")
Back to Journals » International Journal of Nanomedicine » Volume 12
Bacterial membrane vesicles as novel nanosystems for drug delivery
Received 17 March 2017
Accepted for publication 20 June 2017
Published 29 August 2017 Volume 2017:12 Pages 6329—6341
DOI https://doi.org/10.2147/IJN.S137368
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Sapna Jain, Jonathan Pillai
Implants, Devices and Drug Delivery Systems Laboratory, Centre for Biodesign and Diagnostics, Translational Health Science and Technology Institute, Faridabad, Haryana, India
Abstract: Bacterial membrane vesicles (BMVs) are closed spherical nanostructures that are shed naturally and ubiquitously by most bacterial species both in vivo and in vitro. Researchers have elucidated their roles in long-distance transport of a wide array of cargoes, such as proteins, toxins, antigens, virulence factors, microbicidal agents and antibiotics. Given that these natural carriers are important players in intercellular communication, it has been hypothesized that they are equally well attuned for transport and delivery of exogenous therapeutic cargoes. Additionally, BMVs appear to possess specific properties that enable their utilization as drug delivery vehicles. These include their ability to evade the host immune system, protection of the therapeutic payload and natural stability. Using bioengineering approaches, BMVs have been applied as carriers of therapeutic moieties in vaccines and for targeted delivery in cancer. In this article, we explore BMVs from the perspective of understanding their applicability to drug delivery. BMV biology, including biogenesis, physiology and pathology, is briefly reviewed. Practical issues related to bioprocessing, loading of therapeutic moieties and characterization for enabling scalability and commercial viability are evaluated. Finally, challenges to clinical translation and rational design approaches for novel BMV formulations are presented. Although the realization of the full potential of BMVs in drug delivery hinges on the development of scalable approaches for their production as well as the refinement of targeting and loading methods, they are promising candidates for development of a novel generation of drug delivery vehicles in future.
Keywords: bacteria, membrane vesicles, immune system, vaccine, bioengineering, drug delivery
Introduction
Bacteria are known to naturally and ubiquitously secrete nanoscale proteoliposomes from their cellular membranes into the extracellular space during all stages of growth.1 This phenomenon of vesicle secretion may occur under diverse environmental conditions or specifically in response to external stimuli.2 Although this process of vesicle formation (vesiculation) was observed >4 decades ago, researchers have recently realized that this is a general cellular process that appears to be conserved across bacterial species.3 Ranging between 20 and 400 nm in diameter, these nanostructures are generally spherical in morphology. In Gram-negative species, they appear to be typically released from the outer membrane (OM) of bacteria and are hence alternatively named as “outer membrane vesicles”.4 However, since they are known to be produced by all Gram-negative and by a few Gram-positive bacteria, the reference to the OM in their nomenclature may be misleading in the latter case. We therefore refer to these as bacterial membrane vesicles (BMVs) for the remainder of this review. BMVs appear to be produced by their parent cells in a constitutive manner without compromising the integrity of the cell.5 Originally thought to be just “trash bags” and “mere cell artifacts”, these have now been established as important and useful entities of bacteria. Even though our current knowledge of the mechanistic pathways and functional reasons behind their biogenesis and regulation remains fragmentary, their role in intercellular communication, gene transfer, transport of virulence factors, antigens and toxins, as well as in disease pathogenesis, is now well established.6–11
A broad evaluation of the existing literature indicates that, prima facie, the primary role of BMVs appears to be that of transporting biomolecules such as proteins and nucleic acids to distant sites. This consistent observation has led to the hypothesis that BMVs can potentially be developed as a novel class of drug delivery systems (DDSs). Exploiting the properties of these natural entities as in vivo delivery vehicles for small molecules such as proteins or drugs will expand the repertoire of delivery systems available for clinical use. Hence, in this article, we briefly review the recent understanding of membrane vesicle biology as well as their role in disease and intracellular communication. Practical considerations for BMV enrichment, therapeutic loading and characterization are also covered. We also specifically discuss emerging therapeutic opportunities of using BMVs in the field of drug delivery and consider the associated challenges.
BMV biology
Biogenesis
Our current knowledge of MV biogenesis is limited, partly because of the wide variability in originating species and partly because this is still an emerging area of research. The debate regarding the theory of vesicular biogenesis revolves around three well-defined models in the literature. The first model by Wensink and Witholt12 describes the process of BMV biogenesis as the result of the faster growth of the OM vis-a-vis the underlying layer of peptidoglycan. This results in disrupted covalent linkages between the two layers, allowing the OM to protrude out and initiate the vesiculation. The second model correlates vesiculation to the turgor pressure exerted on the OM by the gradual amassing of misfolded proteins or peptidoglycan segments in the periplasm.13 The OM ultimately pinches off, thereby releasing the OM vesicles. According to the third model, BMVs are formed because of an uneven extension of the outer leaflet of the OM.14 This extension is produced by curvature-inducing molecules such as the pseudomonas quinolone signal (PQS) of Pseudomonas aeruginosa. In addition to these, a comprehensive biomechanical model has been presented in a report by Roy (Figure 1).15 According to this model, supramolecular protein rivet complexes (RCs) rivet both the OM and the inner membrane together, thereby mediating the “bubble off” process from pockets of extended bacterial periplasm. This model presents MV biogenesis as analogous to soap bubble release with a bubble tube.
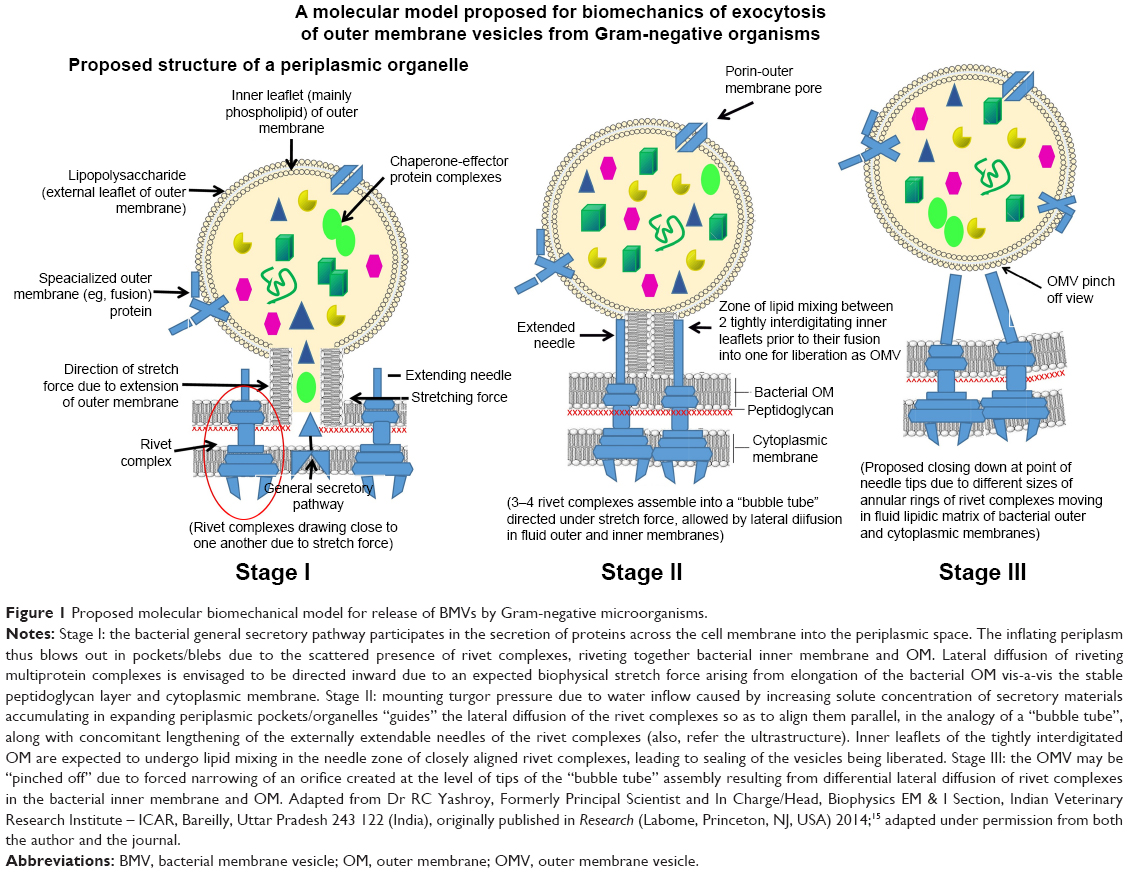 | Figure 1 Proposed molecular biomechanical model for release of BMVs by Gram-negative microorganisms. |
Apart from these models, there is currently no unifying concept describing the biogenesis of BMVs. There also appears to be a lack of conclusive proof about the specific origin of the membrane components that lead to vesicle formation, ie, whether the inner membrane, OM or both are definitively involved, and if so, to what extent. As such, a comprehensive understanding of how the bacterial membrane reorganizes or remodels itself to induce the membrane curvature required to produce a spherical particle from a flat membrane remains elusive.
Structure and characteristic contents
Given their nanoscale and limited resolution with light microscopy, BMVs are best visualized as spherical vesicles bound by a lipid bilayer membrane under an electron microscope. Unraveling of the characteristic composition of BMVs by various molecular biology and analytical techniques has revealed that these bionanoparticles contain a cocktail of bioactive molecules. As summarized in Table 1, these include periplasmic and cytoplasmic proteins, nucleic acids, phospholipids, enzymes and lipopolysaccharides (LPSs), along with other molecules such as ions, metabolites and signaling molecules.16,17 Entrapped proteins include, but are not limited to, adhesins, gingipains, chaperonins, multidrug efflux transporters, OM proteins, flagellin and related hook-associated proteins, ribosomal proteins and OM lipoproteins.18 In addition to proteins, lipids constitute a significant and important component of BMVs. The lipidomic profile of vesicles from Escherichia coli10 and P. syringae19 has revealed the presence of lipids such as phosphatidylethanolamine, phosphatidylglycerol and cardiolipin.
 | Table 1 Summary of BMV contents |
Nucleic acids also form a significant component of these vesicles. Sjöström et al20 have reported the presence of RNA in BMVs harvested from a wild-type Vibrio cholerae O1 El Tor strain, by performing RNA deep sequencing analysis. Their findings suggested that RNA could be packaged and released via membrane vesicles. Recently, small RNAs, similar in size to eukaryotic miRNAs, have been reported in periodontal pathogens, namely, Aggregatibacter actinomycetemcomitans, Porphyromonas gingivalis and Treponema denticola.21
Physiological functions
Variability in genotype and envelope architecture among bacterial species explains the diversity of functions performed by membrane vesicles. In general, it seems that bacteria use these vesicles to enhance their chances of survival within their environment. BMVs are implicated in a wide variety of functions that support bacterial survival, including formation of biofilms, intraspecies transfer of genes and nutrients, protection of ecological niche and intercellular communication. For instance, BMVs released from P. aeruginosa PAO1 are able to lyse competing bacterial species such as Staphylococcus aureus, E. coli and another strain of Pseudomonas itself, such as P. aeruginosa 8803.38 Vesicles from P. gingivalis facilitate its evasion of the host immune system by transferring the enzyme PPAD to the host, which consequently inhibits the complement factor C5a.39 These reports serve as good examples of the role of BMVs in enabling survival of parent bacteria. As an example of horizontal gene transfer between species, vesicles of Acinetobacter baylyi have been shown to transfer plasmid-borne β-lactamase gene to A. baylyi and E. coli populations.40
BMVs not only mediate physiological functions for their parent bacteria but, in some cases, may modulate the host physiology as well. Recent studies have found that BMVs produced by commensal bacteria support the maturation of the host immune system.41 For example, BMVs mediate the delivery of a bacterial homologue of a eukaryotic inositol phosphate signaling enzyme, BtMINPP, by the bacterium Bacteroides thetaiotaomicron into host intestinal epithelial cells.42 This upregulates intracellular calcium signaling and provides nutritional as well as anticarcinogenic benefits to the host.
Pathological functions of BMVs
BMVs from pathogenic species have been implicated in several pathological processes, including virulence, facilitation of infection and inflammation in the host as well as delivery of diverse cargoes such as proteins, toxins and nucleic acids. In the context of infection and inflammation, BMVs have been detected in a range of infected host tissues. For example, they have been reported in the nasopharynx of asymptomatic carriers of Neisseria meningitidis, the cerebrospinal fluid of patients with meningococcal disease, the gastric tissue of individuals infected with Helicobacter pylori, the sinuses of pediatric patients with Moraxella catarrhalis-induced sinusitis and in the lungs of patients with nontypeable Haemophilus influenzae infections.11,43–47 These reports suggest that BMVs do play a significant role in the onset and progression of disease. Further more, BMVs from various pathogens interact with epithelial and immune cells to induce the production of cytokines, chemokines and antimicrobial peptides, thereby modulating the pathology by either upregulating or suppressing the activation of these cells. Mechanisms by which BMVs modulate host immune responses have been reviewed extensively by Kaparakis-Liaskos and Ferrero.48 These vesicles also deliver multiple virulence factors, such as alkaline phosphatase, hemolytic phospholipase C and cystic fibrosis transmembrane regulator (CFTR) inhibitory factor (Cif), to the host, thereby affecting host cell biology.37 In addition, they also trigger immunomodulatory activities, which have been well exploited in the generation of vaccines against N. meningitidis.49,50 BMVs may display antigens on their surface that may elicit potent inflammatory and immune responses in the host. For example, Alaniz et al showed that BMVs of Salmonella enterica serovar Typhimurium stimulate the production of proinflammatory mediators, such as tumor necrosis factor alpha (TNF-alpha) and interleukin-12 (IL-12), along with the activation of CD4+ T-cells.51 Altogether, this body of evidence sheds light on the broad contribution of BMVs in disease pathogenesis. Figure 2 summarizes both the physiological and pathological functions of BMVs.
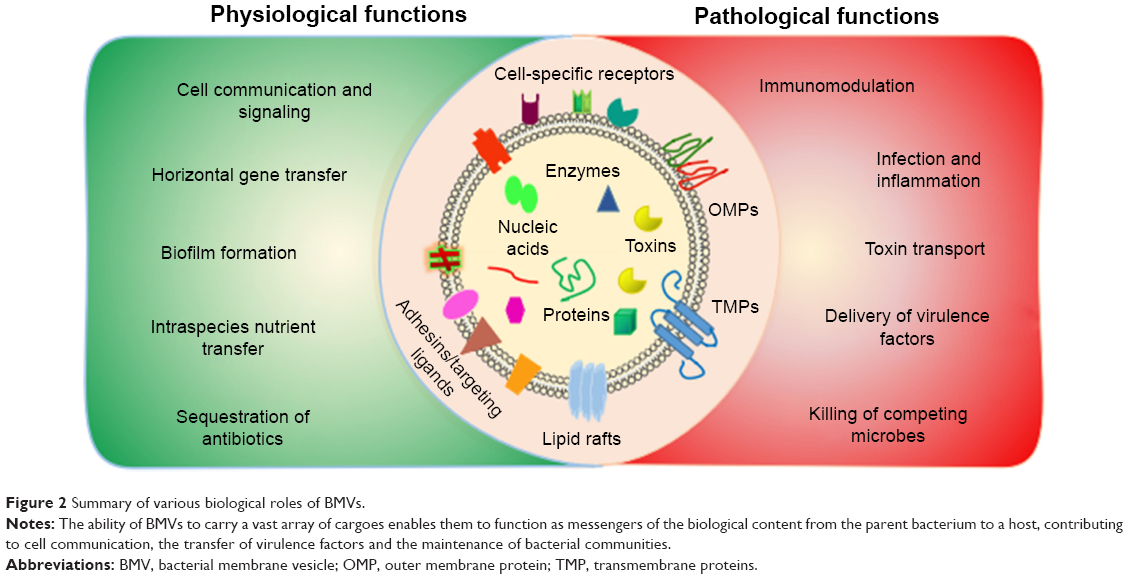 | Figure 2 Summary of various biological roles of BMVs. |
BMVs as nanoplatforms for drug delivery
The field of drug delivery relies on the ability of drug carriers to effectively deliver therapeutic payload to the target. These carriers protect the cargo from the strident host environment as well as release the cargo to the appropriate site, ideally generating an imperceptible host immune response. To rule out the possibility of induction of immunogenic response to such carriers, extracellular vesicles extracted from patients themselves have been developed as clinically relevant DDSs. Among biologically derived drug delivery vehicles, the most commonly explored entities include red blood cells, macrophages, viruses, exosomes, stem cells and lymphocytes.52–61 BMVs have recently been recognized as potential candidates to be included in this list.62
A review of the literature suggests that nature has endowed BMVs with many characteristics and functions desirable of drug delivery vehicles. Specifically, BMVs derived from pathogenic species are adept at subversion or modulation of the host immune system while gaining efficient entry into target cells with high specificity and selectivity. Furthermore, their small size may permit them to travel throughout the body while evading immediate capture and clearance by the host reticuloendothelial system. However, concerns regarding the potential for high immunogenicity, anchoring of immunosuppressive antigens, inter- and intra-strain variations as well as interference with protective immune response highlight the risks related to their safety and effectiveness. LPS, a major component of the OM of most Gram-negative bacteria, and consequently their BMVs, is a very potent activator of immune cells. This response is critical in directing the normal host adaptive immune response, which is advantageous in the development of vaccines or adjuvants. On the other hand, a systemic and potent inflammatory response is undesirable in drug delivery applications, where the vehicle is expected to be nonimmunogenic and noninflammatory. To address these challenges, different approaches can be applied to enhance the clinical acceptance of BMVs as drug delivery vehicles. It is known that triggering of the immune response is mediated strongly by immune recognition of size, surface properties, such as electrostatic charge and the specificity to immune triggers like LPS. These immune triggers are likely minimized for MVs from nonpathogenic bacterial origin. As such, MVs from nonpathogenic species are likely to have the same probability of immune evasion as other comparable minimally immunogenic DDSs, such as liposomes and hydrogel nanoparticles. Hence, steps to minimize the immunogenicity of BMVs may include the following: choice of nonpathogenic strains; removal, addition or alteration of membrane proteins and surface antigens by genetic engineering of BMV-producing strains; or using reagents to decrease LPS content. However, while a couple of instances of BMV-based vaccines and drug delivery have been reported to be well tolerated in patients and animal models, respectively, specific reports on the in vivo fate of BMVs following intravenous or other routes of direct dosing are unavailable to the best of our knowledge. This remains an important area of investigation prior to clinical translation. A generic scheme for how BMVs may be used as drug delivery vehicles for clinical use is presented in Figure 3.
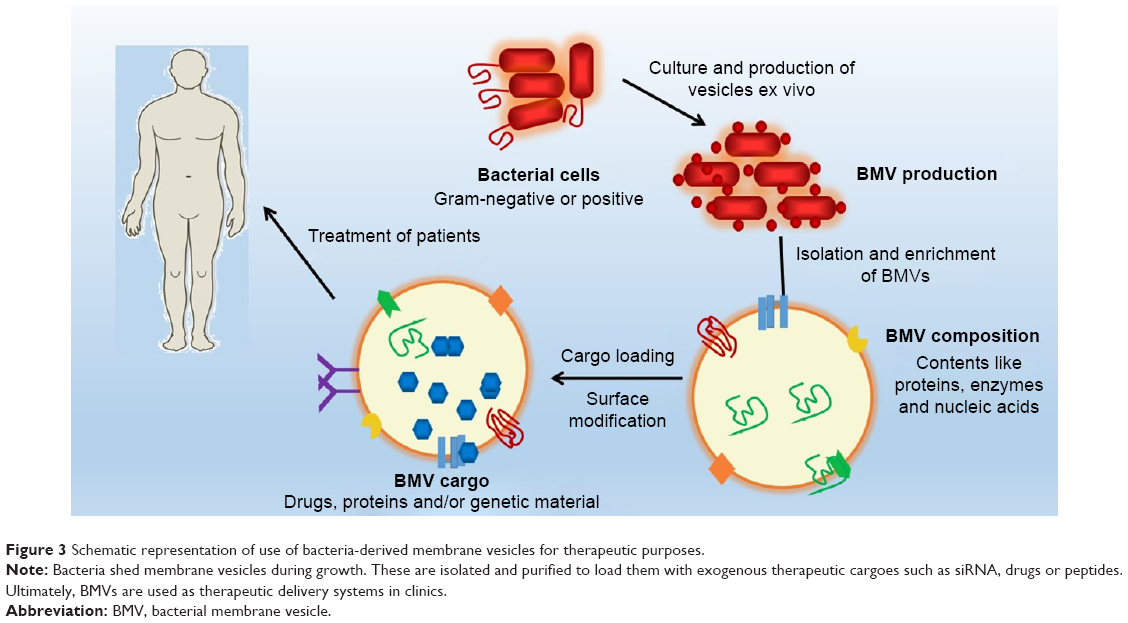 | Figure 3 Schematic representation of use of bacteria-derived membrane vesicles for therapeutic purposes. |
Bacterial packaging of therapeutics in BMVs
In certain cases, it may be possible to directly harness naturally derived BMVs for a therapeutic outcome. BMVs from certain species that package therapeutically relevant cargoes, such as immunomodulatory molecules or antimicrobial peptides against competing species, lend themselves particularly well for this purpose. Vesicles from Shigella, Borrelia, E. coli, Helicobacter, Treponema and Actinomyces perform the function of unloading heterogeneous cargoes into the target cells by adhering or fusing with target cells.9,50,63–65 The fusion delivers proteins that can lyse or kill the target bacteria. For example, Pseudomonas vesicles contain the autolysin murein hydrolase, which is capable of lysing other Gram-negative and Gram-positive bacteria.38 The discovery of these “predatory MVs” clearly indicates that, in an era of antimicrobial resistance, the microbe itself provides the artillery to fight against other pathogens of its kind. This approach, of targeting pathogenic bacteria directly by administration of potent BMVs loaded with biologic therapeutics, is therefore being proposed as a fundamentally novel class of packaged antibiotics.38
Bioengineered vesicles
Of particular interest to bioengineers and formulation scientists is the possibility that BMVs can also be artificially modulated to package therapeutics with high specificity, loading efficiency and stability. Since they originate from different bacterial species, they naturally display a wide array of biological effects and targeting specificities. These can further be enhanced by bioengineering the enriched vesicles, genetically engineering the vesiculating strain itself or a combination of both. These methods may enhance both the BMV surface as well as their packaged cargoes for site- or target-specific delivery.
In the bioengineering approach, the external surface of previously isolated BMVs can be tailored with specific ligands, such as antibodies, cell-targeting peptides (CTPs), aptamers or antigens, by adapting molecular or in vitro bioconjugation techniques routinely employed for synthetically produced targeted nanoparticulate systems. Alternatively, genetic engineering of the vesiculating parent bacteria may be employed, for both cargo enhancement and surface modification. Genetic manipulation of the vesiculating strains may be easily done to encode specific peptide sequences of the surface or other membrane-bound proteins that are expressed on the bacterial membrane. This ligand-enriched membrane, when subsequently incorporated into the BMV surface, can produce BMVs directed to target the corresponding host cells or tissues. For example, in an elegant example of delivering a biological therapeutic, Gujrati et al62 showed that bioengineered BMVs presenting an anti-HER2 affibody on their surface could selectively deliver their siRNA cargo to tumor cells in breast cancer. This resulted in targeted gene silencing and significant regression in tumor growth.
Bioprocessing of native BMVs
In order to exploit membrane vesicles as drug delivery vehicles and to enable their translation for clinical use, they must be produced at a commercially viable scale. In the following section, various processes for isolation, enrichment, purification and storage of these fragile carriers are reviewed.
Biosynthesis from bacterial culture
Our currently limited understanding of BMV biogenesis hampers the optimization of methods that can be applied to produce BMVs at large scales in a reasonably short time. However, investigators in this field have found regulators of vesiculogenesis, which may be a promising development toward commercial production. Usually, this regulation is mediated by some sort of envelope stress response. Hypervesiculation can be induced by modulating these stressors, thereby generating novel phenotypic strains that are optimized for increased BMV production. For instance, the DegP mutant of E. coli, when cultured at high temperatures, led to increased production of BMVs than the wild-type bacterium.13 In this study, a rise in culture temperature (37°C), coupled with the deletion of the DegP gene, which prevents accumulation of proteinaceous waste at the periplasmic periphery, generated a hypervesiculating strain of E. coli. Contrary to this, McMahon et al66 showed that Serratia marcescens hypervesiculates at lower temperatures (22°C or 30°C) and is induced by an active Rcs phosphorelay system. This contradiction can be explained superficially by the specificity of the parent bacterium.
Apart from culture conditions, hypervesiculation may also be induced as a stress response to the introduction of an antibiotic challenge or other environmental stressors such as UV radiations at sublethal doses. Reports indicated that a brief exposure of growing cultures of P. aeruginosa to gentamicin increased the quantity of vesicles produced.38 Similarly, UV exposure to aquatic bacteria resulted in increased production of BMVs.67 Modulating such functionalities of the parent bacteria can help to select strains that can produce vesicles with the desired properties in large quantities. While hypervesiculation seems a good option for scaling up the BMV yield per bacterium, it is important to note that only conditions that do not significantly alter the contents of the BMVs must be adopted.
Isolation and enrichment
Ultracentrifugation remains the gold standard process that is most widely used to isolate BMVs. As shown in the schematic flow in Figure 4, the process consists of multiple steps, making it lengthy, and may result in relatively low yields of BMVs. Moreover, it is more susceptible to coprecipitation of proteins, small molecules, flagella and other membrane fragments from the parent bacterial culture. As a result, the purity and integrity of the extracted vesicle preparations is questionable, particularly if they are destined for translational use or regulatory approval. This issue can be partly overcome by using density-based gradient centrifugation, which resolves vesicle subpopulations based on their buoyant density in a gradient of viscous media of predetermined densities. Sucrose or other viscous solutions of relatively inert molecules, such as inositol, may be used for this purpose. Our experience shows that this additional step can provide a significantly cleaner population of BMVs, devoid of nonvesicular products. Estimation of the total protein content of BMVs purified via density gradient centrifugation clearly indicated the advantage of using this additional step, as the protein content was significantly lower (data not shown). As seen in Figure 5, transmission electron microscopy (TEM) provides a qualitative visual comparison of the extent of extravesicular components and the purity of samples after conventional differential centrifugation and an additional density gradient centrifugation.
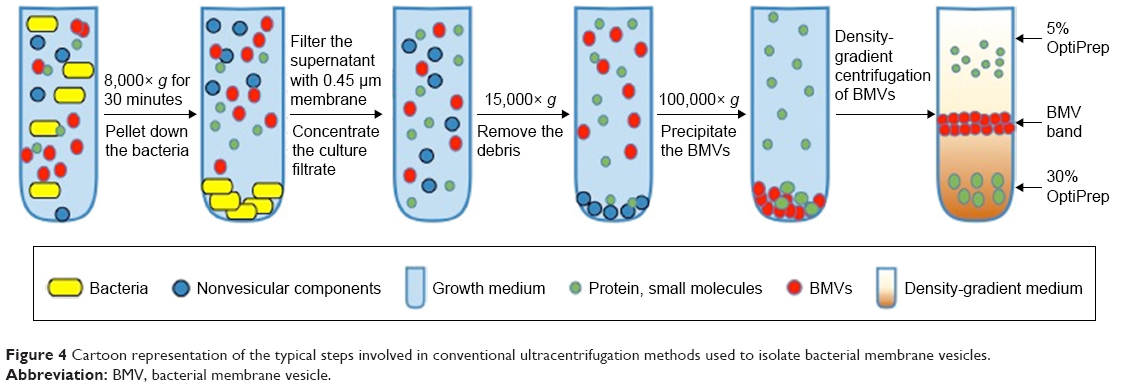 | Figure 4 Cartoon representation of the typical steps involved in conventional ultracentrifugation methods used to isolate bacterial membrane vesicles. |
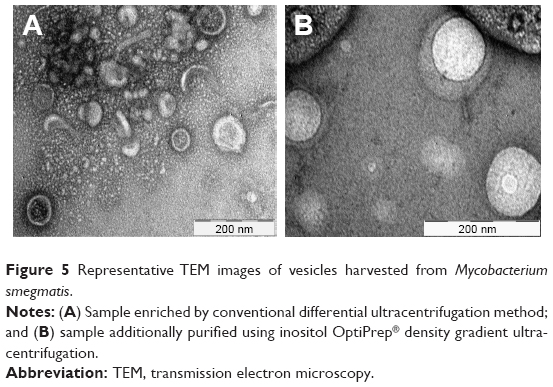 | Figure 5 Representative TEM images of vesicles harvested from Mycobacterium smegmatis. |
Considering that current methods are cumbersome and time consuming, it is important to explore new methods and techniques that are faster, easier and more efficient to isolate high-purity BMV samples. These methods should be able to extract BMVs in a reliable, consistent and reproducible manner. Alternative enrichment techniques may include microfiltration, tangential flow filtration, antibody-coated magnetic beads, immunoaffinity-based separation, microfluidics and column chromatography. For example, Post et al65 used Sephacryl S200 chromatography to get purified and homogeneous preparations of vesicles that were suitable for the successful development of a vaccine against N. meningitidis.
Storage and reconstitution
To preserve BMV integrity and stability, it is essential to store them after extraction under optimal conditions of temperature, duration and pH, while also optimizing use of preservatives or cryoprotectants. It appears that since most researchers tend to evaluate BMVs for understanding their biological significance and for deciphering various mechanistic pathways, their preference is to utilize freshly enriched populations, rather than samples stored for any reasonable length of time. However, for both diagnostic and therapeutic applications, evaluation of optimal processes for storage and reconstitution is critical. For example, the stability of meningococcal OMV vaccine was evaluated following cryogenic storage followed by lyophilization.68 This detailed protocol indicates that optimization of freeze-drying or other preservation protocols, as well as selection of the ideal cryoprotectants or stabilizers, is indispensable for ensuring complete preservation of BMV integrity and cargo during storage and subsequent use.
Synthesis and characterization of drug-loaded BMVs
Small molecule drugs or other therapeutic biologicals can be loaded onto BMVs by encapsulation into the inner lumen, covalent linkage to the surface of vesicles or a combination of both these methods. This approach may improve the pharmacological properties and biological efficacy of the drugs. For example, encapsulating drugs in the BMV lumen may protect them from degradation during their delivery to target sites and from interaction or cross-reactivity with other proteins or drug molecules. Additional effects may include modulation of biological efficacy and induction or suppression of host immune response for the desired therapeutic outcome. While both approaches for drug loading may be employed in vitro, ie, after harvesting BMVs from culture, the former approach of drug entrapment may better suit in vivo loading, ie, during their biogenesis in culture. Figure 6 provides a schematic overview of the in vitro and in vivo drug-loading strategies, both of which are described in detail in the following sections.
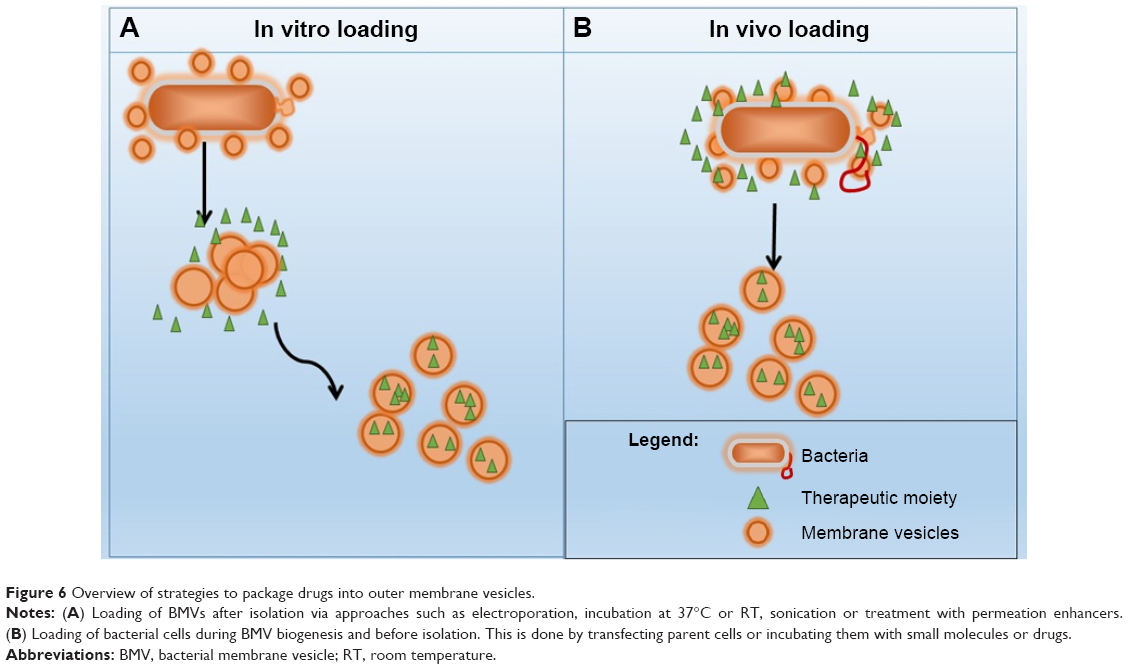 | Figure 6 Overview of strategies to package drugs into outer membrane vesicles. |
In vitro loading
In vitro drug loading refers to loading of drug(s) of interest after initial isolation and enrichment of BMVs from culture. Since BMVs are generalized as lipid-bilayered membranous structures, they could potentially be loaded with lipophilic as well as hydrophilic drugs. The efficiency of drug loading and subsequent retention is dependent on the membrane–drug interaction. This, in turn, is determined by the variability in structure and composition of membranes from different originating species, particularly Gram-positive and Gram-negative bacteria. Drug entrapment can be achieved using passive methods such as diffusion along osmotic gradients. Alternatively, it may be actively driven by techniques such as electroporation or ultrasonication or by enhancing the membrane permeability using cell-penetrating peptides or chemical transfection. Passive diffusion may work well for relatively hydrophobic and positively charged small molecule drugs that readily interact with a lipophilic membrane. However, for hydrophilic molecules such as RNA, the bilayered lipidic membrane may pose a barrier, in that it restricts the passive diffusion of such drugs. In such cases, electroporation might be the method of choice. For example, the successful entrapment of siRNA recently demonstrated by Gujrati et al62 involved the use of electroporation for incorporation of this biomolecule into bioengineered BMVs.
In vivo loading
An alternative approach to BMV loading is to incorporate drugs into BMVs during their biogenesis by the parent bacterium. This method may be preferable for molecules such as high-molecular-weight RNA (mRNA) or aminoglycoside antibiotics, which cannot be easily loaded into BMVs after their isolation and purification. As was previously shown with hypervesiculation, it appears that packaging of the drug incorporated in culture media into BMVs may be a survival mechanism developed by the parent bacteria to a sublethal antibiotic challenge.
Allan and Beveridge69 have demonstrated the incorporation of gentamicin during growth of the PAO1 strain of P. aeruginosa. As proof of the efficacy of this method, BMVs thus harvested were shown to contain gentamicin, and these gentamicin-loaded BMVs could deliver the drug to the target bacterium Burkholderia cepacia. As such, it appears that this mechanism of drug resistance in the parent bacterium may be co-opted for drug delivery purposes, so long as resistance is not mediated by enzymatic degradation of the drug.
This approach carries the inherent risk of inadvertently facilitating the transfer of antibiotic resistance via the drug-loaded BMVs. There are recent reports emerging in literature of species, such as Acinetobacter baylyi and Acinetobacter baumannii in which horizontal gene transfer of resistance is mediated by OMVs.40,70 This packaging approach may be made feasible by carefully choosing the parent species or mutant strains with hyperactive efflux pumps that can easily pump the drug out into BMVs. Stringent purification steps and quality control checks will be necessary following enrichment, to ensure that genetic material is not simultaneously packaged along with the antibiotics.
Characterization
The translation of BMVs into therapeutics in a clinical setting requires them to be well characterized at two stages in their biosynthesis. Preliminary characterization of the physical characteristics of native enriched BMVs is essential, to determine their phenotypical features such as size, morphology, yield, concentration and composition. Collectively, this is done using a suite of analytical techniques, which includes electron microscopy (EM), western blotting, total protein estimation and individual particle tracking. EM provides reasonable qualitative information on the structural morphology, integrity and luminal contents of vesicles, potentially including the successful incorporation of therapeutic moieties. However, it does not provide any quantitative information on the number of the vesicles nor a precise estimation of drug loading. In addition, EM analysis assumes that extrapolation of results from a few random samples is ideally representative of the entire population, while also requiring careful sample preparation. Quantification of total proteins, as a surrogate marker for the mass of isolated BMVs, is currently the gold standard for estimating BMV yields from culture. While this is relevant and adequate for most biological studies, it is an indirect – and potentially misleading – approach, particularly for drug delivery. This is because the method is greatly influenced by extraneous proteins/peptides that may be coprecipitated during ultracentrifugation, leading to an overestimation of BMV yield. Furthermore, dosing of therapeutics is better correlated to the number and luminal volume of individual BMVs in a sample population, rather than to the total protein of the population. Nanoparticle tracking analysis (NTA), in combination with thermogravimetric protocols commonly employed in estimating yields for pharmacological formulations, may therefore be a more direct, accurate and facile measure of BMV yields.
A considerable number of studies have used proteomic, nucleic acid (RNA) and lipidomic analysis of vesicles to characterize their heterogeneous composition. Mass spectrometry appears to be the tool of choice for various “-omics”-based approaches employed for this purpose. Morphologic analysis of individual particles is done by methods such as NTA, dynamic light scattering (DLS) and tunable resistive pulse sensing (TRPS), all of which help to determine their count and size. These techniques are further combined with other technologies such as fluorescent labeling, quantum dots and Raman spectroscopy to reveal the enriched BMV phenotypic profile. Figure 7 provides a typical result from NTA characterization of BMVs enriched in our laboratory, including both a visual image of suspended BMVs and the quantification result of the population distribution as per size.
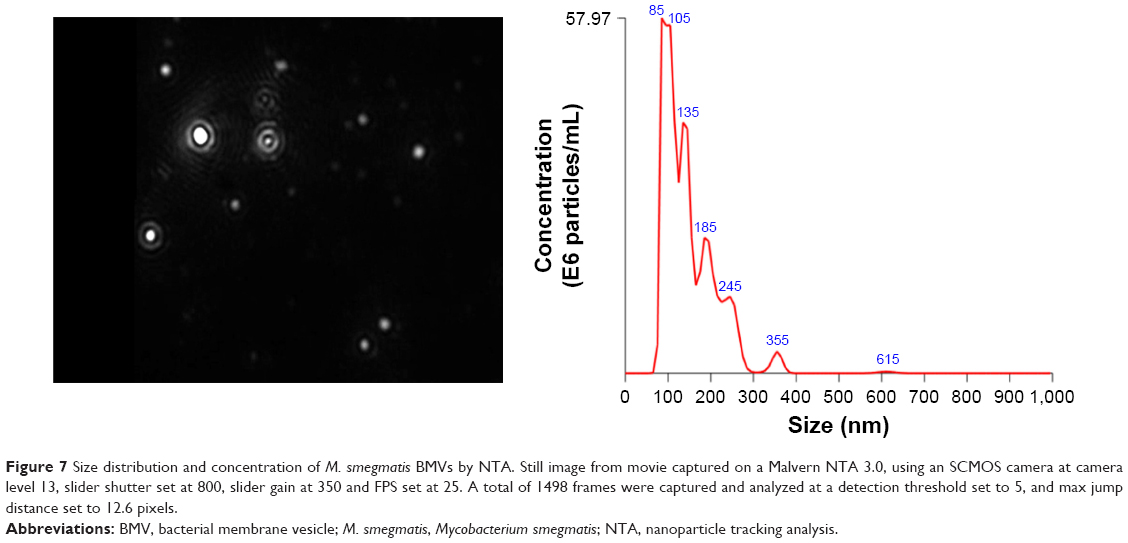 | Figure 7 Size distribution and concentration of M. smegmatis BMVs by NTA. Still image from movie captured on a Malvern NTA 3.0, using an SCMOS camera at camera level 13, slider shutter set at 800, slider gain at 350 and FPS set at 25. A total of 1498 frames were captured and analyzed at a detection threshold set to 5, and max jump distance set to 12.6 pixels. |
In the second stage, the pharmacological characterization of drug-loaded BMVs is essential to validate their safety and efficacy for clinical use. Typically, these studies involve the characterization of drug encapsulation, stability, subsequent drug release, in vitro and in vivo evaluation of safety and efficacy profiles, as well as pharmacokinetic/pharmacodynamic (PK/PD) studies. These need to be appropriately evaluated both in the host as well as against the target pathogenic species as per the needs of the clinical end application. Further more, for BMVs used in vaccine therapy, as well as for those designed to be passive and nonpathogenic drug carriers, an in-depth evaluation of their intrinsic immunogenicity is critical to establish the host immune response to in vivo dosing.
Challenges and strategies for the future
Challenges
As noted herein, many challenges need to be overcome before successful therapeutic application of BMVs in clinical settings. For example, most current enrichment techniques are limited by their dependence on ultracentrifugation. As discussed earlier, these methods are time-consuming and inefficient in isolating vesicles, often suffering from low purity and yields. Given the phenotypic variability in parent bacterial species, there is no single ideal and scalable enrichment protocol for producing highly purified BMVs in substantial quantities for translational and commercial viability. As such, protocols for isolation and enrichment should be customized for each application and species. In addition, analytical methods and parameters for characterization of BMV count, size, morphology, loading capacity, and PK/PD evaluation need to be optimized and improved. Improved understanding of the biological mechanisms of how the native cargo content is sorted and packaged in these vesicles could enable effective modulation of packaged therapeutics and targeted delivery. It is also important to understand how the formation and loading of heterogeneous content into BMVs is regulated and affected by varied growth conditions such as pH, temperature, growth medium, mechanical forces and originating source. Despite recent advances, thorough characterization of BMVs from a clinical use perspective remains a challenging task. While great efforts have been made to accurately measure the vesicle number and their content, there remains a dire need to validate and standardize the BMV enumeration methods across investigators. The extensive heterogeneity and variability of BMVs enriched from even a single originating bacterial species poses questions on their consequent functional identity. For example, it is currently unknown if smaller vesicles <100 nm in diameter are structurally, compositionally and functionally similar or notably different from those that are 200–400 nm in diameter, even if both populations have been isolated from the same batch of bacterial culture.
Strategy for rational design of BMV-based DDSs
Despite the challenges outlined, it is possible, and indeed beneficial, to conceptualize a rational process for the design and development of novel BMV-based nanosystems for drug delivery. This process must include the following major considerations: the target pathology, BMV-producing parent bacterium, exogenous cargo to be loaded (nucleic acids, proteins, therapeutic peptides or small molecule drugs) and any targeting ligands that are naturally or artificially incorporated into their surface. Evolution has endowed bacteria with the power of generating, producing and utilizing these nanostructures for their survival and intrinsic biological functions. The challenge of effective BMV loading may, therefore, partly be overcome by harnessing the bacteria themselves for packaging the exogenous cargo into the vesicles shed by them. This will require appropriate selection of vesiculating bacterial strains capable of producing BMVs compatible with, and optimized for, the cargo they are intended to carry. Subsequently, efficient cargo delivery profoundly depends on the interaction between the target and the vesicles carrying the drugs. As described earlier, BMVs may be targeted to specific sites either passively or actively. Passive targeting is achieved by increasing the fusion and permeability of the drug carrier across the target site without any homing moiety. Assuming that membrane vesicles retain the structural characteristics of the parent bacterial membrane, it may be possible to passively target certain pathogens by selecting vesicles from strains having optimal membrane homology with the target. Alternatively, targeting can be achieved actively by using any homing moiety such as antibodies, coupling ligands or surface antigens. This can be done either by bioengineering the BMV-producing bacterium itself or by modifying the surface of vesicles after purification. Surface-modified BMVs, engineered to display these targeting moieties, may significantly improve the selectivity of vesicles to their targets. Selection of suitable therapeutic molecules is also a major factor for consideration in this rational process design. The criteria of selection include features such as their size or molecular weight, structure and polarity. In addition, for effective encapsulation within vesicles, the candidate molecule to be loaded needs to be evaluated for its interaction and fusion with the membrane of the vesicles, as well as in terms of its subsequent release from the BMV lumen or surface.
Conclusion
The inherent potential of BMVs to evade the host immune system and deliver their cargo selectively to target sites makes them viable candidates for drug delivery vehicles. Indeed, their applicability for modulating host immunity has already been harnessed for clinically approved vaccines, with others being considered for clinical development as well. Currently, they are being proposed as therapeutics for targeting a wide array of pathologies, with applications ranging from use as potentially novel antibiotics against pathogenic bacteria to targeted delivery of nucleic acid therapeutics for cancer. Methods for their bioprocessing and characterization have dramatically improved over the past 2 decades. Nonetheless, the inherent variability in enriched BMVs remains a challenge, along with the additional challenges mentioned herein. Notwithstanding these, the adoption of a rational design strategy, in conjunction with continuous efforts in enhancing our current understanding of the underlying BMV biology, will profoundly enhance the candidature of BMV-based therapeutics.
Acknowledgments
Sapna Jain is funded for her PhD program by a National Eligibility Test (NET) scholarship from the Government of India. We gratefully acknowledge funding provided via a faculty core grant to Jonathan Pillai (PhD) by the Department of Biotechnology, Government of India. The authors thank Drs Krishnamohan Atmakuri (Translational Health Science and Technology Institute) and CV Srikant (Regional Centre for Biotechnology, NCR Biotech Science Cluster, Faridabad, Haryana, India) for proofreading the article and providing helpful comments and suggestions.
Disclosure
The authors report no conflicts of interest in this work.
References
Beveridge TJ. Structures of gram-negative cell walls and their derived membrane vesicles. J Bacteriol. 1999;181(16):4725–4733. | ||
Mashburn-Warren L, Mclean RJ, Whiteley M. Gram-negative outer membrane vesicles: beyond the cell surface. Geobiology. 2008;6(3):214–219. | ||
Adler LW, Rosen BP. Functional mosaicism of membrane proteins in vesicles of Escherichia coli. J Bacteriol. 1977;129(2):959–966. | ||
Kulkarni HM, Jagannadham MV. Biogenesis and multifaceted roles of outer membrane vesicles from Gram-negative bacteria. Microbiology. 2014;160(10):2109–2121. | ||
McBroom AJ, Johnson AP, Vemulapalli S, Kuehn MJ. Outer membrane vesicle production by Escherichia coli is independent of membrane instability. J Bacteriol. 2006;188(15):5385–5392. | ||
Remis JP, Wei D, Gorur A, et al. Bacterial social networks: structure and composition of Myxococcus xanthus outer membrane vesicle chains. Environ Microbiol. 2014;16(2):598–610. | ||
Yaron S, Kolling GL, Simon L, Matthews KR. Vesicle-mediated transfer of virulence genes from Escherichia coli O157: H7 to other enteric bacteria. Appl Environ Microbiol. 2000;66(10):4414–4420. | ||
Mashburn-Warren L, Howe J, Garidel P, et al. Interaction of quorum signals with outer membrane lipids: insights into prokaryotic membrane vesicle formation. Mol Microbiol. 2008;69(2):491–502. | ||
Kato S, Kowashi Y, Demuth DR. Outer membrane-like vesicles secreted by Actinobacillus actinomycetemcomitans are enriched in leukotoxin. Microb Pathog. 2002;32(1):1–3. | ||
Horstman AL, Kuehn MJ. Enterotoxigenic Escherichia coli secretes active heat-labile enterotoxin via outer membrane vesicles. J Biol Chem. 2000;275(17):12489–12496. | ||
Keenan J, Day T, Neal S, et al. A role for the bacterial outer membrane in the pathogenesis of Helicobacter pylori infection. FEMS Microbiol Lett. 2000;182(2):259–264. | ||
Wensink J, Witholt B. Outer membrane vesicles released by normally growing Escherichia coli contain very little lipoprotein. Eur J Biochem. 1981;116(2):331–335. | ||
McBroom AJ, Kuehn MJ. Release of outer membrane vesicles by Gram-negative bacteria is a novel envelope stress response. Mol Microbiol. 2007;63(2):545–558. | ||
Schertzer JW, Whiteley M. A bilayer-couple model of bacterial outer membrane vesicle biogenesis. MBio. 2012;3(2):e297–e311. | ||
Yashroy RC. Membrane vesicle trafficking in prokaryotes: molecular biomechanics of biogenesis of outer membrane vesicles of gram-negative (Salmonella 3, 10: r:-) microbes in chicken ileal invasion model in vivo. Research. 2014;1:1128. | ||
Choi CW, Park EC, Yun SH, et al. Proteomic characterization of the outer membrane vesicle of Pseudomonas putida KT2440. J Proteome Res. 2014;13(10):4298–4309. | ||
Kulp A, Kuehn MJ. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu Rev Microbiol. 2010;64:163–184. | ||
Wessel AK, Liew J, Kwon T, Marcotte EM, Whiteley M. Role of Pseudomonas aeruginosa peptidoglycan-associated outer membrane proteins in vesicle formation. J Bacteriol. 2013;195(2):213–219. | ||
Chowdhury C, Jagannadham MV. Virulence factors are released in association with outer membrane vesicles of Pseudomonas syringae pv. tomato T1 during normal growth. Biochim Biophys Acta. 2013;1834(1):231–239. | ||
Sjöström AE, Sandblad L, Uhlin BE, Wai SN. Membrane vesicle-mediated release of bacterial RNA. Sci Rep. 2015;5:15329. | ||
Choi JW, Kim SC, Hong SH, Lee HJ. Secretable small RNAs via outer membrane vesicles in periodontal pathogens. J Dent Res. 2017;96(4):458–466. | ||
Khandelwal P, Banerjee-Bhatnagar N. Insecticidal activity associated with the outer membrane vesicles of Xenorhabdus nematophilus. Appl Environ Microbiol. 2003;69(4):2032–2037. | ||
Patrick S, McKenna JP, Seamus O, Dermott E. A comparison of the haemagglutinating and enzymic activities of Bacteroides fragilis whole cells and outer membrane vesicles. Microb Pathog. 1996;20(4):191–202. | ||
Chi B, Qi M, Kuramitsu HK. Role of dentilisin in Treponema denticola epithelial cell layer penetration. Res Microbiol. 2003;154(9):637–643. | ||
Shoberg RJ, Thomas DD. Specific adherence of Borrelia burgdorferi extracellular vesicles to human endothelial cells in culture. Infect Immun. 1993;61(9):3892–3900. | ||
Negrete-Abascal E, García RM, Reyes ME, Godínez D, de la Garza M. Membrane vesicles released by Actinobacillus pleuropneumoniae contain proteases and Apx toxins. FEMS Microbiol Lett. 2000;191(1):109–113. | ||
Kadurugamuwa JL, Mayer A, Messner P, Sára M, Sleytr UB, Beveridge TJ. S-layered Aneurinibacillus and Bacillus spp. are susceptible to the lytic action of Pseudomonas aeruginosa membrane vesicles. J Bacteriol. 1998;180(9):2306–2311. | ||
Fiocca R, Necchi V, Sommi P, et al. Release of Helicobacter pylori vacuolating cytotoxin by both a specific secretion pathway and budding of outer membrane vesicles. Uptake of released toxin and vesicles by gastric epithelium. J Pathol. 1999;188(2):220–226. | ||
Dutta S, Iida KI, Takade A, Meno Y, Nair GB, Yoshida SI. Release of Shiga toxin by membrane vesicles in Shigella dysenteriae serotype 1 strains and in vitro effects of antimicrobials on toxin production and release. Microbiol Immunol. 2004;48(12):965–969. | ||
Wai SN, Lindmark B, Söderblom T, et al. Vesicle-mediated export and assembly of pore-forming oligomers of the enterobacterial ClyA cytotoxin. Cell. 2003;115(1):25–35. | ||
Nowotny A, Behling UH, Hammond B, et al. Release of toxic microvesicles by Actinobacillus actinomycetemcomitans. Infect Immun. 1982;37(1):151–154. | ||
Bielaszewska M, Rter C, Bauwens A, et al. Host cell interactions of outer membrane vesicle-associated virulence factors of enterohemorrhagic Escherichia coli O157: intracellular delivery, trafficking and mechanisms of cell injury. PLoS Pathog. 2017;13(2):e1006159. | ||
Dorward DW, Garon CF. DNA is packaged within membrane-derived vesicles of gram-negative but not gram-positive bacteria. Appl Environ Microbiol. 1990;56(6):1960–1962. | ||
Song T, Mika F, Lindmark B, et al. A new Vibrio cholerae sRNA modulates colonization and affects release of outer membrane vesicles. Mol Microbiol. 2008;70(1):100–111. | ||
Kadurugamuwa JL, Beveridge TJ. Virulence factors are released from Pseudomonas aeruginosa in association with membrane vesicles during normal growth and exposure to gentamicin: a novel mechanism of enzyme secretion. J Bacteriol. 1995;177(14):3998–4008. | ||
Li Z, Clarke AJ, Beveridge TJ. A major autolysin of Pseudomonas aeruginosa: subcellular distribution, potential role in cell growth and division and secretion in surface membrane vesicles. J Bacteriol. 1996;178(9):2479–2488. | ||
Bomberger JM, MacEachran DP, Coutermarsh BA, Ye S, O’Toole GA, Stanton BA. Long-distance delivery of bacterial virulence factors by Pseudomonas aeruginosa outer membrane vesicles. PLoS Pathog. 2009;5(4):e1000382. | ||
Kadurugamuwa JL, Beveridge TJ. Bacteriolytic effect of membrane vesicles from Pseudomonas aeruginosa on other bacteria including pathogens: conceptually new antibiotics. J Bacteriol. 1996;178(10):2767–2774. | ||
Bielecka E, Scavenius C, Kantyka T, et al. Peptidyl arginine deiminase from Porphyromonas gingivalis abolishes anaphylatoxin C5a activity. J Biol Chem. 2014;289(47):32481–32487. | ||
Fulsundar S, Harms K, Flaten GE, Johnsen PJ, Chopade BA, Nielsen KM. Gene transfer potential of outer membrane vesicles of Acinetobacter baylyi and effects of stress on vesiculation. Appl Environ Microbiol. 2014;80(11):3469–3483. | ||
Shen Y, Torchia ML, Lawson GW, Karp CL, Ashwell JD, Mazmanian SK. Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe. 2012;12(4):509–520. | ||
Stentz R, Osborne S, Horn N, et al. A bacterial homolog of a eukaryotic inositol phosphate signaling enzyme mediates cross-kingdom dialog in the mammalian gut. Cell Rep. 2014;6(4):646–656. | ||
Craven DE, Peppler MS, Frasch CE, Mocca LF, McGrath PP, Washington G. Adherence of isolates of Neisseria meningitidis from patients and carriers to human buccal epithelial cells. J Infect Dis. 1980;142(4):556–568. | ||
Stephens DS, Edwards KM, Morris F, McGee ZA. Pili and outer membrane appendages on Neisseria meningitidis in the cerebrospinal fluid of an infant. J Infect Dis. 1982;146(4):568. | ||
Vidakovics ML, Jendholm J, Mörgelin M, et al. B cell activation by outer membrane vesicles – a novel virulence mechanism. PLoS Pathog. 2010;6(1):e1000724. | ||
Schaar V, Nordstrom T, Morgelin M, Riesbeck K. Moraxella catarrhalis outer membrane vesicles carry β-lactamase and promote survival of Streptococcus pneumoniae and Haemophilus influenzae by inactivating amoxicillin. Antimicrob Agents Chemother. 2011;55(8):3845–3853. | ||
Ren D, Nelson KL, Uchakin PN, Smith AL, Gu XX, Daines DA. Characterization of extended co-culture of non-typeable Haemophilus influenzae with primary human respiratory tissues. Exp Biol Med. 2012;237(5):540–547. | ||
Kaparakis-Liaskos M, Ferrero RL. Immune modulation by bacterial outer membrane vesicles. Nat Rev Immunol. 2015;15(6):375–387. | ||
Lapinet JA, Scapini P, Calzetti F, Perez O, Cassatella MA. Gene expression and production of tumor necrosis factor alpha, interleukin-1beta (IL-1beta), IL-8, macrophage inflammatory protein 1alpha (MIP-1alpha), MIP-1beta, and gamma interferon-inducible protein 10 by human neutrophils stimulated with group B meningococcal outer membrane vesicles. Infect Immun. 2000;68(12):6917–6923. | ||
Durand V, MacKenzie J, De Leon J, et al. Role of lipopolysaccharide in the induction of type I interferon-dependent cross-priming and IL-10 production in mice by meningococcal outer membrane vesicles. Vaccine. 2009;27(13):1912–1922. | ||
Alaniz RC, Deatherage BL, Lara JC, Cookson BT. Membrane vesicles are immunogenic facsimiles of Salmonella typhimurium that potently activate dendritic cells, prime B and T cell responses, and stimulate protective immunity in vivo. J Immunol. 2007;179(11):7692–7701. | ||
Talwar N, Jain NK. Erythrocytes as carriers of metronidazole: in-vitro characterization. Drug Dev Ind Pharm. 1992;18(16):1799–1812. | ||
Annese V, Latiano A, Rossi L, et al. Erythrocytes-mediated delivery of dexamethasone in steroid-dependent IBD patients – a pilot uncontrolled study. Am J Gastroenterol. 2005;100(6):1370–1375. | ||
Dou H, Grotepas CB, McMillan JM, et al. Macrophage delivery of nanoformulated antiretroviral drug to the brain in a murine model of neuroAIDS. J Immunol. 2009;183(1):661–669. | ||
Choi MR, Stanton-Maxey KJ, Stanley JK, et al. A cellular Trojan Horse for delivery of therapeutic nanoparticles into tumors. Nano Lett. 2007;7(12):3759–3765. | ||
Studeny M, Marini FC, Dembinski JL, et al. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J Natl Cancer Inst. 2004;96(21):1593–1603. | ||
Roger M, Clavreul A, Venier-Julienne MC, et al. Mesenchymal stem cells as cellular vehicles for delivery of nanoparticles to brain tumors. Biomaterials. 2010;31(32):8393–8401. | ||
Schmidt U, Günther C, Rudolph R, Böhm G. Protein and peptide delivery via engineered polyomavirus-like particles. FASEB J. 2001;15(9):1646–1648. | ||
Zhuang X, Xiang X, Grizzle W, et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol Ther. 2011;19(10):1769–1779. | ||
Haney MJ, Klyachko NL, Zhao Y, et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J Control Release. 2015;207:18–30. | ||
Sun D, Zhuang X, Xiang X, et al. A novel nanoparticle drug delivery system: the anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol Ther. 2010;18(9):1606–1614. | ||
Gujrati V, Kim S, Kim SH, et al. Bioengineered bacterial outer membrane vesicles as cell-specific drug-delivery vehicles for cancer therapy. ACS Nano. 2014;8(2):1525–1537. | ||
Gankema H, Wensink J, Guinée PA, Jansen WH, Witholt B. Some characteristics of the outer membrane material released by growing enterotoxigenic Escherichia coli. Infect Immun. 1980;29(2):704–713. | ||
Koeppen K, Hampton TH, Jarek M, et al. A novel mechanism of host-pathogen interaction through sRNA in bacterial outer membrane vesicles. PLoS Pathog. 2016;12(6):e1005672. | ||
Post DM, Zhang D, Eastvold JS, Teghanemt A, Gibson BW, Weiss JP. Biochemical and functional characterization of membrane blebs purified from Neisseria meningitidis serogroup B. J Biol Chem. 2005;280(46):38383–38394. | ||
McMahon KJ, Castelli ME, Vescovi EG, Feldman MF. Biogenesis of outer membrane vesicles in Serratia marcescens is thermoregulated and can be induced by activation of the Rcs phosphorelay system. J Bacteriol. 2012;194(12):3241–3249. | ||
Gamalier JP, Silva TP, Zarantonello V, Dias FF, Melo RC. Increased production of outer membrane vesicles by cultured freshwater bacteria in response to ultraviolet radiation. Microbiol Res. 2017;194:38–46. | ||
Arigita C, Jiskoot W, Westdijk J, et al. Stability of mono-and trivalent meningococcal outer membrane vesicle vaccines. Vaccine. 2004;22(5):629–642. | ||
Allan ND, Beveridge TJ. Gentamicin delivery to Burkholderia cepacia group IIIa strains via membrane vesicles from Pseudomonas aeruginosa PAO1. Antimicrob Agents Chemother. 2003;47(9):2962–2965. | ||
Rumbo C, Fernández-Moreira E, Merino M, et al. Horizontal transfer of the OXA-24 carbapenemase gene via outer membrane vesicles: a new mechanism of dissemination of carbapenem resistance genes in Acinetobacter baumannii. Antimicrob Agents Chemother. 2011;55(7):3084–3090. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.