")
Back to Journals » Drug Design, Development and Therapy » Volume 15
Bardoxolone-Methyl Prevents Oxidative Stress-Mediated Apoptosis and Extracellular Matrix Degradation in vitro and Alleviates Osteoarthritis in vivo
Authors Pang Z, Jiang Z, Zhu R, Song C, Tang H, Cao L, Guo C
Received 8 April 2021
Accepted for publication 19 August 2021
Published 4 September 2021 Volume 2021:15 Pages 3735—3747
DOI https://doi.org/10.2147/DDDT.S314767
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jianbo Sun
Zhiying Pang,* Zengxin Jiang,* Runwen Zhu, Chunfeng Song, Han Tang, Lu Cao, Changan Guo
Department of Orthopedic Surgery, Zhongshan Hospital, Fudan University, Shanghai, 200032, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Changan Guo; Lu Cao
Department of Orthopedic Surgery, Zhongshan Hospital, Fudan University, NO. 180 Feng Lin Road, Xuhui District, Shanghai, 200032, People’s Republic of China
Tel +86 13681975321
; +86 13482794964
Email [email protected]; [email protected]
Purpose: Oxidative stress-induced chondrocyte apoptosis and extracellular matrix (ECM) degradation plays an important role in the progression of osteoarthritis (OA). Bardoxolone methyl (BM), a semisynthetic triterpenoid, exerts strong effect against oxidative stress. The purpose of the present study was to determine the effectiveness of bardoxolone-methyl (BM) in preventing oxidative stress-induced chondrocyte apoptosis and extracellular ECM degradation in vitro and the role of alleviating OA progression in vivo.
Methods: Oxidative damage was induced by 25 mM tert-butyl hydroperoxide (TBHP) for 24 h in rat chondrocytes. 0.025 and 0.05 μM bardoxolone-methyl (BM) were used in vitro treatment. Ex-vivo cartilage explant model was established to evaluate the effect of BM on oxidative stress-induced ECM degradation. The mouse OA model was induced by surgical destabilization of the medial meniscus.
Results: In vitro, 0.025 and 0.05 μM BM reduced TBHP-induced excessive ROS generation, improved cell viability, increased malondialdehyde level and decreased superoxide dismutase level. 0.025 and 0.05 μM BM prevented TBHP-induced mitochondrial damage and apoptosis in chondrocytes BM activated heme oxygenase-1 (HO-1)/NADPH quinone oxidoreductase 1 (NOQ1) signaling pathway through targeting nuclear factor erythroid derived-2-related factor 2 (Nrf2). Additionally, BM treatment enhanced the expression levels of aggrecan and collagen II and inhibited the expression levels of matrix metalloproteinase 9 (MMP 9), MMP 13, Bax and cleaved-caspase-3. BM increased proteoglycan staining area and IOD value in ex vivo cultured experiment cartilage explants and improved the OARSI score, stands, max contact mean intensity, print area and duty cycle in mouse OA model.
Conclusion: BM prevented oxidative stress-induced chondrocyte apoptosis and ECM degradation in vitro and alleviated OA in vivo, suggesting that BM serves as an effective drug for treatment with OA.
Keywords: osteoarthritis, chondrocyte apoptosis, extracellular matrix degradation, oxidative stress, bardoxolone-methyl
Introduction
Osteoarthritis (OA), a chronic and progressive degenerative joint disease characterized by articular cartilage degeneration, affects more than 240 million people. Pathological changes in OA include inflammation, oxidative stress, chondrocytes apoptosis, cartilage extracellular matrix degradation, subchondral bone sclerosis and osteophyte formation. OA severely affects patients’ quality of life and cause a huge socioeconomic burden.1,2
Chondrocytes are the only cell type in articular cartilage. Chondrocytes secrete a series of cytokines to regulate the growth, distribution and reconstruction of cartilage matrix. Increasing evidence has shown that chondrocyte apoptosis serves an important role in OA.3–5 Multiple pathological factors including inflammatory cytokines, mechanical stress leads to excessive reactive oxygen species (ROS) production. High concentrations of ROS triggers intracellular oxidative stress and thus promote cell apoptosis. Chondrocytes are hypothesized to be an important site of ROS generation. Excessive ROS production induce chondrocytes cell death by apoptosis and causes irreversible damage to the articular cartilage. Chondrocyte apoptosis induced by ROS is one of the primary causes of OA.6–8
Currently, non-steroidal anti-inflammatory drugs (NSAIDs) are mainly used for clinical treatment of OA. However, NSAIDs can only provide short-term relief without preventing or reversing cartilage degradation. Previous study showed that oxidative stress-induced chondrocyte apoptosis contributes to the occurrence and development of OA. Inhibiting oxidative-stress apoptosis in chondrocytes may be a suitable method for preventing cartilage degradation.9
Nuclear factor erythroid derived-2-related factor 2 (Nrf2) functions as the master regulator of intracellular antioxidant response. Kelch-like ECH-associated protein 1 (Keap1) suppressed the activation of Nrf2 signaling pathway in homeostatic conditions. In response to pathological factors such as oxidative stress caused by excessive ROS. Nrf2 transfer to the nucleus and then activate the antioxidant response element (ARE). ARE is known as an important cis element necessary to the expression of many antioxidant genes, including heme oxygenase-1 (HO-1) and NADPH quinone oxidoreductase 1 (NQO1). Nrf2 transfers into the nucleus and then activates the downstream HO-1/NQO1 signaling pathway to exert antioxidant effects.10
Bardoxolone methyl (BM) is a semisynthetic triterpenoid based on natural product oleanolic acid scaffold. BM is a semisynthetic triterpenoid based on natural product oleanolic acid scaffold. BM is proven strong antioxidant effects through activating Keap1/Nrf2/ARE pathway in animal models or clinical study in some diseases such as acute lung injury and diabetic kidney disease.11–16 However, it is still unclear whether BM may inhibit chondrocyte apoptosis through preventing oxidative stress. Therefore, we attempted to investigate the effect of BM on oxidative stress-induced apoptosis of chondrocytes and the underlying molecular mechanisms by the regulation of Keap1/Nrf2/ARE signaling pathway. In this study, TBHP was applied to induce oxidative stress, which could mimic the pathological mechanisms of mitochondrial dysfunction and apoptosis.
Materials and Methods
Cell Isolation and Culture
All animal experiments were approved by the ethics committee on animal Experiments of Fudan University Zhongshan hospital (Shanghai, China). The operations were conducted in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Three 4-week-old male Sprague-Dawley rats were euthanized by intraperitoneal injection of 1% sodium pentobarbital (40 mg/kg; Sigma-Aldrich; Merck KGaA, MO, USA). Death was confirmed by checking breathing and heartbeat. Cartilages were harvested from the bilateral hip joints of rats and then carefully cut into 1 mm3 pieces. The cartilage pieces were digested with 0.25% trypsin (Sigma-Aldrich; Merck KGaA) for 1 h followed by treatment with 0.02% collagenase type II (Sigma-Aldrich; Merck KGaA) at 37°C for 12 h. Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 10% foetal bovine serum (FBS; Thermo Fisher Scientific, Inc., USA) at 37°C and 5% CO2. The chondrocytes were sub-cultured on 6-well plates (5×105 cells/well) or 96-well plate (1×104 cells/well) for in vitro experiments.
Cell Viability Assay
The viability of chondrocytes was assessed using a Cell Counting Kit-8 (CCK-8) assay (Dojindo Co., Kumamoto, Japan). Briefly, cells were sub-cultured in 96-well plates (1×104 cells/well). According to the experimental design, cells were treated with TBHP and different concentrations of BM. After 24 h, the cells were incubated with 10 µL CCK-8 solution added in 100 µL serum-free DMEM at 37°C for 2 h. The absorbance was measured at 450 nm using a microplate reader (Epoch; BioTek Instruments, Inc.).
Intracellular ROS Quantification
Intracellular ROS levels in chondrocytes were evaluated using 20,70-Dichlorodihydrofluorescein diacetate (DCFH-DA; Sigma-Aldrich; Merck KGaA). Cells were collected and washed with phosphate-buffered saline (PBS, Gibco) twice. Cells were stained with 10 mM DCFH-DA solution with serum-free DMEM and incubated at 37°C for 25 min. The cells were then washed three times with serum-free DMEM to completely remove extracellular DCFH-DA. ROS levels were evaluated by BD Accuri C6 plus flow cytometer (BD Biosciences).
Mitochondrial Membrane Potential Determination
JC-1 staining (Abcam, Cambridge, MA) was used to determinate the mitochondrial membrane potential of chondrocytes. Cells were washed twice with PBS and stained with JC-1 working buffer at 37°C for 25 min. The cells were then washed twice with PBS and observed under confocal microscopy (Olympus, FV3000) (magnification, x200). Moreover, the mitochondrial membrane potential of cells was analysed using a BD Accuri C6 Plus flow cytometer.
Cell Apoptosis Evaluation
Annexin V-fluorescein isothiocyanate (FITC)/ propidium iodide (PI) staining (BD Biosciences) was used to evaluate chondrocyte apoptosis. Cells were collected and washed twice with ice-cold PBS. Cells were incubated with u Annexin V-FITC combined with100 μL binding buffer for 25min and then stained with 5 μL PI for 10 min at room temperature in the dark. Chondrocyte apoptosis was analysed using a BD Accuri C6 Plus flow cytometer within 1 h.
Malondialdehyde (MDA) and Superoxide Dismutase (SOD) Quantification
MDA was evaluated using a lipid peroxidation MDA assay kit (Beyotime Institute of Biotechnology, Jiangsu, China) while SOD was determined using a WST-8 assay kit (Beyotime). Briefly, cell lysates were incubated with working buffer for 30 min at 37°C. The absorbance was measured to quantified SOD (450 nm) and MDA (523 nm) using a microplate reader. Total protein concentration was measured to normalize the MDA and SOD levels using bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, Inc.).
Real-Time PCR Assay
Total RNA of the chondrocytes was extracted and isolated using TRIzol reagent. The concentration and quality of the extracted RNA were measured by a Nanodrop spectrophotometer (DeNovix, Wilmington, DE, United States). The cDNA was synthesized from 1µg RNA using Reverse Transcript Master Mix (Thermo Fisher Scientific, Inc.). qPCR amplification was performed using an Applied Biosystems QuantStudio 5 Real-Time PCR System with qPCR Master Mix Kit. The following thermocycling conditions were used: Melting at 95°C for 10 sec, annealing at 95°C for 5 sec, and extension at 60°C for 20 sec for 45 cycles. GAPDH expression was used to normalize the Ct values. The sequences of forward and reverse primer involved in this study are listed in Table 1.
 |
Table 1 Primers’ Sequences Used in the Real-Time PCR (5ʹ - 3ʹ) |
Western Blot Assay
After 24 h of treatment, total proteins of chondrocytes were extracted using radioimmunoprecipitation assay buffer (beyotime) while nuclear proteins were extracted using nuclear protein extraction kit (beyotime). Protein concentrations were measured using a BCA protein assay kit. Total protein (20 µg per sample) was separated by SDS-PAGE on a 12% gel and subsequently transferred to a nitrocellulose membrane (EMD Millipore). The membranes were incubated overnight at 4°C with the following antibodies: Nrf2 (1:1000; Cell Signaling Technology Inc., USA), NQO1 (1:1000; Proteintech, Wuhan, China), Keap1 (1:1000; Proteintech), Matrix metalloproteinase 9 (MMP 9, 1:1000; Proteintech), Matrix metalloproteinase 13 (MMP13, 1:1000; Proteintech), aggrecan (1:1000; CST), collagen II (1:1000; CST), cleaved caspase-3 (1:1000; CST), Bax (1:1000; CST), Bcl-2 (1:1000; CST), GAPDH (1:2000; Proteintech), Tublin (1:2000; Proteintech), LaminB (1:2000; Proteintech). Membranes were washed with TBS-Tween (0.05%) for 30 min, followed by incubation with anti-rabbit secondary antibody (1:5000; CST) for 2h at room temperature. The protein signals were visualized using Enhanced chemiluminescence Plus (Tanon Science and Technology Co., Ltd.).
Ex-vivo Cartilage Explants Model
This study was approved by the he Ethics Committee of Fudan University Zhongshan Hospital. Intact osteochondral tissue harvested from hip joints of SD Rat were used for ex-vivo cartilage explants culture. The detailed procedure was described in a published protocol.17 Initially, the explants were cultured in DMEM containing 10% FBS and 0.25% penicillin-streptomycin at 37°C with 5% CO2 for 2 days. Then, the explants were changed by medium containing THBP and/or THBP with BM for 14 days. Next, explants were collected and fixed in 4% paraformaldehyde, sectioned at 6 μm, and stained with hematoxylin and eosin (H&E) and Safranine O-Fast Green. Further, collagen type II and aggrecan immunochemistry were performed and the percentages of Collagen II, Aggrecan positive cells in each section were quantified by Image Pro Plus. All stained sections were imaged using an upright microscope (Olympus).
Animal Model
This study was approved by the he Ethics Committee of Fudan University Zhongshan Hospital. The study was performed in three groups of eighteen 8-week-old C57BL/6 mice (Shanghai SLAC laboratory animal CO.LTD). The osteoarthritis model was induced by surgical destabilization of the medial meniscus (DMM) as previously described.18 The right knee joint is the side of the operation. 6 mice with simple incision and suture of the knee joint were used as sham group. After surgery, the rats were randomly divided into the following groups: sham group, DMM group, DMM+BM treatment group. BM or vehicle was administered at 10 mg/kg by supplement BM or vehicle into the drinking water. All mice were sacrificed after four weeks.
Behavioral Evaluation (Gait Analysis)
4 weeks after the operation, gaits of rats were analysed in detail using the CatWalk system (Noldus Information Technology, Wageningen, The Netherlands). The light beam from the fluorescent lamp is reflected inside the glass plate. When a mouse’s paw touches the glass plate, the light beam is refracted in the opposite direction, resulting in a bright image of the paw print. The whole process was recorded by the video camera. Data was collected and analyzed using catwalk software.
Histopathological Analysis
After the treatment of BM for 4 weeks, the harvested knee joint of mice in different groups were fixed in 4% paraformaldehyde for 24 h and then decalcified in 10% EDTA for 4 weeks. The tissues were then dehydrated, embedded in paraffin and cut into 5 µm slices. Slices were stained using hematoxylin-eosin (H&E) and Safranin O/Fast Green. The degradation change was evaluated by a Osteoarthritis Research Society International (OARSI) scoring system.
Statistical Analysis
Each experiment was independently carried out at least three repeats. Data are represented as the mean ± standard deviation. Statistical analysis was performed using SPSS software (version 20.0; IBM Corp.). Statistical comparisons among multiple groups were performed using one-way ANOVA followed by Bonferroni’s multiple comparison test. Data of OARSI score was analysed using Mann–Whitney test. P-value < 0.05 was considered statistically significant.
Results
Effects of BM on the Cell Viability of Chondrocytes
The structural formula of BM is shown in Figure 1H. Firstly, the cytotoxic effects of different concentrations of BM (0.01, 0.025, 0.05, 0.075, 0.1, and 0.2 µM) on chondrocytes were evaluated by CCK8 assay. The results showed that a concentration of 0.2 µM showed cell toxicity while BM concentrations lower than 0.2µM presents no significant cytotoxicity at 24 h (Figure 1A). According to this result, the BM concentrations of 0.025 and 0.05 µM were used in vitro experiments. 25 µM TBHP was used to induce oxidative stress in chondrocytes. The results indicated that TBHP treatment decreased viability of chondrocytes. 0.025 and 0.05 µM BM significantly prevented the decrease of cell viability caused by TBHP (Figure 1B).
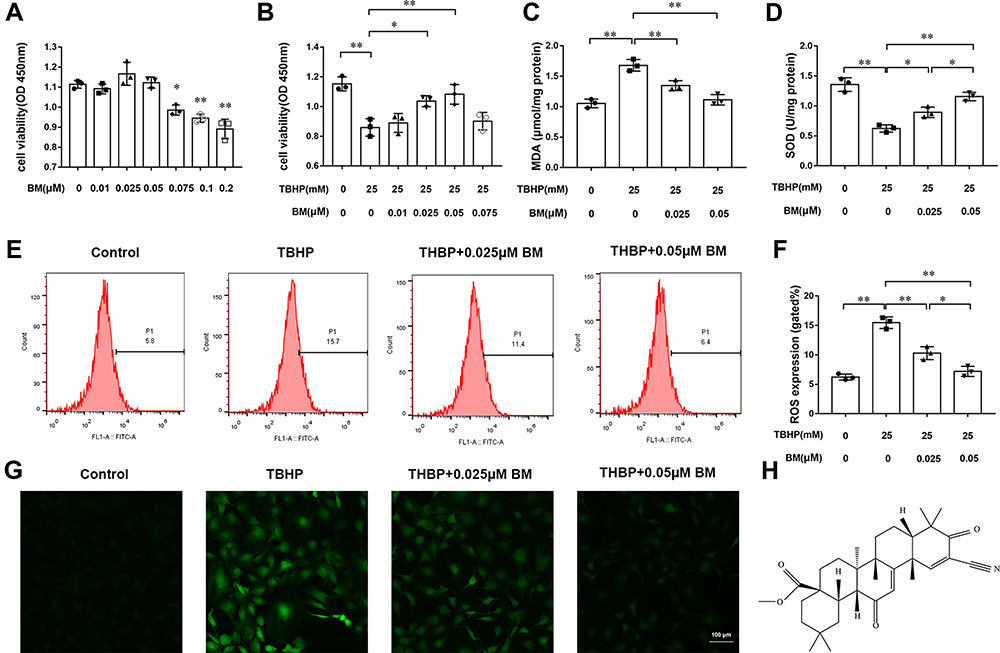 |
Figure 1 BM prevented TBHP-induced oxidative stress in rat chondrocytes. (A) Effects of different concentrations (0, 0.025, 0.05, 0.075, 0.1, and 0.2 µM) of BM on rat chondrocyte viability. (B) 0.01, 0.025, 0.05, and 0.075 µM BM increased viability of rat chondrocytes treated with 25 mM THBP. (C and D) 0.025 and 0.05 µM effectively reduced MDA level and increased SOD level in rat chondrocytes treated with 25 mM THBP. (E and F) Chondrocytes were stained with DCFH-DA and measured by flow cytometry to determine the ROS level. The results showed that 0.025 and 0.05 µM BM effectively decreased ROS level in rat chondrocytes treated with 25 mM THBP. (G) Represent images of chondrocytes stained with DCFH-DA under confocal microscopy. Scale bar, 100 µm. (H) The structural formula of BM. *P < 0.05 and **P < 0.01. |
BM Increases SOD Levels and Decreases MDA Levels
SOD and MDA are commonly used indicators to evaluate oxidative damage. THBP treatment increased MDA levels and decreased SOD levels in chondrocytes. Compared with TBHP treatment alone, two concentrations of BM could both significantly reduced MDA levels and increased SOD levels, suggesting that BM protected chondrocytes from oxidative stress (Figure 1C and D).
BM Inhibits ROS Generation
DCFH-DA staining was used to quantify the ROS levels in chondrocytes. Flow cytometry analysis showed that the 25 µM TBHP treatment leaded to excessive ROS generation in chondrocytes. The BM concentrations of 0.025 and 0.05 µM effectively decreased ROS levels (P<0.01). 0.05 µM concentration of BM is more effective than 0.05 µM to inhibit ROS production (P<0.05). The results demonstrated that BM effectively inhibited excessive ROS generation as a result of the THBP treatment (Figure 1E–G).
BM Prevents TBHP-Induced Mitochondrial Damage
Depolarized mitochondrial membrane potential is a primary event of cell apoptosis. JC-1 staining was used to evaluate the mitochondrial membrane potential of chondrocytes. The TBHP treatment group showed increased monomers (green fluorescence) while 0.025 and 0.05 µM BM treatment decreased monomers and increased aggregates (red fluorescence) when observed under confocal microscopy (Figure 2E). Flow cytometry analysis also showed that TBHP treatment leaded to the increasing mitochondrial membrane potential depolarization cell ratio. While 0.025 and 0.05 µM BM treatment significantly alleviated mitochondrial membrane potential depolarization cell ratio compared to TBHP treatment group (Figure 2A and B). The results suggested that BM protected chondrocytes against TBHP induced mitochondrial damage.
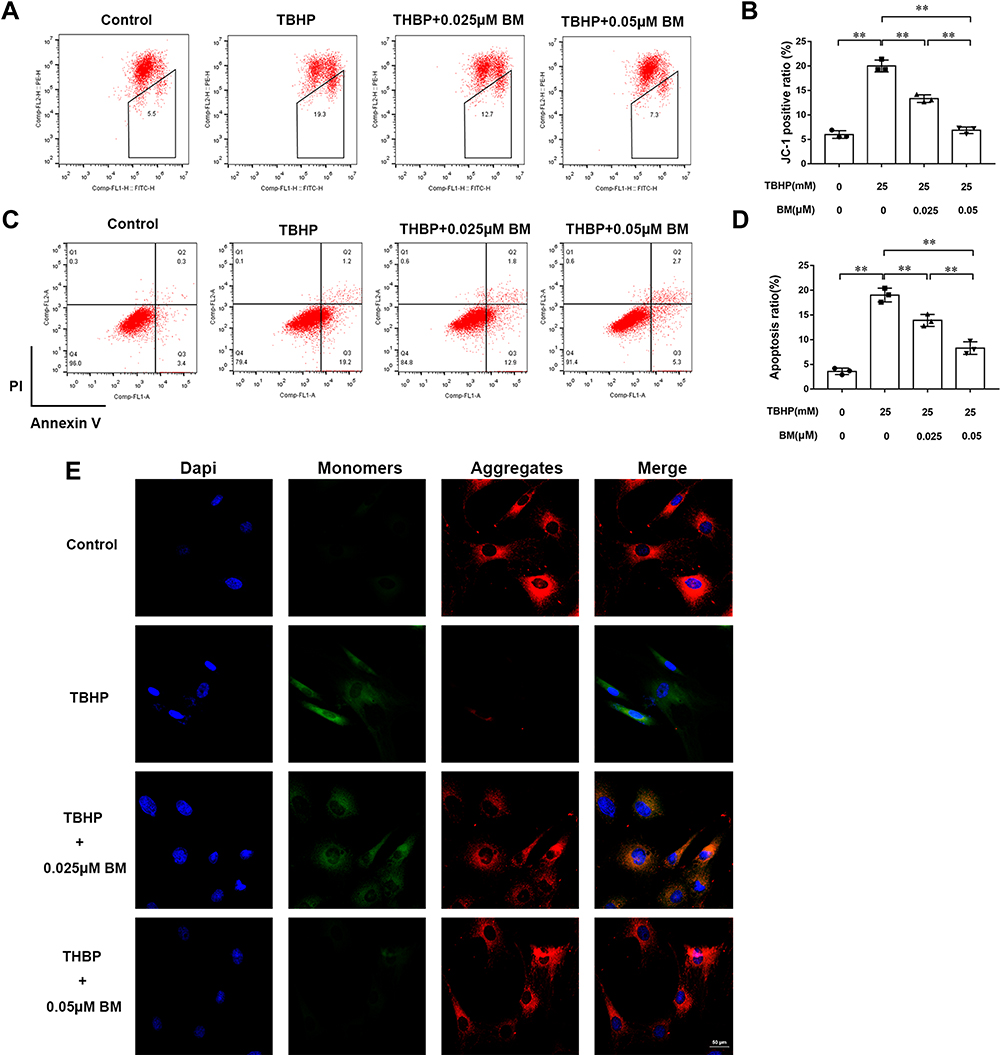 |
Figure 2 BM prevented apoptosis in rat chondrocytes treated with 25 mM TBHP. (A and B) Chondrocytes were stained with JC-1 and measured by flow cytometry to determine mitochondrial membrane potential. The results showed that 0.025 and 0.05 µM BM effectively protected mitochondrial damage in rat chondrocytes treated with 25 mM THBP. (E) Represent images of chondrocytes stained with JC-1 under confocal microscopy. Scale bar, 50 µm. Red fluorescence represented normal membrane potential, and green fluorescence represented mitochondrial membrane potential depolarization. (C and D) Chondrocytes were stained with Annexin V/PI and measured by flow cytometry to detect cell apoptosis. The results showed that 0.025 and 0.05 µM BM significantly inhibited apoptosis of rat chondrocytes treated with 25 mM THBP. *P < 0.05 and **P < 0.01. |
BM Inhibits Oxidative Stress-Induced Apoptosis
Annexin V-FITC/PI staining was used to detect chondrocyte apoptosis (Figure 2C and D). Flow cytometry analysis showed that the ratio of chondrocyte apoptosis in the TBHP treatment group was significantly increased compared with the control group. 0.025 and 0.05 µM BM treatment significantly reduced apoptosis compared with the TBHP treatment group (P<0.01), indicating that BM protected against apoptosis induced by oxidative stress in chondrocytes.
BM Ameliorates Oxidative Stress-Induced ECM Degradation
Real-time PCR and Western blotting were used to measure the expression of MMP 9, MMP 13, aggrecan and collagen II at the mRNA and protein levels. The results showed that TBHP treatment promoted MMP13 expression and reduced aggrecan and collagen II expression at the mRNA (Figure 3A) and protein (Figure 3B) levels. 0.025 and 0.05 µM BM treatment inhibited MMP13 mRNA expression but enhanced aggrecan and collage II expression both at the mRNA and protein levels compared with the TBHP treatment group. Western blot analysis was used to measure the expression of Bax, Bcl-2 and cleaved caspase-3. The results showed that TBHP treatment promoted apoptosis, while 0.025 and 0.05 µM BM treatment inhibited Bax and cleaved caspase-3 at protein levels compared with the TBHP treatment group. The results demonstrated that BM treatment ameliorates oxidative stress-induced ECM degradation in vitro.
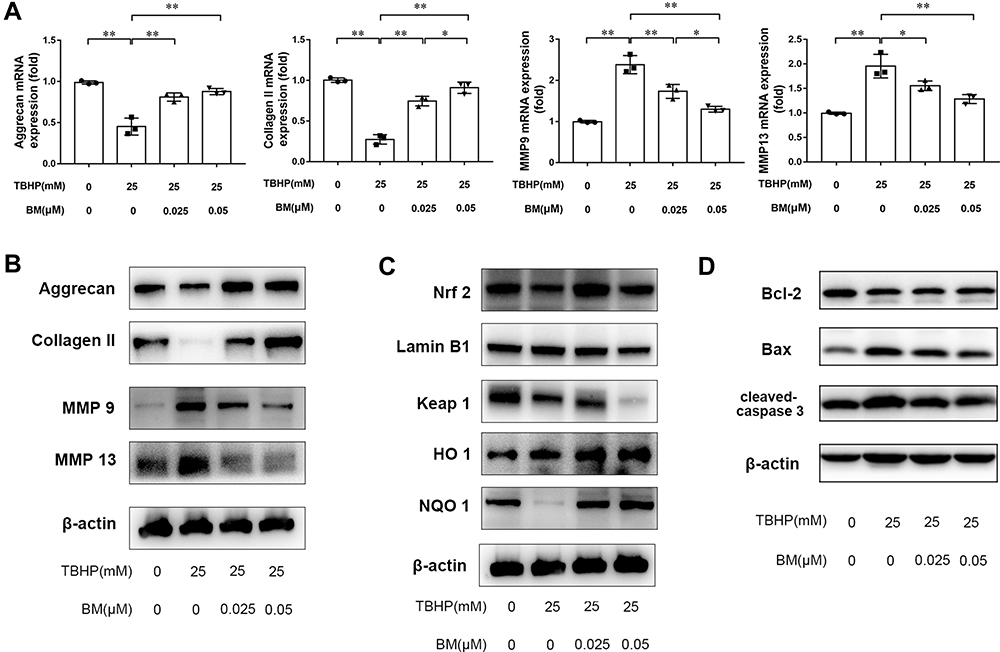 |
Figure 3 BM alleviated TBHP-induced ECM degradation in rat chondrocytes. (A and B) PCR and Western blot results showed 0.025 and 0.05 µM BM increased mRNA and protein levels of aggrecan and Collagen II and reduced mRNA and protein levels of MMP 9 and MMP 13. (C) 0.025 and 0.05 µM BM increased Nrf2 level in the nucleus, inhibited keap1 expression and enhanced HO-1 and NQO1 expression, suggesting that BM activated Keap1/Nrf2/ARE signaling pathway. (D) Western blot results showed 0.025 and 0.05 µM BM inhibited protein levels of Bax and Cleaved-Caspase-3. *P < 0.05 and **P < 0.01. |
BM Activates Keap1/Nrf2/ARE Signaling Pathway in Chondrocytes
Western blot analysis was used to measure the expression of Nrf2, Keap1, NQO1, HO-1. The results showed that TBHP treatment increased Nrf2 level in nucleus. In addition, TBHP treatment increased Keap1 expression level and decreased NQO1 and HO-1 expression level. 0.025 and 0.05 µM BM treatment inhibited Keap1 expression but enhanced Nrf2, NQO1 and HO-1 expression compared with TBHP treatment group (Figure 3C) … The results suggested that BM activated Keap1/Nrf2/ARE signaling pathway and thus exerted a potential antioxidant effect.
BM Ameliorates ECM Degradation in Cartilage Explant Culture
We used an ex vivo culture model of cartilage explants from nine 4-week-old rats to evaluate effect of BM on cartilage degradation. The cartilage explants were cultured in medium supplemented with THBP and/or BM for 3 days. The explants were divided into four groups: (1) Control, (2) THBP stimulated (25 μm), (3) THBP (25 μm) plus BM (0.05 μm) and (4) THBP (25 μm) plus BM (0.05 μm). Histopathological changes in cartilage were evaluated by H&E, Safranine O-Fast Green and immunochemistry of collagen II, aggrecan. As shown in Figure 4, H&E and S-O Fast Green staining revealed normal structure of cartilage including smooth and intact surfaces and normal morphology and numbers of well-organized chondrocytes in the control group. However, the THBP treated group had apparent morphological changes including rough surfaces, clustered and disorganized chondrocytes, obvious hypocellularity, loss of Safranin-O staining and fewer cells positive for collagen II and aggrecan immunochemistry compared with the control group. Notably, 0.025 and 0.05 µM BM treatment recovered the percentage of Safranin-O staining area and immunochemistry cell number positive for of collagen II and aggrecan immunochemistry (Figure 5), suggesting that cartilage damage was significantly attenuated by treatment with BM.
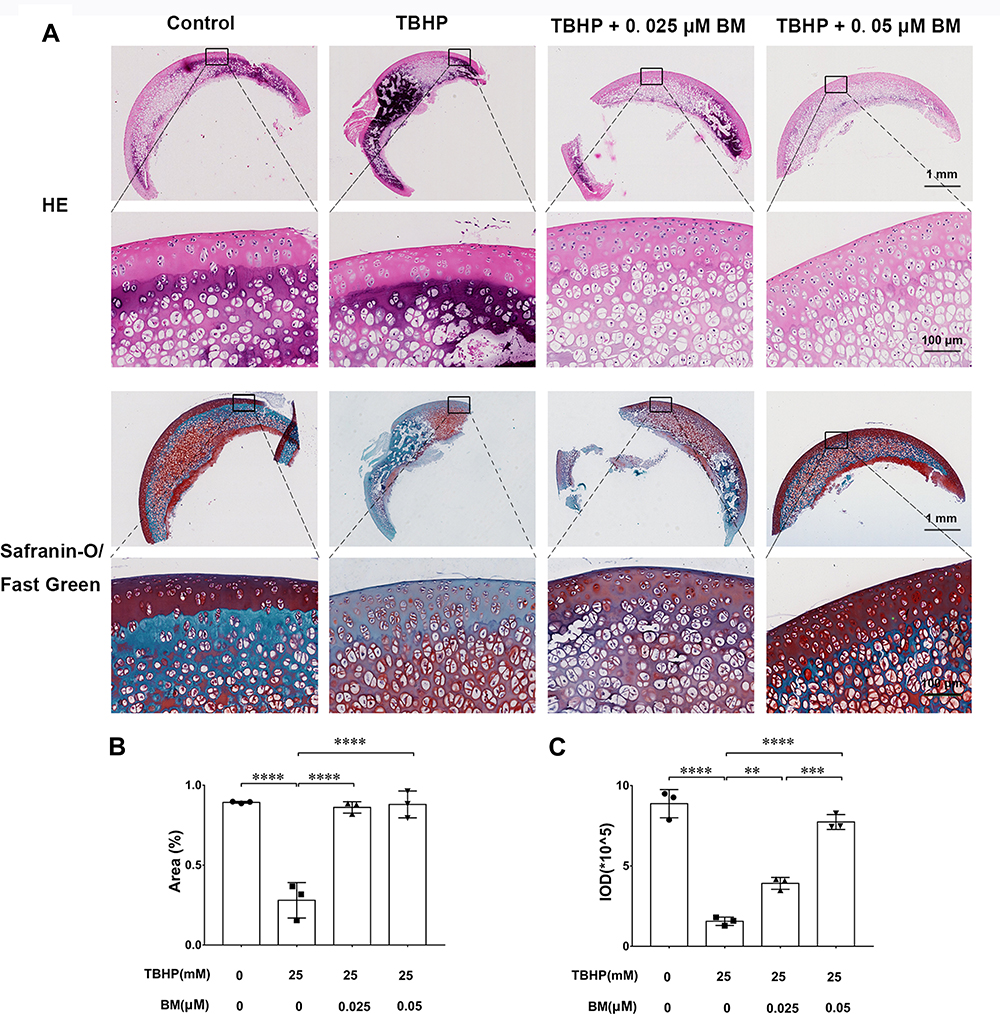 |
Figure 4 Histopathological analysis of the cartilage explants cultured ex vivo. (A) Representative images of HE and Safranine O-Fast Green staining at magnification of 10× (scale bar, 1mm) and 100× (scale bar, 50 µm). (B) The integrated optical density (IOD) value for proteoglycan content determined by safranin O-fast green staining (n = 3 for each group; one-way ANOVA). (C) Proteoglycan staining area (%) determined by safranin O-fast green staining (n = 3 for each group; one-way ANOVA;). **P < 0.01,***P < 0.005 and ****P < 0.001. |
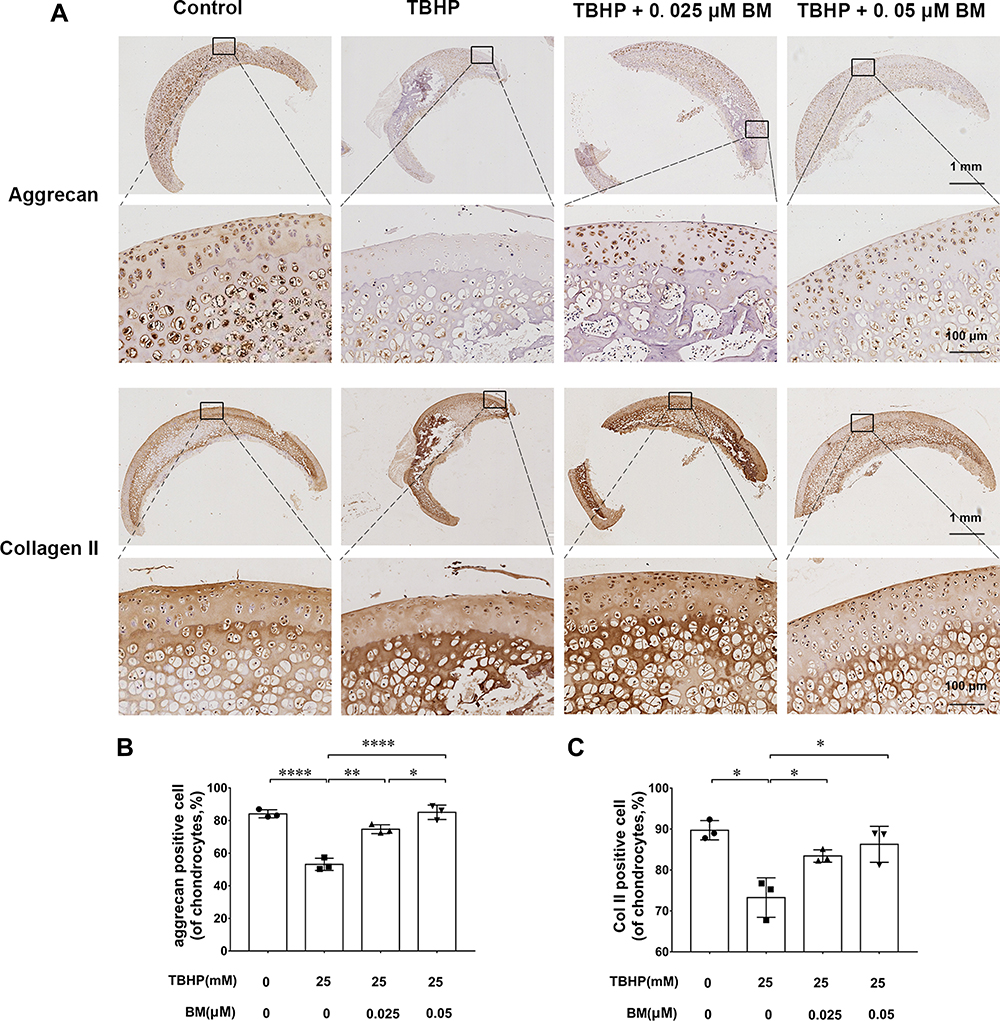 |
Figure 5 Immunohistochemical analysis of the cartilage explants cultured ex vivo. (A) Representative images of collagen II and aggrecan immunochemistry at magnification of 10× (scale bar, 1mm) and 100× (scale bar, 50 µm). (B) 0.025 and 0.05 µM BM significantly increased collagen II and aggrecan positive cell ratio, suggesting BM alleviated TBHP-induced ECM degradation in cartilage explants cultured ex vivo. *P < 0.05, **P < 0.01 and ****P < 0.001. |
BM Ameliorates OA Development in vivo
To explore the protective effects of BM on OA in vivo, DMM mouse OA models were established. Decreased chondrocytes, severe cartilage destruction and massive loss of proteoglycans were observed in DMM group using H&E and Safranine O-Fast Green staining. However, BM treatment ameliorated these changes and prevented the progression of OA in DMM mice (Figure 6A, Supplement Figure 1). The OARSI score in BM group is significantly higher than that in DMM group (Figure 6B). IHC The results of gait analysis showed that BM improved the stands, max contact mean intensity, print area and duty cycle in DMM mice (Figure 6C) Supplement Figure 1. The results of histological and immunohistochemical analysis and gait analysis demonstrated that BM ameliorates OA development in mouse DMM model.
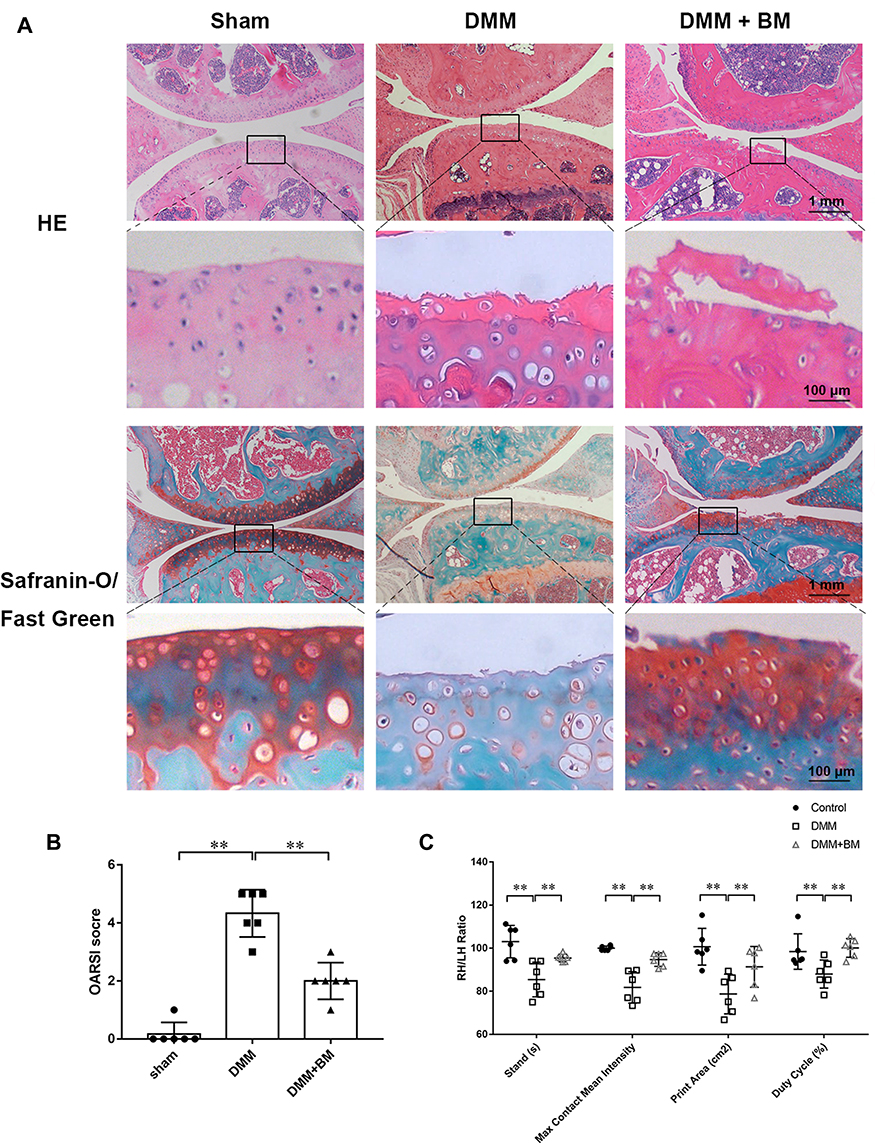 |
Figure 6 Gait analysis of mice and histopathological analysis of mouse knee joint. (A) Representative images of HE and Safranine O-Fast Green staining at magnification of 10× (scale bar, 1mm) and 100× (scale bar, 50 µm). (B) OARSI scores of the knee cartilage from different groups. The results demonstrated that BM effectively alleviates osteoarthritis in mouse DMM models in 4 weeks post-operation. (C) The results of gait analysis using catwalk system showed that BM improved the stands, max contact mean intensity, print area and duty cycle in DMM mice. *P < 0.05 and **P < 0.01. Abbreviations: RH, right hind; LH, left hind. |
Discussion
Previous studies have demonstrated that oxidative damages contribute to the progression of OA. Effective drugs to prevent oxidative stress-induced chondrocyte apoptosis and ECM degradation of SCs are potential and valuable approaches to relieve and reverse cartilage injury in OA. Our study showed that 0.025 and 0.05 µM BM effectively improved the viability of rat chondrocytes treated with 25 mM TBHP. 0.025 and 0.05 µM BM prevented TBHP-induced excessive ROS generation in rat chondrocytes. The present study also showed that 0.025 and 0.05 µM BM prevented oxidative stress-induced mitochondrial damage and apoptosis in chondrocytes. BM treatment promoted the transfer of nuclear factor erythroid derived-2-related factor 2 (Nrf2) into the nucleus. BM treatment enhanced the expression of aggrecan and collagen II and inhibited the expression of MMP 9 and MMP 13 at mRNA and protein levels, suggesting BM effectively prevented ECM degradation. BM treatment increased the Nrf2 expression in chondrocyte nucleus, indicating that BM promoted Nrf2 transferring into cell nucleus. BM treatment increased the expression levels of NOQ1 and HO-1 and decreased the expression levels of Keap1. BM activated Keap1/Nrf2/ARE signaling pathway, which may underlie the beneficial antioxidant effects of BM. BM increased proteoglycan staining area and IOD value in ex vivo cultured experiment cartilage explants, indicating that BM ameliorated TBHP-induced ECM degradation in cartilage tissue. In vivo study, BM improved the OARSI score, stands, max contact mean intensity, print area and duty cycle in DMM mice, demonstrating that BM can ameliorate OA development in mouse DMM model.
Articular cartilage is only composed of extracellular matrix synthesized by sparsely distributed resident chondrocytes. Therefore, the survival of chondrocytes is critical to maintaining a proper cartilage matrix. Any factor that can damage the function and survival of cartilage cells may cause the degeneration of articular cartilage.19,20 Although the report on the ratio of apoptotic chondrocytes in osteoarthritis cartilage is still controversial (from 1% to about 20%), many studies have confirmed that chondrocyte apoptosis contributes to the occurrence and progression of OA. Excessive apoptosis of chondrocytes will make cartilage unable to regenerate and remodel.21,22 Oxidative stress is proved a primary cause of chondrocyte apoptosis and ECM degradation. Lipid peroxidation products including oxidized low-density lipoprotein (ox-LDL), nitrite (NO2-) products were increased while antioxidant enzymes, such as superoxide dismutase (SOD) were decreased in the cartilage of OA patients and OA animal models, confirming that oxidative stress contributes to OA pathogenesis. Therefore, preventing oxidative stress-induced chondrocyte apoptosis and ECM degradation may be an effective method for treating OA.
Antioxidant response element is known as an important cis element necessary to the expression of many antioxidant genes, including HO-1 and NQO1. Activating HO-1/NQO1 signaling pathway is an effective way to exert anti-oxidant effect. Nrf2 functions as the master regulator of intracellular antioxidant response. The binding of Keap1-Nrf2 suppresses the activation of Nrf2 in homeostatic conditions but will become unstable after specific stimulation.10 Promoting the Nrf2 separating from keap1 and transferring into the nucleus to activate downstream HO-1/NQO1 signaling pathways may be an effective means to combat oxidative damage.23 BM, a semisynthetic triterpenoid based on natural product oleanolic acid scaffold, works as Nrf2 activator and subsequently enhances expression of genes encoding antioxidants. BM has been proved effective in patients and animal models of certain diseases due to its antioxidant properties including acute kidney injury, acute lung injury, chronic heart failure, and diabetic kidney disease.12,13,24,25 Therefore, BM may potentially prevent chondrocyte apoptosis through inhibiting oxidative stress.
The present study demonstrated that TBHP induced oxidative stress promoted rat chondrocyte apoptosis and ECM degradation. Two different concentrations of BM (0.025 and 0.05 µM) based on CCK8 results were selected. The results showed that TBHP treatment promoted ROS generation, increased MDA levels and decreased SOD levels in rat chondrocytes, suggesting that TBHP treatment induced oxidative stress in rat chondrocytes. Furthermore, TBHP treatment depolarized the mitochondrial membrane potential, demonstrating the mitochondrial damage in rat chondrocytes. Oxidative stress reduced cell viability and increased apoptosis of rat chondrocytes treated with TBHP in the present study. TBHP inhibited the expression of aggrecan and collagen II and enhanced the expression of MMP13 at mRNA and protein levels, marking the occurrence of ECM degradation. 0.025 and 0.05 µM BM treatment significantly prevented ROS generation, reduced MDA levels and increased SOD levels in rat chondrocytes treated with TBHP. Mitochondria play a key role in maintaining cell survival and normal function. The damage of mitochondrial function caused by excessive ROS is an important event of cell apoptosis. 0.025 and 0.05 µM BM treatment alleviated mitochondrial damage, and thus improved cell viability and prevented cell apoptosis. Besides, the results of PCR and Western blot analysis of MMP 9, MMP 13, aggrecan and collagen II, as well as the results of ex vivo study, suggest that BM can protect ECM from degradation. The current study also showed that 0.05 µM concentration of BM exhibited stronger antioxidant capacity compared with 0.025 µM concentration of BM. Finally, BM ameliorates OA development in mouse DMM model, further proves its therapeutic potential.
In conclusion, our study showed that BM effectively prevents oxidative stress induced by TBHP and thus inhibits cell apoptosis and ECM degradation through activating Keap1/Nrf2/ARE pathway in rat chondrocytes. Except from chondrocyte apoptosis and ECM degradation, synovial inflammation, subchondral bone remodelling and the formation of osteophytes all play certain roles in the development of OA. Whether BM can alleviate these pathological changes is still worth studying. Further works in vivo are required to identify whether BM may effectively alleviate the development of OA additionally. However, the current study proved BM is a promising therapeutic agent for treatment of OA.
Conclusions
In summary, the current study showed that BM alleviated oxidative stress-induced chondrocyte apoptosis and ECM degradation through activating the Keap1/Nrf2/ARE signaling pathway. Additionally, BM ameliorated the degenerative process in a mouse OA model in vivo. These results demonstrated that BM may serve as a potential treatment for OA.
Abbreviations
OA, osteoarthritis; ROS, reactive oxygen species; ECM, extracellular matrix; BM: Bardoxolone-methyl; Nrf2, nuclear factor erythroid derived-2-related factor 2; Keap1, Kelch-like ECH-associated protein 1; NQO1, NADPH quinone oxidoreductase 1; HO-1, heme oxygenase-1; MMP, matrix metalloproteinase; MDA, Malondialdehyde; SOD, superoxide dismutase.
Data Sharing Statement
The datasets used during the present study are available from the corresponding author upon reasonable request.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
The work was supported by grants from National Natural Science Foundation of China (81672142), “Rising Stars of Medical Talent” Youth Development Program (Shangahai Municipal Health Commission 2019-72), and grants from Shanghai Science and Technology Commission (21ZR1412100).
Disclosure
Dr Lu Cao reports grants from Shanghai Municipal Health Commission, during the conduct of the study. The author reports no conflicts of interest in this work.
References
1. Latourte A, Kloppenburg M, Richette P. Emerging pharmaceutical therapies for osteoarthritis. Nat Rev Rheumatol. 2020;16(12):673–688. doi:10.1038/s41584-020-00518-6
2. Hu Y, Chen X, Wang S, Jing Y, Su J. Subchondral bone microenvironment in osteoarthritis and pain. Bone Res. 2021;9(1):20. doi:10.1038/s41413-021-00147-z
3. Fang T, Zhou X, Jin M, Nie J, Li X. Molecular mechanisms of mechanical load-induced osteoarthritis. Int Orthop. 2021;1:548.
4. McClurg O, Tinson R, Troeberg L. Targeting cartilage degradation in osteoarthritis. Pharmaceuticals. 2021;14(2):584.
5. Jiang W, Liu H, Wan R, Wu Y, Shi Z, Huang W. Mechanisms linking mitochondrial mechanotransduction and chondrocyte biology in the pathogenesis of osteoarthritis. Ageing Res Rev. 2021;67:101315. doi:10.1016/j.arr.2021.101315
6. Hwang HS, Kim HA. Chondrocyte apoptosis in the pathogenesis of osteoarthritis. Int J Mol Sci. 2015;16(11):26035–26054. doi:10.3390/ijms161125943
7. Hosseinzadeh A, Kamrava SK, Joghataei MT, et al. Apoptosis signaling pathways in osteoarthritis and possible protective role of melatonin. J Pineal Res. 2016;61(4):411–425. doi:10.1111/jpi.12362
8. Charlier E, Relic B, Deroyer C, et al. Insights on molecular mechanisms of chondrocytes death in osteoarthritis. Int J Mol Sci. 2016;17(12):2146. doi:10.3390/ijms17122146
9. Pérez-Lozano ML, Cesaro A, Mazor M, et al. Emerging natural-product-based treatments for the management of osteoarthritis. Antioxidants. 2021;10(2):796.
10. Cuadrado A, Manda G, Hassan A, et al. Transcription Factor NRF2 as a therapeutic target for chronic diseases: a systems medicine approach. Pharmacol Rev. 2018;70(2):348–383. doi:10.1124/pr.117.014753
11. Tian C, Gao L, Zhang A, Hackfort BT, Zucker IH. Therapeutic effects of Nrf2 activation by bardoxolone methyl in chronic heart failure. J Pharmacol Exp Ther. 2019;371(3):642–651. doi:10.1124/jpet.119.261792
12. Pei X, Zhang XJ, Chen HM. Bardoxolone treatment alleviates lipopolysaccharide (LPS)-induced acute lung injury through suppressing inflammation and oxidative stress regulated by Nrf2 signaling. Biochem Biophys Res Commun. 2019;516(1):270–277.
13. Kanda H, Yamawaki K. Bardoxolone methyl: drug development for diabetic kidney disease. Clin Exp Nephrol. 2020;24(10):857–864. doi:10.1007/s10157-020-01917-5
14. Nagasu H, Sogawa Y, Kidokoro K, et al. Bardoxolone methyl analog attenuates proteinuria-induced tubular damage by modulating mitochondrial function. FASEB J. 2019;33(11):12253–12263. doi:10.1096/fj.201900217R
15. Kalvala AK, Kumar R, Sherkhane B, Gundu C, Arruri VK, Kumar A. Bardoxolone Methyl Ameliorates Hyperglycemia Induced Mitochondrial Dysfunction by Activating the keap1-Nrf2-ARE Pathway in Experimental Diabetic Neuropathy. Mol Neurobiol. 2020;57(8):3616–3631. doi:10.1007/s12035-020-01989-0
16. Kamel AM, Bowlin S, Anwar B, Reichard H, Argus J, Blair IA. In vitro biotransformation of the Nrf2 activator bardoxolone: formation of an epoxide metabolite that undergoes two novel glutathione-mediated metabolic pathways: epoxide reduction and oxidative elimination of nitrile moiety. Chem Res Toxicol. 2019;32(11):2268–2280. doi:10.1021/acs.chemrestox.9b00289
17. Ding SL, Pang ZY, Chen XM, et al. Urolithin a attenuates IL-1β-induced inflammatory responses and cartilage degradation via inhibiting the MAPK/NF-κB signaling pathways in rat articular chondrocytes. J Inflammation. 2020;17:13. doi:10.1186/s12950-020-00242-8
18. Xu C, Zhu S, Wu M, Zhao Y, Han W, Yu Y. The therapeutic effect of rhMK on osteoarthritis in mice, induced by destabilization of the medial meniscus. Biol Pharm Bull. 2014;37(11):1803–1810. doi:10.1248/bpb.b14-00470
19. Tateiwa D, Yoshikawa H, Kaito T. Cartilage and bone destruction in arthritis: pathogenesis and treatment strategy: a literature review. Cells. 2019;8(8). doi:10.3390/cells8080818
20. Boehme KA, Rolauffs B. Onset and progression of human osteoarthritis-can growth factors, inflammatory cytokines, or differential miRNA Expression Concomitantly Induce Proliferation, ECM Degradation, and Inflammation in Articular Cartilage? Int J Mol Sci. 2018;19(8):2282. doi:10.3390/ijms19082282
21. Bolduc JA, Collins JA, Loeser RF. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic Biol Med. 2019;132:73–82. doi:10.1016/j.freeradbiomed.2018.08.038
22. Zahan OM, Serban O, Gherman C, Fodor D. The evaluation of oxidative stress in osteoarthritis. Med Pharm Rep. 2020;93(1):12–22.
23. Sun K, Luo J, Jing X, et al. Astaxanthin protects against osteoarthritis via Nrf2: a guardian of cartilage homeostasis. Aging. 2019;11(22):10513–10531. doi:10.18632/aging.102474
24. Kadioglu E, Teksen Y, Kocak C, Kocak FE. Beneficial effects of bardoxolone methyl, an Nrf2 activator, on crush-related acute kidney injury in rats. Eur J Trauma Emerg Surg. 2021;47(1):241–250. doi:10.1007/s00068-019-01216-z
25. Wu QQ, Wang Y, Senitko M, et al. Bardoxolone methyl (BARD) ameliorates ischemic AKI and increases expression of protective genes Nrf2, PPARγ, and HO-1. Am J Physiol Renal Physiol. 2011;300(5):F1180–1192. doi:10.1152/ajprenal.00353.2010
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.