")
Back to Journals » Journal of Blood Medicine » Volume 5
Back pain: the sole of presentation of sickle cell disease
Authors Osman S, Khan S, Hendaus M
Received 17 February 2014
Accepted for publication 26 March 2014
Published 8 May 2014 Volume 2014:5 Pages 49—53
DOI https://doi.org/10.2147/JBM.S62570
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Samar Osman, Shabina Khan, Mohamed A Hendaus
General Pediatrics, Department of Pediatrics, Hamad Medical Corporation, Doha, Qatar
Abstract: Diagnosing back pain in children and adolescents can be a challenge to health care providers. Although studies show that more than half of the cases of back pain in children are of non-organic cause, missing the right diagnosis could be detrimental. We present a case of lower back pain in a ten-year-old male whom we eventually diagnosed with hemoglobin SE mutation, which responded well to pain management. Hence, sickle cell disease with vaso-occlusive crisis should be incorporated into the list of differential diagnoses in children with back pain.
Keywords: hemoglobin, HbSE, case report, paediatrics
Introduction
Back pain in children used to be a dreaded symptom; however, currently more than half of these cases are being labeled as non specific musculoskeletal pain, or pain with no organic cause.1–4
The incidence of back pain in children increases with age, from a mere 7% of children reporting back pain at 12 years of age, to 50% complaining of this symptom by the age of 20 in both sexes.5
Nevertheless, it should be emphasized that despite the fact that most cases have a benign etiology, it is important to effectively evaluate these children for any underlying serious health problems like malignancy, infection, or rheumatological diseases. Children with sickle cell disease might present, although rarely, with back pain as a part of a vaso-occlusive crisis.
We describe a ten-year-old Arab male child, who presented to the emergency department with intermittent back pain of 12 months’ duration. A thorough investigation revealed that the patient was heterozygous for hemoglobin S and hemoglobin E (HbSE), which had not been previously diagnosed.
Case presentation
A ten-year-old Qatari boy, of Arab ethnicity, presented to our emergency department with a history of intermittent lower back pain of 12 months’ duration. The pain was reported by the child as severe in nature, intermittent, restricting his daily routine activity, and with partial response to anti-inflammatory medications like 200 mg of ibuprofen. There was no history of joint involvement. The child’s family had sought medical advice repeatedly in the past on an outpatient basis, without any specific work up or diagnosis. The family denied any history of fever, trauma, vigorous excercise, weight loss, or any gastro-intestinal symptoms. There was also a negative history of any raw milk ingestion, chronic cough, or contact with patients known to have tuberculosis. The patient had no previous hospitalizations or blood transfusions. Despite parental consanguinity, there was no family medical history suggestive of sickle cell disease symptoms in any of the first or second degree relatives, nor were the parents aware of any known carrier for Hemoglobin S or thalassemia in the family.
On examination, the child was noted to be in pain that was assessed by numerical rating scale as 8/10. General examination did not show any signs of pallor or jaundice. The patient was afebrile and hemodynamically stable with a temperature of 37.5C orally, heart rate of 100 beats per minute, respiratory rate of 20 breaths per minute, and blood pressure of 120 mmHg/85 mmHg.
The patient’s height and weight were at the 50th percentile for his age and sex. Muskuloskeletal examination revealed tenderness upon palpation of the entire lumbo-sacral area, with no other external signs of inflammation such as erythema or swelling. He had limited movement of the trunk, with pain elicited during forward and lateral flexion. There was no spine deformity. There was no hepatosplenomegaly or lymphadenopathy, and the rest of the physcial exam was unremarkbale.
The patient was admitted to the general pediatric ward for pain management and investigation of his recurrent back pain. The admitting differential diagnosis included chronic osteomyelitis involving the lumbosacral vertebrae, primary and secondary bone neoplams, and possible undiagnosed rheumatological conditions. The patient was started on regular intravenous acetaminophen along with oral ibuprofen to manage his pain, to which he responded rapidly.
Initial laboratory results were as follows: white blood cells 9,500/uL (neutrophils 90%; lymphocytes 6.1%; monocytes 3.8%; and basophils 0.1%); hemoglobin 12.5 g/dL; platelets 217,000/uL, mean corpuscular volume of 70.8 fL and mean corpuscular hemoglobin of 23.6 picograms/cell, with a normal reticulocyte count. The peripheral smear showed microcyic hyppochromic red blood cells with mild neutrophil leukocytosis, with rare immature granulocytes and some toxic features.
C-reactive protein was less than 5 mg/L, and erythrocyte sedimentation rate was 9 mm/hour. Serum electrolytes were normal. Urine and blood culture were negative as well as the QuantiFERON test, salmonella, and brucella serology.
Antero–posterior spine radiograph revealed straightening of the lumbar lordosis with multiple fish mouth-appearing vertebral bodies from L2–L5, diffusely porotic bone with accentuated medullary pattern, and normal intervertebral spaces with slight widening of the sacro-iliac joint space. AP chest radiograph was essentially normal, with no mediastinal mass or hilar lymphadenopathy.
Magnetic resonance imaging (MRI) of the spine (Figure 1) and pelvis with contrast was requested on the second day post-admission. It showed multiple bone marrow-replacing lesions involving the thoracic, lumbar, and sacral vertebrae extending from T2 vertebral level downwards to the lower sacral segment. These displayed a predominantly low signal intensisty in T1-weighted images and high signal intensity in T12 fluid-attenuated inversion recovery-weighted images, with a patchy homogenous pattern of enhancement. There was an associated reduction of the height of the vertebral bodies, mainly in L3, L4, L5, and S1 levels.
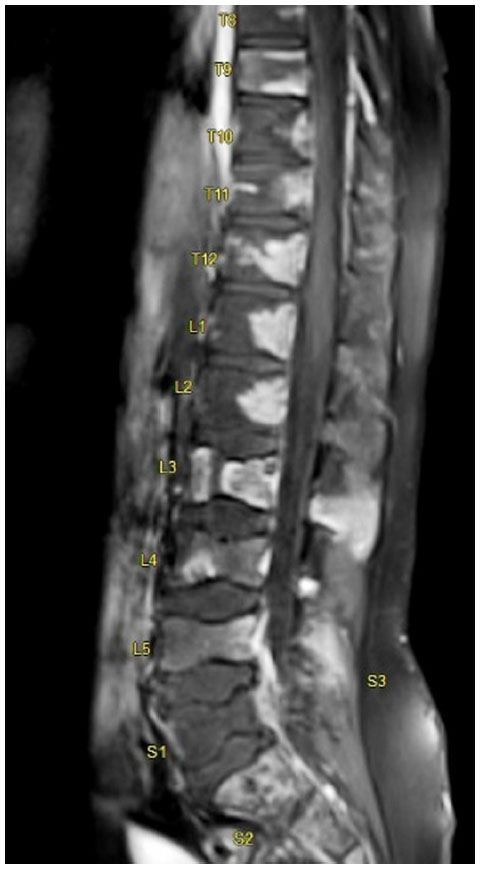 | Figure 1 Post contrast saggital magnetic resonance imaging (MRI) picture of the whole spine showing multiple collapsed lumbar vertebral bodies (L3–L5) and non-enhancing areas involving vertebral bodies at dorsal and lumbar levels, suggesting infarcts. |
The MRI findings prompted a consultation with the pediatric hematology and oncology services to discuss the possibility of malignant inflitration, such as with leukemia and lymphoma.
A bone marrow biopsy and an aspirate were conducted, both of which were normal.
MRI of the abdomen was conducted to exclude the possibility of an underlying malignancy. The image showed mild hepatomegaly, with a liver span of 15 cm with moderate splenomegaly, no focal lesions, and no evidence of any abdominal lympahdeopathy or mass lesion in the retroperotenium or adrenal glands.
On day 7 post-admission, the result of the hemoglobin electrophoresis that was requested on admission revealed HbF =2.9%, HbA2 =2%, HbA =0%, HbS =66.8%, and HbE =28.3%, which confirmed the diagnosis of sickle cell disease with HbSE component.
Our patient was discharged with a diagnosis of sickle cell disease (HbSE subtype) on oral folic acid 1 mg once a day and ibuprofen 400 mg per mouth every six hours if needed. In addition, our patient was started on oral penicillin VK 250 mg every 12 hours at the hematologist’s discretion, although there are no specific guidelines clarifying the need for penicillin prophylaxis in HbSE disease.6 The patient’s parents refused hemoglobin electrophoresis for themselves as well as for the patient’s siblings.
Follow up
The patient was seen in the pediatric out patient clinic after 2 months and was in a stable condition. In addition, he has been seen twice since then in the pediatric hematology clinic and reportedly has good pain control with ibuprofen as needed. The patient’s parents stated that patient had used ibuprofen only twice in 2 months.
Discussion
Benign musculoskeletal disease and trauma are responsible for the majority of cases of back pain in children and adolescents. The other causes of back pain in the pediatric age group include infection, neoplastic lesions, rheumatologic diseases, and miscellaneous causes like vaso-occlusive crisis, nephrolithiasis, and others (Table 1).2,4
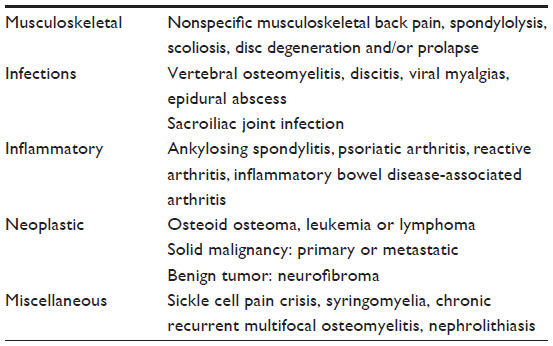 | Table 1 Causes of back pain in children and adolescents |
Previous studies have established a positive correlation between female sex, increased time watching television and playing sports, and family history with back pain in children.7,8 However, the relationship between back pain and heavy school backpacks remains controversial.9,10
Back pain in adolescents needs a special mention, as the recent literature has emphasized a unique set of etiologies which need to be considered in this age group, including spondylolysis, disc herniation, and back pain in adolescent athletes.4
Sickle cell disease is an autosomal recessive disorder that includes all patients who have the sickle mutation (HbS) plus a second beta globin gene mutation, the combination of which results in clinical sickling.11
Sickle cell anemia, or HbSS disease, is the most common form of SCD, in which the patient is homozygous for HbS. The other types of SCD include, but are not limited to, sickle thalassemias, HbSC, HbSD, and HbSE.12
Hemoglobin (Hb)E is well known is some parts of the world and its frequency can reach up to 60% in many regions of Laos, Thailand, and Cambodia. The mutation usually leads to HbE trait and HbEE, which are both considered mild diseases. However, the combination of HbE and HbS (HbSE) leads to a syndrome similar to sickle β+ thalassemia.13
Affected persons with HbSE disease might develop vaso-occlusive crisis resembling SCD.6 The exact pathophysiology of sickling in patients with HbSE is not yet well known, but it has been postulated that it depends on the high percentage of the HbS component (more than 60%).14
The hemoglobin electrophoresis of our patient showed a component of 66.8% of HbS. Moreover, the bone marrow is prone to infarction due to the occlusion caused by the circulation of sickle cells.15,16
In patients with sickle cell disease, recurrent vaso-occlusive crisis coupled with chronic tissue hypoxia manifest as various skeletal tissue changes, which include bone and bone marrow infarction, bone marrow hyperplasia, osteomyelitis, and secondary growth defects.17
MRI of the spine is a crucial investigation in sickle cell patients presenting with back pain as it enables the clinician to detect any early subtle skeletal changes.18 Magnetic resonance imaging with contrast enhancement is an indispensable mode of imaging because it can differentiate between osteomyelitis and bone infarction.19
Chronic bone infarcts in MRI of the spine will manifest as focal areas of decreased signal intensity on both T1- and T2-weighted images.20
On a plain X-ray of the spine, despite its inferiority to MRI, the following changes may be visualized in SCD patients: H-shaped vertebrae due to bone marrow infarction, biconcave vertebrae and pathological fractures secondary to de-ossification as a result of marrow hyperplasia, and decreased vertebral body height as a result of abnormal bone maturation.17
Our patient’s primary suspected diagnosis at admission was osteomyelitis versus malignancy. However, the index of suspicion for the possibility of painful crisis secondary to an undiagnosed sickle cell disease remained high, as our patient’s back pain responded rapidly to supportive and pain management, and also since the patient was of at risk Arab ethnicity. Our patient’s diagnosis of sickle cell disease of HbSE subtype was confirmed by hemoglobin electrophoresis.
There is a lack of sufficient literature available worldwide on HbSE disease. Patients with HbSS disease, which is far more prevalent than HbSE, are diagnosed easily as they are typically anemic and often present early, with frequent admissions for recurrent painful vaso-occlusive episodes.21 In contrast, HbSE is more likely to present with painful crisis in adulthood, and is relatively asymptomatic in children.14,22
However, despite a more benign course compared to HbSS, there have been reports of severe clinical presentations in HbSE patients, such as vaso-occlusive crisis and bone marrow infarctions.23
A study in Oman, where HbSE is the second most common hemoglobinopathy, revealed that 50% of the studied population with the disease were asymptomatic, while the other half had a course similar to HbSS disease, including severe vaso-occlusive skeletal pain.23
Remarkably, sickling crisis in HbSE disease can occur in the absence of sickle cells in the peripheral smear,22 which was the case in our patient; hence the significance of conducting hemoglobin electrophoresis for patients suspected of suffering from HbSE disease needs to be emphasized.
Conclusion
Sickle cell disease with vaso-occlusive crisis is one of the differential diagnoses that needs to be considered in children with back pain, even in a previously healthy child, as certain subtypes of SCD like HbSE disease can have a late presentation.
Disclosure
No funds were provided to conduct the study. However, the Medical Research Center at Hamad Medical Corporation covers publication fees as well as abstract presentation if required. The authors report no conflicts of interest in this work.
References
Combs JA, Caskey PM. Back pain in children and adolescents: a retrospective review of 648 patients. South Med J. 1997;90:789–792. | |
Feldman DS, Hedden DM, Wright JG. The use of bone scan to investigate back pain in children and adolescents. J Pediatr Orthop. 2000;20(6):790–795. | |
Bhatia NN, Chow G, Timon SJ, Watts HG. Diagnostic modalities for the evaluation of pediatric back pain: a prospective study. J Pediatr Orthop. 2008;28(2):230–233. | |
Kim HJ, Green DW. Adolescent back pain. Curr Opin Pediatr. 2008;20(1):37–45. | |
Leboeuf-Yde C, Kyvik KO. At what age does low back pain become a common problem? A study of 29,424 individuals aged 12–41 years. Spine (Phila Pa 1976). 1998;23(2):228–234. | |
Rayburg M, Kalinyak KA, Towbin AJ, Baker PB, Joiner CH. Fatal bone marrow embolism in a child with hemoglobin SE disease. Am J Hematol. 2009;85(3):182–184. | |
Balagué F, Skovron ML, Nordin M, Dutoit G, Pol LR, Waldburger M. Low back pain in schoolchildren. A study of familial and psychological factors. Spine (Phila Pa 1976). 1995;20(11):1265–1270. | |
Troussier B, Davoine P, de Gaudemaris R, Fauconnier J, Phelip X. Back pain in school children. A study among 1178 pupils. Scand J Rehabil Med. 1994;26(3):143–146. | |
Jones GT, Watson KD, Silman AJ, Symmons DP, Macfarlane GJ. Predictors of low back pain in British schoolchildren: a population-based prospective cohort study. Pediatrics. 2003;111(4 Pt 1):822–828. | |
Reneman MF, Poels BJ, Geertzen JH, Dijkstra PU. Back pain and backpacks in children: biomedical or biopsychosocial model? Disabil Rehabil. 2006;28(20):1293–1297. | |
McCavit TL. Sickle cell disease. Pediatr Rev. 2012;33(5):195–204. | |
Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010; 376(9757):2018–2031. | |
Vichinsky E. Hemoglobin e syndromes. Hematology Am Soc Hematol Educ Program. 2007:79–83. | |
Masiello D, Heeney MM, Adewoye AH, et al. Hemoglobin SE disease: a concise review. Am J Hematol. 2007;82(7):643–649. | |
Rossi P, Curiel M, Demoux AL, et al. Bone marrow necrosis and sickle cell crisis associated with double heterozygosity for HbS and HbOARAB. Am J Hematol. 2011;86(3):309–310. | |
Milner PF, Brown M. Bone marrow infarction in sickle cell anemia: correlation with hematologic profiles. Blood. 1982;60(6):1411–1419. | |
Ramirez I, Choi MH, Asrat W. Sickle Cell Anemia Skeletal Imaging [webpage on the Internet]. Medscape [cited August 5, 2013]. Available from: http://emedicine.medscape.com/article/413542-overview. Accessed March 18, 2014. | |
Pratesi A, Medici A, Bresci R, Micheli A, Barni S, Pratesi C. Sickle cell-related bone marrow complications: the utility of diffusion-weighted magnetic resonance imaging. J Pediatr Hematol Oncol. 2013;35(4):329–330. | |
Umans H, Haramati N, Flusser G. The diagnostic role of gadolinium enhanced MRI in distinguishing between acute medullary bone infarct and osteomyelitis. Magn Reson Imaging. 2000;18(3):255–262. | |
Watanabe M, Saito N, Nadgir RN, et al. Craniofacial bone infarcts in sickle cell disease: clinical and radiological manifestations. J Comput Assist Tomogr. 2013;37(1):91–97. | |
Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease. Rates and risk factors. N Eng J Med. 1991;325(1):11–16. | |
Rey KS, Unger CA, Rao SP, Miller ST. Sickle cell-hemoglobin E disease: clinical findings and implications. J Pediatr. 1991;119(6):949–951. | |
Knox-Macaulay HH, Ahmed MM, Gravell D, Al-Kindi S, Ganesh A. Sickle cell-haemoglobin E (HbSE) compound heterozygosity: a clinical and haematological study. Int J Lab Hematol. 2007;29(4):292–301. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.