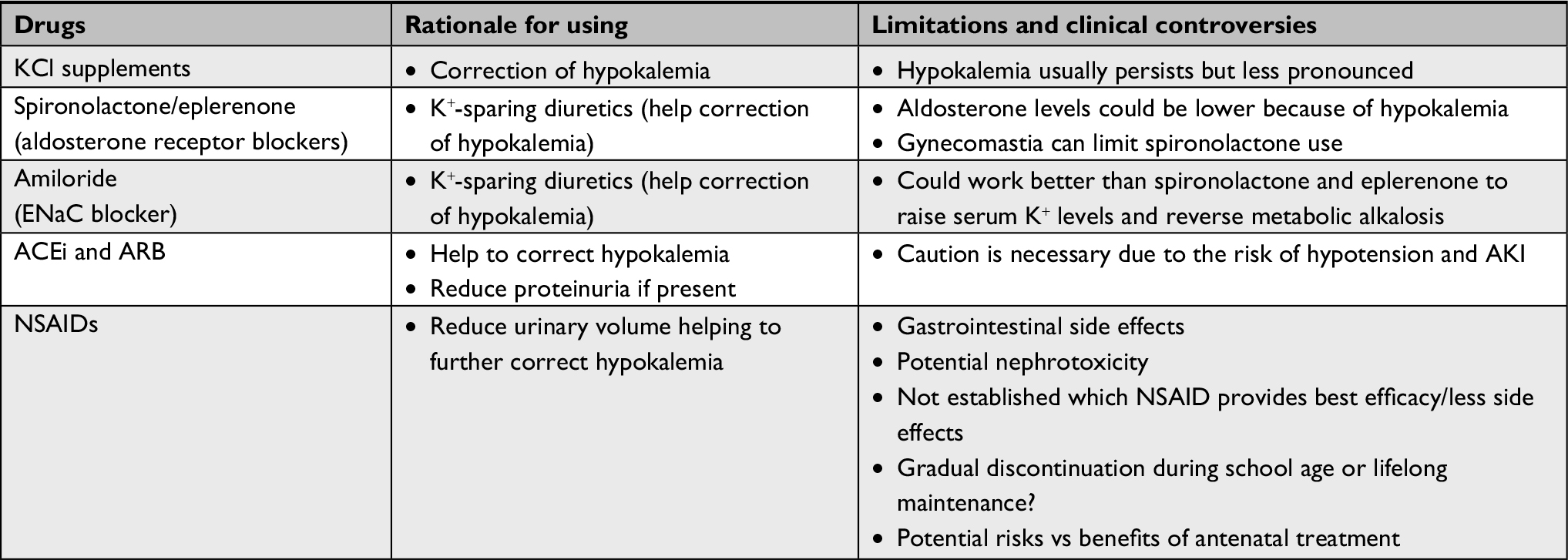 | Table 2 Treatment options and clinical controversies in Bartter syndrome Abbreviations: ACEi, angiotensin-converting enzyme inhibitor; AKI, acute kidney injury; ARB, angiotensin receptor blocker; ENaC, epithelial Na+ channel; KCl, potassium chloride; NSAIDs, nonsteroidal anti-inflammatory drugs. |
There have been reports of patients who developed focal segmental glomerulosclerosis (FSGS) during the course of BS due to chronic stimulation of the renin–angiotensin system with secondary chronic glomerular hyperfiltration.76 FSGS as well as recurrent episodes of dehydration, long-term use of NSAIDs, and presence of nephrocalcinosis were all considered risk factors for renal damage. Chronic hypokalemia itself was believed to lead to a fall in glomerular filtration rate. However, recent data suggest that the degree of hypokalemia does not correlate with a decrease in glomerular filtration rate and actually represents just the result of a secondary hyperaldosteronism.77
Patients with BS who developed FSGS and underwent renal transplantation were previously described.78,79 However, most patients with BS do not develop ESRD.
Osteopenia is an established finding in BS, probably due to the hypercalciuric phenotype. Therefore, bone mineral density must be performed and monitored in such patients.80 For the treatment of short stature and growth failure, administration of growth hormone (GH) is required.81 However, previous studies show that GH does not stimulate growth when severe hypokalemia is present.82
Little information is currently available on the progress of these patients, but most of them tend to present a satisfactory prognosis after a follow-up of at least 10 years although late manifestations such as proteinuria and impaired renal function have been also described.83,84
In summary, BS is a rare salt-losing tubulopathy resulting from several gene defects and distinct types of mutations, with a poor phenotype–genotype relationship due to interaction with other cotransporters and different degrees of compensation through alternative pathways. A handful of information has been learned about BS due to the recognition of the underlying gene mutations, which reinforces the importance of performing genetic tests. A better understanding of the mutated proteins could possibly generate targets for specific treatment. Future therapeutic strategies should focus on drugs aiming to correct deficiencies in folding of mutated proteins or helping to avoid the retention of mutant forms in the endoplasmic reticulum, thus enhancing the plasma membrane expression of the mutant protein, such as in cases of BS II and IV, among others. Despite all recent insights, controversy about treatment and lack of experience with long-term follow-up of these patients still exist given the rarity of the disease.
Disclosure
The authors report no conflicts of interest in this work.
References
Bartter FC, Pronove P, Gill JR, Maccardle RC. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome. Am J Med. 1962;33:811–828. | ||
Lee BH, Cho HY, Lee H, et al. Genetic basis of Bartter syndrome in Korea. Nephrol Dial Transplant. 2012;27(4):1516–1521. | ||
Calò L, Davis PA, Semplicini A. Reduced content of alpha subunit of Gq protein content in monocytes of Bartter and Gitelman syndromes: relationship with vascular hyporeactivity. Kidney Int. 2002;61(1):353–354. | ||
Kurtz I, Cohen JJ, Harrington JT, Madias NE, Zusman CJ. Molecular pathogenesis of Bartter’s and Gitelman’s syndromes. Kidney Int. 1998;54(4):1396–1410. | ||
Seyberth HW. An improved terminology and classification of Bartter-like syndromes. Nat Clin Pract Nephrol. 2008;4(10):560–567. | ||
Ares GR, Caceres PS, Ortiz PA. Molecular regulation of NKCC2 in the thick ascending limb. Am J Physiol Renal Physiol. 2011;301(6): F1143–F1159. | ||
Yang SS, Lo YF, Wu CC, et al. SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J Am Soc Nephrol. 2010;21(11):1868–1877. | ||
Rodan AR, Huang C-L. An emerging role for SPAK in NCC, NKCC, and blood pressure regulation. J Am Soc Nephrol. 1966;8(21):323–351. | ||
Carmosino M, Procino G, Svelto M. Na+-K+-2Cl-cotransporter type 2 trafficking and activity: the role of interacting proteins. Biol Cell. 2012;104(4):201–212. | ||
Kleta R, Bockenhauer D. Salt-Losing Tubulopathies in Children: What’s New, What’s Controversial? J Am Soc Nephrol. 2018;29(3)727–739. | ||
Matsumura Y, Uchida S, Kondo Y, et al. Overt nephrogenic diabetes insipidus in mice lacking the CLC-K1 chloride channel. Nat Genet. 1999;21(1):95–98. | ||
Hennings JC, Andrini O, Picard N, et al. The ClC-K2 chloride channel is critical for salt handling in the distal nephron. J Am Soc Nephrol. 2017;28(1):209–217. | ||
Seyberth HW, Schlingmann KP. Bartter- and Gitelman-like syndromes: salt-losing tubulopathies with loop or DCT defects. Pediatr Nephrol. 2011;26(10):1789–1802. | ||
Deschênes G, Fila M. Primary molecular disorders and secondary biological adaptations in Bartter syndrome. Int J Nephrol. 2011;2011:1–8. | ||
Kömhoff M, Reinalter SC, Gröne HJ, Seyberth HW. Induction of microsomal prostaglandin E2 synthase in the macula densa in children with hypokalemic salt-losing tubulopathies. Pediatr Res. 2004;55(2):261–266. | ||
Friis UG, Stubbe J, Uhrenholt TR, et al. Prostaglandin E2 EP2 and EP4 receptor activation mediates cAMP-dependent hyperpolarization and exocytosis of renin in juxtaglomerular cells. Am J Physiol Renal Physiol. 2005;289(5):989–F997. | ||
Jensen BL, Schmid C, Kurtz A. Prostaglandins stimulate renin secretion and renin mRNA in mouse renal juxtaglomerular cells. Am J Physiol. 1996;271(3 Pt 2):659–F669. | ||
Simon DB, Karet FE, Rodriguez-Soriano J, et al. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat Genet. 1996;14(2):152–156. | ||
Simon DB, Bindra RS, Mansfield TA, et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat Genet. 1997;17(2):171–178. | ||
Birkenhäger R, Otto E, Schürmann MJ, et al. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet. 2001;29(3):310–314. | ||
Schlingmann KP, Konrad M, Jeck N, et al. Salt wasting and deafness resulting from mutations in two chloride channels. N Engl J Med. 2004;350(13):1314–1319. | ||
Fahlke C, Fischer M. Physiology and pathophysiology of ClC-K/barttin channels. Front Physiol. 2010;1:1–12. | ||
Vargas-Poussou R, Huang C, Hulin P, et al. Functional characterization of a calcium-sensing receptor mutation in severe autosomal dominant hypocalcemia with a Bartter-like syndrome. J Am Soc Nephrol. 2002;13(9):2259–2266. | ||
Laghmani K, Beck BB, Yang SS, et al. Polyhydramnios, transient antenatal Bartter’s syndrome, and MAGED2 mutations. N Engl J Med. 2016;374(19):1853–1863. | ||
Simon DB, Karet FE, Hamdan JM, Dipietro A, Sanjad SA, Lifton RP. Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet. 1996;13(2):183–188. | ||
Sun M, Ning J, Xu W, et al. Genetic heterogeneity in patients with Bartter syndrome type 1. Mol Med Rep. 2017;15(2):581–590. | ||
Starremans PG, Kersten FF, Knoers NV, van den Heuvel LP, Bindels RJ. Mutations in the human Na-K-2Cl cotransporter (NKCC2) identified in Bartter syndrome type I consistently result in nonfunctional transporters. J Am Soc Nephrol. 2003;14(6):1419–1426. | ||
Seyberth HW, Königer SJ, Rascher W, Kühl PG, Schweer H. Role of prostaglandins in hyperprostaglandin E syndrome and in selected renal tubular disorders. Pediatr Nephrol. 1987;1(3):491–497. | ||
Wongsaengsak S, Vidmar AP, Addala A, et al. A novel SLC12A1 gene mutation associated with hyperparathyroidism, hypercalcemia, nephrogenic diabetes insipidus, and nephrocalcinosis in four patients. Bone. 2017;97:121–125. | ||
Rachid ML, Dreux S, Pean de Ponfilly G, et al. Prenatal diagnosis of Bartter syndrome: amniotic fluid aldosterone. Prenat Diagn. 2016;36(1):88–91. | ||
Bhat YR, Vinayaka G, Sreelakshmi K. Antenatal Bartter syndrome: a review. Int J Pediatr. 2012;2012(Table 1):857136. | ||
Proesmans W, Massa G, Vandenberghe K, van Assche A. Prenatal diagnosis of Bartter syndrome. Lancet. 1987;1(8529):394. | ||
Garnier A, Dreux S, Vargas-Poussou R, et al. Bartter syndrome prenatal diagnosis based on amniotic fluid biochemical analysis. Pediatr Res. 2010;67(3):300–303. | ||
Huang L, Luiken GP, van Riemsdijk IC, Petrij F, Zandbergen AA, Dees A. Nephrocalcinosis as adult presentation of Bartter syndrome type II. Neth J Med. 2014;72(2):91–93. | ||
Gollasch B, Anistan YM, Canaan-Kühl S, Gollasch M. Late-onset Bartter syndrome type II. Clin Kidney J. 2017;10(5):594–599. | ||
Welling PA, Ho K. A comprehensive guide to the ROMK potassium channel: form and function in health and disease. Am J Physiol Renal Physiol. 2009;297(4):849–863. | ||
Fretzayas A, Gole E, Attilakos A, Daskalaki A, Nicolaidou P, Papadopoulou A. Expanding the spectrum of genetic mutations in antenatal Bartter syndrome type II. Pediatr Int. 2013;55(3):371–373. | ||
Nozu K, Fu XJ, Kaito H, et al. A novel mutation in KCNJ1 in a Bartter syndrome case diagnosed as pseudohypoaldosteronism. Pediatr Nephrol. 2007;22(8):1219–1223. | ||
O’Donnell BM, Mackie TD, Subramanya AR, Brodsky JL. Endoplasmic reticulum-associated degradation of the renal potassium channel, ROMK, leads to type II Bartter syndrome. J Biol Chem. 2017;292(31):12813–12827. | ||
Ji W, Foo JN, O’Roak BJ, et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet. 2008;40(5):592–599. | ||
Castaño AG, de Nanclares GP, Madariaga L, et al. Poor phenotype-genotype association in a large series of patients with Type III Bartter syndrome. PLoS One. 2017;12(3):1–11. | ||
Konrad M, Vollmer M, Lemmink HH, et al. Mutations in the chloride channel gene CLCNKB as a cause of classic Bartter syndrome. J Am Soc Nephrol. 2000;11(8):1449–1459. | ||
Seys E, Andrini O, Keck M, et al. Clinical and genetic spectrum of Bartter syndrome type 3. J Am Soc Nephrol. 2017;28(8):2540–2552. | ||
Andrini O, Keck M, Briones R, Lourdel S, Vargas-Poussou R, Teulon J. ClC-K chloride channels: emerging pathophysiology of Bartter syndrome type 3. Am J Physiol Renal Physiol. 2015;308(12):F1324–F1334. | ||
García Castaño A, Pérez de Nanclares G, Madariaga L, et al. Genetics of type III Bartter syndrome in Spain, proposed diagnostic algorithm. PLoS One. 2013;8(9):1–9. | ||
Cheng CJ, Lo YF, Chen JC, Huang CL, Lin SH. Functional severity of CLCNKB mutations correlates with phenotypes in patients with classic Bartter’s syndrome. J Physiol. 2017;595(16):5573–5586. | ||
Vergine G, Fabbri E, Pedini A, Tedeschi S, Borsa N. Bartter syndrome type 1 presenting as nephrogenic diabetes insipidus. Case Rep Pediatr. 2018;2018:1–3. | ||
Janssen AG, Scholl U, Domeyer C, Nothmann D, Leinenweber A, Fahlke C. Disease-causing dysfunctions of barttin in Bartter syndrome type IV. J Am Soc Nephrol. 2009;20(1):145–153. | ||
Heilberg IP, Tótoli C, Calado JT. Adult presentation of Bartter syndrome type IV with erythrocytosis. Einstein. 2015;13(4):604–606. | ||
Brum S, Rueff J, Santos JR, Calado J. Unusual adult-onset manifestation of an attenuated Bartter’s syndrome type IV renal phenotype caused by a mutation in BSND. Nephrol Dial Transplant. 2007;22(1):288–289. | ||
Miyamura N, Matsumoto K, Taguchi T, et al. Atypical Bartter syndrome with sensorineural deafness with G47R mutation of the beta-subunit for ClC-Ka and ClC-Kb chloride channels, barttin. J Clin Endocrinol Metab. 2003;88(2):781–786. | ||
García-Nieto V, Flores C, Luis-Yanes MI, Gallego E, Villar J, Claverie-Martín F. Mutation G47R in the BSND gene causes Bartter syndrome with deafness in two Spanish families. Pediatr Nephrol. 2006;21(5):643–648. | ||
Estévez R, Boettger T, Stein V, et al. Barttin is a Cl- channel beta-subunit crucial for renal Cl- reabsorption and inner ear K+ secretion. Nature. 2001;414(6863):558–561. | ||
Nomura N, Kamiya K, Ikeda K, Yui N, Chiga M, Sohara E. Biochemical and biophysical research communications treatment with 17-allylamino-17-demethoxygeldanamycin ameliorated symptoms of Bartter syndrome type IV caused by mutated Bsnd in mice. 2013;441:544–549. | ||
Allison SJ. Renal physiology: MAGED2 mutations in transient antenatal Bartter syndrome. Nat Rev Nephrol. 2016;12(7):377. | ||
Legrand A, Treard C, Roncelin I, et al. Prevalence of novel MAGED2 mutations in antenatal Bartter syndrome. Clin J Am Soc Nephrol. 2018;13(2):242–250. | ||
Carmosino M, Gerbino A, Hendy GN, et al. NKCC2 activity is inhibited by the Bartter’s syndrome type 5 gain-of-function CaR-A843E mutant in renal cells. Biol Cell. 2015;107(4):98–110. | ||
Fremont OT, Chan JC. Understanding Bartter syndrome and Gitelman syndrome. World J Pediatr. 2012;8(1):25–30. | ||
Lin SH, Cheng NL, Hsu YJ, Halperin ML. Intrafamilial phenotype variability in patients with Gitelman syndrome having the same mutations in their thiazide-sensitive sodium/chloride cotransporter. Am J Kidney Dis. 2004;43(2):304–312. | ||
Colussi G, Bettinelli A, Tedeschi S, et al. A thiazide test for the diagnosis of renal tubular hypokalemic disorders. Clin J Am Soc Nephrol. 2007;2(3):454–460. | ||
Eldin TK, Tosone G, Capuano A, Orlando R. Reversible hypokalemia and Bartter-like syndrome during prolonged systemic therapy with colistimethate sodium in an adult patient. Drug Saf Case Rep. 2017;4(1):5–7. | ||
Al-Balawi MM, Al-Mobaireek K, Alotaibi W, Al-Shamrani A, Ahmad KS, Al-Saleh S. Clinical presentation of air leak in an infant with undiagnosed cystic fibrosis: a case report. J Med Case Rep. 2015;9(1):10–12. | ||
Kose M, Pekcan S, Ozcelik U, et al. An epidemic of pseudo-Bartter syndrome in cystic fibrosis patients. Eur J Pediatr. 2008;167(1):115–116. | ||
Jamison RL, Ross JC, Kempson RL, Sufit CR, Parker TE. Surreptitious diuretic ingestion and pseudo-Bartter’s syndrome. Am J Med. 1982;73(1):142–147. | ||
Colussi G, Rombolà G, Airaghi C, de Ferrari ME, Minetti L. Pseudo-Bartter’s syndrome from surreptitious diuretic intake: differential diagnosis with true Bartter’s syndrome. Nephrol Dial Transplant. 1992;7(9):896–901. | ||
Verberckmoes R, van Damme B, Clement J, Amery A, Michielsen P. Bartter’s syndrome with hyperplasia of renomedullary cells: successful treatment with indomethacin. Kidney Int. 1976;9(3):302–307. | ||
Griffing GT, Komanicky P, Aurecchia SA, Sindler BH, Melby JC. Amiloride in Bartter’s syndrome. Clin Pharmacol Ther. 1982;31(6):713. | ||
Hené RJ, Koomans HA, Dorhout Mees EJ, vd Stolpe A, Verhoef GE, Boer P. Correction of hypokalemia in Bartter’s syndrome by enalapril. Am J Kidney Dis. 1987;9(3):200–205. | ||
Morales JM, Ruilope LM, Praga M, et al. Long-term enalapril therapy in Bartter’s syndrome. Nephron. 1988;48(4):327. | ||
Nagao R, Suzuki S, Kawashima H, Nozu K, Iijima K. Acute kidney injury in type 3 Bartter syndrome: angiotensin-converting enzyme inhibitors as a cause. Pediatr Int. 2016;58(12):1373–1374. | ||
Vaisbich MH, Fujimura MD, Koch VH. Bartter syndrome: benefits and side effects of long-term treatment. Pediatr Nephrol. 2004;19(8):858–863. | ||
Reinalter SC, Jeck N, Brochhausen C, et al. Role of cyclooxygenase-2 in hyperprostaglandin E syndrome/antenatal Bartter syndrome. Kidney Int. 2002;62(1):253–260. | ||
White WB, West CR, Borer JS, et al. Risk of cardiovascular events in patients receiving celecoxib: a meta-analysis of randomized clinical trials. Am J Cardiol. 2007;99(1):91–98. | ||
Nascimento CLP, Garcia CL, Schvartsman BGS, Vaisbich MH. Treatment of Bartter syndrome, unsolved issue. J Pediatr. 2014;90(5):512–517. | ||
Tourne G, Collet F, Varlet MN, et al. Prenatal Bartter’s syndrome. Report of two cases. J Gynecol Obs Biol Reprod. 2003;32(8):751. | ||
Su IH, Frank R, Gauthier BG, et al. Bartter syndrome and focal segmental glomerulosclerosis: a possible link between two diseases. Pediatr Nephrol. 2000;14(10-11):970–972. | ||
Walsh SB, Unwin E, Vargas-Poussou R, Houillier P, Unwin R. Does hypokalaemia cause nephropathy? An observational study of renal function in patients with Bartter or Gitelman syndrome. QJM. 2011;104(11):939–944. | ||
Lee SE, Han KH, Jung YH, et al. Renal transplantation in a patient with Bartter syndrome and glomerulosclerosis. Korean J Pediatr. 2011;54(1):36–39. | ||
Kim JY, Kim GA, Song JH, et al. A case of living-related kidney transplantation in Bartter’s syndrome. Yonsei Med J. 2000;41(5):662. | ||
Rodríguez-Soriano J, Vallo A, Aguirre M. Bone mineral density and bone turnover in patients with Bartter syndrome. Pediatr Nephrol. 2005;20(8):1120–1125. | ||
Al Shibli A, Narchi H. Bartter and Gitelman syndromes: spectrum of clinical manifestations caused by different mutations. World J Methodol. 2015;5(2):55. | ||
Hochberg Z, Amit T, Flyvbjerg A, Dørup I. Growth hormone (GH) receptor and GH-binding protein deficiency in the growth failure of potassium-depleted rats. J Endocrinol. 1995;147(2):253–258. | ||
Puricelli E, Bettinelli A, Borsa N, et al. Long-term follow-up of patients with Bartter syndrome type I and II. Nephrol Dial Transplant. 2010;25(9):2976–2981. | ||
Bettinelli A, Borsa N, Bellantuono R, et al. Patients with biallelic mutations in the chloride channel gene CLCNKB: long-term management and outcome. Am J Kidney Dis. 2007;49(1):91. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.