")
Back to Journals » ImmunoTargets and Therapy » Volume 3
B cell receptor pathway in chronic lymphocytic leukemia: specific role of CC-292
Received 3 October 2013
Accepted for publication 13 November 2013
Published 24 January 2014 Volume 2014:3 Pages 29—38
DOI https://doi.org/10.2147/ITT.S37419
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Jon E Arnason,1 Jennifer R Brown2
1Beth Israel Deaconess Medical Center, 2CLL Center, Department of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA, USA
Abstract: Chronic lymphocytic leukemia (CLL) is the most common adult leukemia. The current treatment paradigm involves the use of chemoimmunotherapy, when patients develop an indication for therapy. With this strategy, a majority of patients will obtain a remission, though cure remains elusive. While treatable, the majority of CLL patients will die of complications of their disease. Recent advances in the understanding of the importance of the B cell receptor (BCR) pathway in CLL have led to the development of a number of agents targeting this pathway. In this review, we discuss recent developments in the targeting of the BCR pathway, with a focus on CC-292. CC-292 covalently binds to Bruton's tyrosine kinase, a key mediator of BCR signaling, and has demonstrated preclinical and clinical activity in CLL, with acceptable tolerability. Based on the success of CC-292 and other inhibitors of the BCR pathway, these agents are being investigated in combination with standard therapy, with the hope that they will increase the depth and length of response, without significant toxicity.
Keywords: Bruton's tyrosine kinase inhibitor, ibrutinib
Introduction
Chronic lymphocytic leukemia (CLL) is the most common adult leukemia, with an estimated incidence in the United States of 15,680 in 2013.1 The disease is characterized by the proliferation and accumulation in the periphery, lymph nodes, and bone marrow, of a clonal B cell population expressing cluster of differentiation (CD5), CD19, and CD23.2 The course of the disease is highly variable, and while many patients may go for years from diagnosis until treatment, the majority of patients will eventually die of complications related to CLL.
Patients with an indication for treatment, as defined by the 2008 International Workshop on CLL Guidelines,3 are usually managed with chemoimmunotherapy, most commonly with rituximab in combination with fludarabine and cyclophosphamide (FCR),4,5 or rituximab in combination with bendamustine (BR).6 While a majority of patients will respond to upfront treatment (95% overall response rate with FCR), the disease will invariably recur. At recurrence, patients are retreated with their initial regimen, if the response duration was sufficient, or receive a different, second-line regimen. The cumulative effects of persistent CLL and recurrent therapy result in an increased risk of infection, the leading cause of death in CLL. With the above strategy, CLL can be controlled, but toxicity related to therapy can be significant, and cure remains elusive. The only current therapy that results in long-term remission is the use of allogeneic bone marrow transplantation.7 However, the morbidity and mortality of allogeneic transplantation is prohibitive, and its use is generally limited to patients with high-risk cytogenetics or very refractory disease.
New targeted therapies for CLL with limited toxicity are needed to better treat this chronic condition. The recent evolution of treatment in many other malignancies has been the use of rationally designed agents that target the cell signaling pathways essential to tumor growth. The B cell receptor signaling pathway (BCR) represents an exciting potential target in CLL. Signaling through the BCR is thought to be deregulated in CLL and a key mediator of CLL survival and proliferation.8 In this review, we will examine the role of BCR signaling in CLL and the development of recent therapies targeting BCR signaling, with a focus on CC-292, a specific inhibitor of Bruton’s tyrosine kinase (BTK), currently being investigated in CLL and other B cell lymphoproliferative disorders.
B cell receptor signaling background
BTK is an essential mediator of BCR signaling, expressed in normal healthy B but not T lymphocytes.9 Mutation in BTK results in the human primary immune deficiency disease, X-linked agammaglobulinemia (XLA).10 The B cells in patients with XLA cannot differentiate, resulting in a reduction of mature B cells and the inability to produce immunoglobulins. This disease demonstrates the necessity of BTK in B cell development and function. In healthy B cells, antigenic stimulation of the BCR results in CD79a- and CD79b-recruitment, and activation of the spleen tyrosine kinase (SYK) and LYN kinases, which phosphorylate immunoreceptor tyrosine-based activation motifs (ITAMs) on the cytoplasmic immunoglobulin domains of the receptor. ITAM phosphorylation begins a cascade of activation, including the activation of BTK and phosphoinositide 3′-kinase (PI3K). Activated BTK phosphorylates, and thereby activates, phospholipase C gamma 2 (PLCγ2), which, through multiple mediators, promotes the downstream release of intracellular Ca2+ stores and propagation of the BCR signal, resulting in increased proliferation, survival, and avoidance of apoptosis. These processes are mediated by the upregulation of transcription factors, including nuclear-factor κB (NF-κB), resulting in a number of cellular processes, including proliferation, chemokine-mediated migration, and integrin activation (Figure 1).11
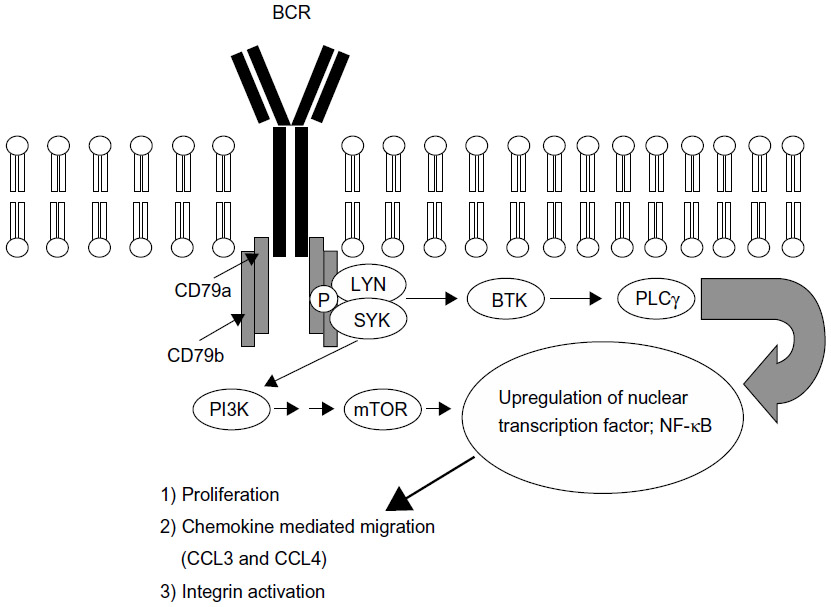 | Figure 1 Antigenic stimulation of the BCR recruits CD79a and CD79b, and activates SYK and LYN kinase, resulting in the phosphorylation of cytoplasmic ITAMs on the immunoglobulin domains of the receptor. The ITAM phosphorylation begins a cascade of activation involving BTK and PI3K. Activated BTK promotes the downstream release of intracellular Ca2+ stores and propagation of the BCR signal, resulting in increased proliferation, survival, and avoidance of apoptosis, mediated by the upregulation of transcription factors, including NF-κB. |
CLL is dependent on signaling through the BCR for the avoidance of apoptosis and promotion of proliferation and activation. The mechanism by which BCR signaling is activated remains to be determined, but there may be antigen-independent12 and -dependent pathways,13−16 secondary to microbial or autologous antigens. Consistent with this hypothesis, in about one-third of CLL cases, there is a limited repertoire of B cell receptors.17,18 In addition to its role in B cell survival, BTK is involved in pathways of B cell migration, through the expression of the adhesion molecules chemokine receptor (CXCR)4 and CXCR5 and their interaction with the chemokines CXCL12 and CXCL13, respectively.19 BTK is important for the activation of integrin-mediated adhesion, which promotes the migration of B cells into the lymph node follicles and germinal center organization.20
Because of its central role in BCR signaling and importance for B cell development and function, BTK has been identified as a promising target for drug development in both B cell malignancies and autoimmune diseases.21
Targeting BCR in CLL
In the last 5 years, many new agents targeting the BCR pathway have been investigated in clinical trials for CLL (Figure 2). Here we briefly summarize the most recent data related to many of these agents. All of these agents have demonstrated reasonable toxicity and clinical responses in CLL patients.
| Figure 2 The BCR pathway has been targeted in CLL, at multiple different sites. |
Ibrutinib
Ibrutinib targets BTK and has been investigated clinically in a number of B cell malignancies. Ibrutinib binds covalently to cysteine-481 in the active site of BTK, inhibiting its activity at a half maximal inhibitory concentration (IC50) of 0.5 nM.22 In addition to its inhibitory activity against BTK, ibrutinib has shown measurable activity against 19 other kinases.22 In a biochemical study, ibrutinib has demonstrated strong inhibition of Tec family members with a cognate cysteine as well as significant inhibition, likely noncovalent, of a number of kinases that do not have a cognate cysteine (Table 1). In a cellular study, ibrutinib appeared to selectively inhibit BTK, consistent with preferential covalent binding to BTK. In an in vitro study, ibrutinib decreased CLL migration, had a small effect on stimulated CLL proliferation, and demonstrated improved survival in a CLL animal model.23 In the initial Phase I trial of ibrutinib, 56 patients with B cell malignancies, including CLL, were enrolled to receive treatment with either intermittent dosing (28 days on, 7 days off) or continuous dosing. The agent was well tolerated, with no dose limiting toxicities. In the 14 patients with CLL, the overall response rate (ORR) was 79%.24 Given the success of the Phase I trial, ibrutinib is being investigated as a single agent in relapsed and refractory CLL and in combination studies with rituximab (NCT01520519),25 lenalidomide (NCT01886859),26 BR or FCR (the FCR arm closed early due to poor accrual after only three patients were enrolled) (NCT01611090),27,28 and ofatumumab (NCT01217749).29
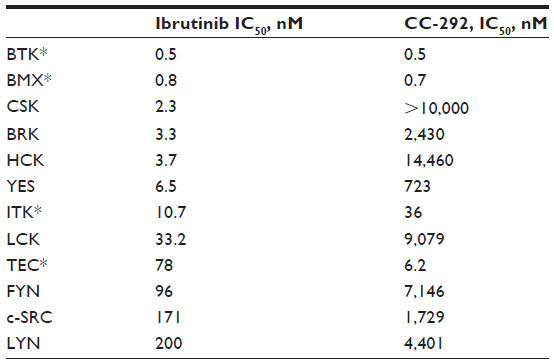 | Table 1 Kinase inhibitory biochemical activity of ibrutinib and CC-29222,47 |
Most recently, the results of a Phase IB−II multicenter study of ibrutinib in patients with relapsed and refractory CLL were reported in the New England Journal of Medicine.30 A total of 85 patients were enrolled. The toxicity was modest, with the majority of adverse events being grade 1 or 2 including diarrhea, fatigue, and upper respiratory infections. Only two patients stopped treatment related to toxicities. The overall response rate (ORR) was 71%, with an additional 18% of patients obtaining a partial nodal response, with lymphocytosis. The responses did not vary according to the traditional high-risk prognostic features. While the responses have been significant, several patients have developed resistance to ibrutinib. Ribonucleic acid (RNA) and whole-exome sequencing performed on patients progressing after ibrutinib demonstrated single-nucleotide variations at cysteine 481 on BTK (the binding site of ibrutinib) and a potential gain of function in PLCγ, downstream of BTK.31
One interesting aspect of the responses to ibrutinib in CLL was a transient lymphocytosis observed in the setting of decreasing lymphadenopathy. It is thought that BTK inhibition results in a migration of CLL cells from the protected lymph node32 and bone marrow niches into the periphery, where the cell survival is shortened. This phenomenon has been observed, to a greater or lesser degree, with all agents targeting the BCR signaling pathway and has led to efforts to refine the definition of response in CLL to include a category for partial response (PR) with lymphocytosis, to account for this phenomenon.33,34
Based on the promising early experiences with ibrutinib in CLL and mantle cell lymphoma, on July 10th, 2013, a New Drug Application was submitted to the Food and Drug Administration (FDA) for use in patients with relapsed and refractory CLL and previously treated mantle cell lymphoma. Additionally, CLL with the high-risk deletion of 17p has been granted breakthrough designation by the FDA.
Idelalisib
The delta isoform of PI3K is selectively expressed in hematopoietic cells and has increased enzymatic activity in CLL. Idelalisib, a PI3K delta specific inhibitor, has demonstrated significant in vitro inhibition of PI3K in CLL, leading to apoptosis of ex vivo CLL cells.35,36 Data from a Phase I trial in 54 patients with relapsed and refractory CLL demonstrated a nodal response rate of 81%, with an ORR of 72%: 39% with a PR and 33% meeting the revised criteria of PR with treatment-induced lymphocytosis.37 Idelalisib has been investigated in Phase I combination trials with bendamustine with or without rituximab, with nodal responses in 82%−87% of patients,38 as well as in combination with ofatumumab and with chlorambucil. Ongoing Phase III trials are investigating idelalisib in combination with rituximab versus rituximab alone (NCT01539512),39 in combination with ofatumumab versus ofatumumab alone, and in combination with BR versus BR alone (NCT01569295).40
SAR245409
SAR245409, a pan PI3K inhibitor and mammalian target of rapamycin (mTOR) inhibitor, is being investigated in a Phase II clinical trial in CLL and follicular and other non-Hodgkin lymphomas, and has demonstrated acceptable tolerability.41
IPI-145
IPI-145, a gamma- and delta-specific PI3K inhibitor, is being investigated in a Phase I trial in patients with advanced hematologic malignancies, and in a preliminary analysis, IPI-145 appeared to be well tolerated, with a nodal or PR in nine of eleven (82%) CLL patients.42
Dasatinib
LYN, the switch molecule that couples the B cell receptor to downstream signaling, is overexpressed in CLL and appears to be constitutively active.43 Fifteen CLL patients who had failed at least one fludarabine-containing regimen were enrolled to treatment with dasatinib, in an effort to target LYN. A PR was noted in three patients (20%), and five additional patients had nodal responses. Pharmacodynamic studies indicated apoptosis in the peripheral blood CLL cells, with an associated downregulation of spleen tyrosine kinase (SYK).44
Fostamatinib and everolimus
SYK and mTOR have been targeted in CLL patients with relapsed and refractory disease, with fostamatinib and everolimus, respectively, with nodal response rates of 45%−55%.45,46
CC-292, a specific BTK inhibitor
CC-292, previously known as AVL-292, is a potent, selective inhibitor of BTK that is currently being investigated as a treatment for CLL. Like ibrutinib, CC-292 was rationally designed to covalently bind with high affinity to cysteine 481 in BTK, blocking the adenosine triphosphate (ATP) binding pocket of the enzyme. A series of in vitro and animal experiments has demonstrated CC-292’s ability to covalently and selectively inhibit BTK, resulting in the decreased viability of CLL cells in response to anti-IgM stimulation and nurse like cell (NLC) stimulation, and decreased migration in response to chemotactic signals. A Phase IA trial in healthy volunteers demonstrated acceptable safety and tolerability, with rapid oral absorption and BTK occupancy that persisted beyond the normalization of plasma concentration. The early results from the Phase IB trial demonstrate tolerability, nodal responses, and lymphocytosis, as has been observed with other inhibitors of the BCR signaling pathway.47
In vitro experience
In vitro studies have demonstrated that CC-292 is a potent, selective inhibitor of BTK.47 Biochemically, CC-292 was tested against full-length recombinant BTK, using the Omnia® (Life Technologies, Carlsbad, CA, USA) kinase assay to measure CC-292’s effect on BTK kinase activity and demonstrated an IC50 of less than 0.5 nM. The inhibitory activity against BTK was significantly greater than the activity against other kinases in the BCR signaling pathway and appeared to have greater specificity for BTK than did ibrutinib (Table 1). Using multiple cell lines, CC-292 demonstrated dose-dependent inhibition of BTK phosphorylation and inhibition of phosphorylation of the BTK substrate PLCγ2. In the cellular setting, CC-292 was found not to inhibit several other kinases, including epidermal growth factor receptor (EGFR), Janus kinase 3 (Jak3), and IL-2 inducible T cell kinase (ITK).48
Cells from the Ramos lymphoma cell line, which expresses an intact BCR signaling pathway, were treated with increasing concentrations of CC-292 for 1 hour and lysed, and the percent occupancy of BTK was determined with enzyme-linked immunosorbent assay (ELISA) by using a specific biotinylated probe for uninhibited BTK. The concentration of CC-292 required for 50% occupancy was 5.9 nM, which correlated with the concentration needed for the inhibition of BTK signaling. Additionally, in these cellular experiments, it was demonstrated that CC-292 did not inhibit the kinases LYN and SYK, upstream of BTK.
CC-292 abrogated IgM-mediated BCR triggering and induced apoptosis in CLL cells ex vivo. CLL cells isolated from patients were exposed to anti-IgM antibodies (the BCR ligand) in the presence or absence of CC-292. The CLL cells exposed to anti-IgM had improved viability relative to the untreated CLL cells. However, the CLL cells exposed to anti-IgM in the presence of CC-292 had decreased viability and went through apoptosis. Immunoblots from this experiment demonstrated an absence of phosphorylation of BTK in the CC-292-exposed cells.
CC-292 was shown to reduce the viability of CLL cells cocultured with NLCs. NLCs, expressing CLL costimulatory molecules, are an ex vivo model of the protective niche of the lymph node microenvironment. Incubating CLL cells with NLC results in improved CLL viability. This was abrogated by the addition of CC-292. Furthermore, the NLC-stimulated production of CCL3 and CCL4, the cell surface receptors for the chemotactic signals CXCL12 and CXCL13, was decreased in CLL cells in the presence of CC-292.49
CC-292 has been shown to inhibit the migration of CLL cells towards CXCL12 and CXCL13. Using Transwell® inserts, the migration of CLL in the presence and absence of CXCL12 and CXCL13 was measured. CC-292 significantly decreased the number of CLL cells migrating toward these chemotactic signals.
Animal studies
CC-292 was found to be effective in a number of arthritis animal models, consistent with its proposed effects on B cell function and number. CC-292 was similar to dexamethasone in its ability to prevent the development of arthritis in the peptidoglycan-polysaccharide arthritis model in rats. Female Lewis rats received peptidoglycan-polysaccharide 15 ug/g on day 0. CC-292 was administered daily beginning on day 6 and was found to be equivalent to dexamethasone in its ability to reduce the development of inflammation, as measured by ankle swelling.
CC-292 was also similar to dexamethasone in its ability to improve arthritis scores in a semi established collagen-induced arthritis model in mice. Male DBA/1 mice were injected with bovine type II collagen in Freund’s complete adjuvant on arthritis day 0 and day 21. CC-292 in increasing doses, dexamethasone, or a control vehicle was injected on day 21, and the mice were followed for the development of arthritis. CC-292 10 mg/kg was similar to dexamethasone in its ability to prevent the development of arthritis, as measured by a clinical arthritis score. The CC-292-treated animals did not appear to have developed joint damage, inflammation, or invasive pannus, when the joints were examined microscopically.
Finally, CC-292 was found to be equally efficacious to dexamethasone in an established collagen induced arthritis model. Male DBA/1 mice were injected with bovine type II collagen in Freund’s complete adjuvant on day 0 and day 21. Randomization of the mice occurred after swelling was established in at least one paw. The mice treated with CC-292 10 mg/kg demonstrated similar improvements in a clinical arthritis score compared with the mice treated with dexamethasone. They also had similar improvements in cartilage damage, bone damage, inflammation, and pannus, when investigated pathologically.
Phase IA clinical trial in healthy normal volunteers
Six subjects in each of five different dose cohorts ranging from 0.5 mg/kg to 7 mg/kg were investigated to determine the safety and tolerability, pharmacokinetics, and pharmacodynamics of CC-292.49 CC-292 was found to be generally safe and well tolerated, with no serious adverse effects and no apparent dose-related trends in adverse events.
CC-292 was highly orally available and rapidly absorbed, with the peak plasma concentrations occurring 20−60 minutes after dosing, with minimal subject-to-subject variability. The averaged area-under-the curve values across the cohorts demonstrated a linear and dose proportional exposure of CC-292. Complete BTK target occupancy was achieved with doses equal to or greater than 2 mg/kg CC-292, with greater than 97% occupancy at 4 hours and recovery toward 50% predose BTK values at 48 hours. The mean plasma concentration reached its maximum value 20−60 minutes after dosing and then rapidly decreased. However, the mean BTK occupancy reached maximal levels at 2−4 hours after dosing and was maintained for at least 8 hours, consistent with covalent binding of CC-292 to BTK. The rapid clearance of CC-292 from the plasma and sustained BTK inhibition is a clinical demonstration of the benefits of a covalently bound therapeutic. In theory, the rapid clearance from the plasma will decrease the potential for off-target effects of CC-292, and the sustained BTK inhibition will decrease the need for frequent repeat dosing.
Phase IB trial in CLL patients and B cell non-Hodgkin lymphoma
Patients with previously treated CLL were administered CC-292 in escalating doses from 125 mg to 1,000 mg daily, and 375 mg and 500 mg twice daily (BID) on a continuous dosing 28-day cycle, until progressive disease or intolerable toxicity. A maximum tolerated dose was not established, and CLL patients have been enrolled in an early dose expansion cohort of 750 mg daily and a second cohort of 500 mg BID.
A total of 78 patients with relapsed or refractory CLL have been enrolled as of May 28, 2013. Within the group, the median age was 67 years (range 34−89), 54% had Rai stage 3 or 4 disease, the median number of prior therapies was three, and 33% had been refractory to their most recent prior treatment. Poor risk factors were common, including the presence of del11q (23%), del17p (22%), and unmutated immunoglobulin variable heavy chain (IGVH) (53%).
The CC-292 was well tolerated, with a limited number of grade 3 or 4 adverse events, including neutropenia (23%), thrombocytopenia (17%), pneumonia (9%), and anemia (9%). Other common adverse events were diarrhea, fatigue, and nausea.
Nodal responses were seen at all dose levels, with an increase in absolute lymphocyte count. At the dose of 750 mg daily, the proportion with total response, PR, and nodal response with increased absolute lymphocyte count was 48%, 31%, and 17%, respectively. Similar responses were seen with 1,000 mg daily (57% total response, 43% PR, and 14% nodal response with increased absolute lymphocyte count), 375 mg BID dosing (67% total response and 67% PR), and 500 mg BID dosing (70% total response, 20% PR, and 50% nodal response with increased absolute lymphocyte count). In patients receiving BID dosing, the treatment response improved from cycle 2 to cycle 5, consistent with a continued treatment effect. The response rate was similar, despite the presence of poor risk factors, including 17p deletion, 11q deletion, and unmutated IGHV. In summary, despite a limited duration of follow up, the Phase IB experience with CC-292 demonstrated acceptable tolerability and a significant response rate. Based on these findings the recommended dose for the Phase II study of CLL is 500 mg BID.50
Ongoing trials
Based on the promising Phase IB experience in CLL, CC-292 is currently being investigated in combination trials in relapsed and refractory CLL. A multicenter, Phase IB, open label study is currently looking at the safety and activity of CC-292 in combination with lenalidomide in subjects with relapsed and refractory CLL (NCT01732861).51 Enrollment began November 2012, and the preliminary data has not yet been presented. Additionally, another Phase IB, multicenter, open label study is investigating the safety and activity of CC-292 in combination with rituximab in the same CLL population (NCT01744626).52 Enrollment began in December 2012, and the preliminary data of this study has also not yet been presented.
Discussion
Multiple inhibitors of the BCR signaling pathway are in development for the treatment of CLL and other lymphoid malignancies. The early clinical work with these agents has seen remarkable responses, with limited toxicity. In this review, we focused on the recent experience with CC-292, in the context of the evolving experience with BCR signaling inhibition in CLL. CC-292 is a promising inhibitor of BTK, with preclinical data demonstrating specific, covalent binding and inhibition of BTK. The Phase IA study demonstrated excellent oral bioavailability and in vivo inhibition of BTK with acceptable tolerability. In the Phase IB trial in patients with CLL, the safety of CC-292 was confirmed in this population and its activity established, as demonstrated by transient lymphocytosis and a decrease in lymph node size at all dose levels. Of particular note, responses have been seen in patients with poor-risk cytogenetics (17p, 11q, or unmutated IGVH), following the use of BCR pathway inhibitors, including CC-292. With conventional therapy, these patients are often refractory to treatment and at high risk for early relapse, which makes these inhibitors an exciting alternative treatment option for this population with limited options upon disease relapse.
Interestingly the Phase IB study in CLL patients has required higher doses to achieve strong lymph node responses and consistent lymphocytosis. In particular, the change to a BID dosing schedule has enhanced activity.53 At this early stage of CC-292 development, it remains unclear whether the degree of nodal response in patients with CLL treated with CC-292 is as dramatic as that seen with ibrutinib. Certainly, higher doses have been required. The reason for this difference remains unclear. Possible reasons for the differences may relate to the shorter half-life of the drug, leading to a requirement for BID dosing or to differences in tissue penetration between the two agents – correlative studies, which will address this question, are part of ongoing CC-292 trials. Another possibility is CC-292’s increased specificity for BTK, relative to ibrutinib (Table 1). CC-292 was rationally designed to covalently bind to BTK and has limited effects on other kinases. While ibrutinib is also specific for BTK, it inhibits other kinases at lower concentrations than CC-292. Perhaps, one of these off-target kinases contributes to ibrutinib’s efficacy in CLL. Interleukin-2 inducible kinase (ITK) was recently identified as a potential additional therapeutic target of ibrutinib.54 ITK is an important mediator of T cell receptor signaling, which is essential to the development of a T helper (Th)2 T cell response. In vitro studies and samples obtained from patients with CLL treated with ibrutinib have demonstrated ITK inhibition, with subsequent skewing of the T cell repertoire to a Th1 response. Th1 responses are important for T cell immunity against malignancy and may be directing a T cell response against CLL. It is possible that this or another off-target effect of ibrutinib is contributing to its marked activity.
Following the demonstration of single-agent activity of the BCR pathway-inhibiting agents, recent clinical trials have focused on combining these agents with established chemotherapeutic or immunotherapeutic agents. The rationale of these combinations is to use the BCR signaling inhibitors to force a migration of the malignant cells from their protected lymph node niches so that they can be more effectively targeted with the standard agents. However, given the significant activity of inhibition of a single component of the BCR pathway, there is also a strong rationale for the combination of multiple agents that target different components of the BCR pathway, in order to result in more complete pathway inhibition. The observed toxicity with CC-292 and the other BCR pathway inhibitors has been limited thus far, but the long-term toxic effects of BCR inhibition have not been established. Furthermore, as clinical trials focus on combining multiple different inhibitors of the BCR pathway, the toxicity may increase, or unexpected toxicity may emerge. The increased specificity of CC-292 for BTK may therefore be of benefit in limiting emergent toxic side effects, in combination trials. Frequent grade 1 or 2 ecchymosis and contusions are a common adverse event with ibrutinib, although significant bleeding events are rarely seen. One proposed mechanism is the involvement of BTK signaling in platelets.55 Early data suggest that patients receiving ibrutinib have a significant inhibition of platelet aggregation in response to collagen but not in response to arachidonic acid or thrombin-receptor activating peptide.56 The exact mechanism for the increased contusions continues to be investigated and may be related to an off-target effect of ibrutinib. As combination trials proceed, the increased risk of bleeding with ibrutinib may become a more significant clinical issue, and in that context, an agent with increased specificity for BTK that does not appear to increase the bleeding risk may become more attractive. In combination studies, CC-292’s very specific inhibition of BTK may become its most significant strength, allowing for directed inhibition of BTK with limited effect on other kinases.
As we learn more about the BCR and signaling in CLL, agents like CC-292 will likely become a standard part of the armamentarium in the treatment of CLL. CC-292 binds specifically to BTK and has demonstrated safety and efficacy, in still very early data. Its role as a BCR signaling pathway inhibitor, in what is rapidly becoming a crowded market, is still to be determined in future studies. The primary characteristic that differentiates CC-292 from ibrutinib, the other BTK inhibitor most studied in CLL, is its increased specificity for BTK. However, as the field evolves and more combinatorial trials are performed, CC-292’s specificity may become its greatest strength, allowing for the precise inhibition of BTK with limited off-target effects.
Disclosure
JEA reports no conflicts of interest in this work. JRB reports that she has served as a consultant for Celgene, Pharmacyclics, Gilead and Sanofi.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. | |
Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med. 2005;352(8):804–815. | |
Hallek M, Cheson BD, Catovsky D, et al; International Workshop on Chronic Lymphocytic Leukemia. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111(12):5446–5456. | |
Hallek M, Fischer K, Fingerle-Rowson G, et al; International Group of Investigators; German Chronic Lymphocytic Leukaemia Study Group. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376(9747):1164–1174. | |
Tam CS, O’Brien S, Wierda W, et al. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood. 2008;112(4):975–980. | |
Fischer K, Cramer P, Busch R, et al. Bendamustine combined with rituximab in patients with relapsed and/or refractory chronic lymphocytic leukemia: a multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol. 2011;29(26):3559–3566. | |
Kater AP, van Oers MHJ, Kipps TJ. Cellular immune therapy for chronic lymphocytic leukemia. Blood. 2007;110(8):2811–2818. | |
Bernal A, Pastore RD, Asgary Z, et al. Survival of leukemic B cells promoted by engagement of the antigen receptor. Blood. 2001;98(10):3050–3057. | |
Genevier HC, Hinshelwood S, Gaspar HB, et al. Expression of Bruton’s tyrosine kinase protein within the B cell lineage. Eur J Immunol. 1994;24(12):3100–3105. | |
Hendriks RW, Bredius RG, Pike-Overzet K, Staal FJ. Biology and novel treatment options for XLA, the most common monogenetic immunodeficiency in man. Expert Opin Ther Targets. 2011;15(8):1003–1021. | |
Mohamed AJ, Yu L, Bäckesjö CM, et al. Bruton’s tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev. 2009;228(1):58–73. | |
Dühren-von Minden M, Übelhart R, Schneider D, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature. 2012;489(7415):309–312. | |
Chiorazzi N, Efremov DG. Chronic lymphocytic leukemia: a tale of one or two signals? Cell Res. 2013;23(2):182–185. | |
Lanemo Myhrinder A, Hellqvist E, Sidorova E, et al. A new perspective: molecular motifs on oxidized LDL, apoptotic cells, and bacteria are targets for chronic lymphocytic leukemia antibodies. Blood. 2008;111(7):3838–3848. | |
Catera R, Silverman GJ, Hatzi K, et al. Chronic lymphocytic leukemia cells recognize conserved epitopes associated with apoptosis and oxidation. Mol Med. 2008;14(11–12):665–674. | |
Chu CC, Catera R, Zhang L, et al. Many chronic lymphocytic leukemia antibodies recognize apoptotic cells with exposed nonmuscle myosin heavy chain IIA: implications for patient outcome and cell of origin. Blood. 2010;115(19):3907–3915. | |
Messmer BT, Albesiano E, Efremov DG, et al. Multiple distinct sets of stereotyped antigen receptors indicate a role for antigen in promoting chronic lymphocytic leukemia. J Exp Med. 2004;200(4):519–525. | |
Agathangelidis A, Darzentas N, Hadzidimitriou A, et al. Stereotyped B-cell receptors in one-third of chronic lymphocytic leukemia: a molecular classification with implications for targeted therapies. Blood. 2012;119(19):4467–4475. | |
de Rooij MF, Kuil A, Geest CR, et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood. 2012;119(11):2590–2594. | |
de Gorter DJ, Beuling EA, Kersseboom R, et al. Bruton’s tyrosine kinase and phospholipase Cgamma2 mediate chemokine-controlled B cell migration and homing. Immunity. 2007;26(1):93–104. | |
Herman SE, Gordon AL, Hertlein E, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117(23):6287–6296. | |
Honigberg LA, Smith AM, Sirisawad M, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010;107(29):13075–13080. | |
Ponader S, Chen SS, Buggy JJ, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119(5):1182–1189. | |
Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31(1):88–94. | |
M.D. Anderson Cancer Center. Phase 2 Study of the Combination of Bruton’s Tyrosine Kinase Inhibitor PCI-32765 and Rituximab in High-Risk Chronic Lymphocytic Leukemia and Small Lymphocytic Lymphoma Patients. Available from: http://clinicaltrials.gov/show/NCT01520519. NLM identifier: NCT01520519. Accessed November 26, 2013. | |
National Cancer Institute (NCI). Lenalidomide and Ibrutinib in Treating Patients With Relapsed or Refractory Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma. Available from: http://clinicaltrials.gov/show/NCT01886859. NLM identifier: NCT01886859. Accessed November 26, 2013. | |
Janssen Research and Development, LLC. A Study of Ibrutinib in Combination With Bendamustine and Rituximab in Patients With Relapsed or Refractory Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma. Available from: http://clinicaltrials.gov/show/NCT01611090. NLM identifier: NCT01611090. Accessed November 26, 2013. | |
Brown J, Barrientos J, Flinn I, et al. The Bruton’s Tyrosine Kinase (BTK) inhibitor ibrutinib combined with bendamustine and rituximab is active and tolerable in patients with relapsed/refractory CLL, interim results of a phase Ib/II study. In: Program and abstracts of the 17th Congress of the European Hematology Association; June 14–17, 2012; Amsterdam, The Netherlands. Abstract 0543. | |
Pharmacyclics. Efficacy and Safety Study of PCI-32765 Combine With Ofatumumab in CLL (PCYC-1109-CA). Available from: http://clinicaltrials.gov/show/NCT01217749. NLM identifier: NCT01217749. Accessed November 26, 2013. | |
Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32–42. | |
Chang BY, Furman RR, Zapatka M, et al. Use of tumor genomic profiling to reveal mechanisms of resistance to the BTK inhibitor ibrutinib in chronic lymphocytic leukemia (CLL). In: Program and abstracts of the ASCO Annual Meeting; November 1–2, 2013; San Diego, CA. Abstract 7014. | |
Herishanu Y, Pérez-Galán P, Liu D, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood. 2011;117(2):563–574. | |
Cheson BD, Byrd JC, Rai KR, et al. Novel targeted agents and the need to refine clinical end points in chronic lymphocytic leukemia. J Clin Oncol. 2012;30(23):2820–2822. | |
Hallek M, Cheson BD, Catovsky D, et al. Response assessment in chronic lymphocytic leukemia treated with novel agents causing an increase of peripheral blood lymphocytes. Blood. 2012. | |
Herman SE, Gordon AL, Wagner AJ, et al. Phosphatidylinositol 3-kinase-δ inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010;116(12):2078–2088. | |
Lannutti BJ, Meadows SA, Herman SE, et al. CAL-101, a p110 delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117(2):591–594. | |
Brown JR, Furman RR, Flinn I, et al. Final results of a phase I study of idelalisib (GS-1101) a selective inhibitor of PI3Kδ, in patients with relapsed or refractory CLL. In: Program and abstracts of the 2013 ASCO Annual Meeting; November 1–2, 2013; San Diego, CA. Abstract 7003. | |
Coutre SE, Leonard JP, Furman RR, et al. Combinations of the selective phosphatidylinositol 3-kinase-delta (PI3K delta) inhibitor GS-1101 (CAL-101) with rituximab and/or bendamustine are tolerable and highly active in patients with relapsed or refractory chronic lymphocytic leukemia (CLL): Results From a Phase I Study. In: Program and abstracts of the 54th ASH Annual Meeting and Exposition; December 7–11, 2012; Atlanta, GA. Abstract 191. | |
Gilead Sciences. A Randomized, Double-Blind and Placebo-Controlled Study of Idelalisib in Combination With Rituximab for Previously Treated Chronic Lymphocytic Leukemia (CLL). Available from: http://clinicaltrials.gov/show/NCT01539512. NLM identifier: NCT01539512. Accessed November 26, 2013. | |
Gilead Sciences. A Randomized, Double-Blind and Placebo-Controlled Study of Idelalisib in Combination With Bendamustine and Rituximab for Previously Treated Chronic Lymphocytic Leukemia (CLL). Available from: http://clinicaltrials.gov/show/NCT01569295. NLM identifier: NCT01569295. Accessed November 26, 2013. | |
Papadopoulos KP, Abrisqueta P, Chambers G, et al. A Phase I dose expansion cohort study of the safety, pharmacokinetics and pharmacodynamics of SAR245409 (S09), an orally administered PI3K/mTOR Inhibitor, in patients with lymphoma. In: Program and abstracts of the 53rd ASH Annual Meeting and Exposition; December 10–13, 2011; San Diego, CA. | |
Flinn IW, Horwitz SM, Patel M, et al. Clinical safety and activity in a Phase 1 trial of IPI-145, a potent inhibitor of phosphoinositide-3-kinase-δ,γ, in patients with advanced hematologic malignancies. In: Program and abstracts of the 54th ASH Annual Meeting and Exposition; December 7–11; 2012; Atlanta, GA. Abstract 3663. | |
Contri A, Brunati AM, Trentin L, et al. Chronic lymphocytic leukemia B cells contain anomalous Lyn tyrosine kinase, a putative contribution to defective apoptosis. J Clin Invest. 2005;115(2):369–378. | |
Amrein PC, Attar EC, Takvorian T, et al. Phase II study of dasatinib in relapsed or refractory chronic lymphocytic leukemia. Clin Cancer Res. 2011;17(9):2977–2986. | |
Friedberg JW, Sharman J, Sweetenham J, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. 2010;115(13):2578–2585. | |
Zent CS, LaPlant BR, Johnston PB, et al. The treatment of recurrent/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL) with everolimus results in clinical responses and mobilization of CLL cells into the circulation. Cancer. 2010;116(9):2201–2207. | |
Evans EK, Tester R, Aslanian S, et al. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J Pharmacol Exp Ther. 2013;346(2):219–228. | |
Karp R, Evans E, Aslanian S, et al. Inhibition of BTK with AVL-292 translates to protective activity in rodent models of rheumatoid arthritis. In: Program and abstracts of the Inflammation Research Association Sixteenth International Conference; September 26–29, 2010; Chantilly, VA. Abstract A113. | |
Evans E, Ponader S, Karp R, et al. AVL-292: A targeted therapy for Bruton’s tyrosine kinase in B cell malignancies. Poster presented at: the 16th Congress of the European Hematology Association; June 9–12, 2011; London, UK. | |
Brown JR, Harb WA, Sharman JP, et al. Phase 1 study of single agent CC-292, a highly selective Bruton’s tyrosine kinase inhibitor (BTK), in relapsed/refractory chronic lymphocytic leukemia (CLL). In: Program and abstracts of the 15th International Workshop on CLL (IWCLL); September 9–11, 2013; Cologne, Germany. Abstract. | |
Celgene Corporation. Safety Study of CC-292 and Lenalidomide in Subjects With Chronic Lymphocytic Leukemia/ Small Lymphocytic Lymphoma. Available from: http://clinicaltrials.gov/show/NCT01732861. NLM identifier: NCT01732861. Accessed November 26, 2013. | |
Celgene Corporation. Safety Study of CC-292 and Rituximab in Subjects With Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma. Available from: http://clinicaltrials.gov/show/NCT01744626. NLM identifier: NCT01744626. Accessed November 26, 2013. | |
Pierce DW, Heise C, Nacht M, et al. Preclinical and phase 1 translational studies of CC-292, a potent and selective inhibitor of Bruton’s tyrosine kinase, in CLL patients. In: Program and abstracts of the 15th International Workshop on CLL (IWCLL); September 9–11, 2013; Cologne, Germany. Abstract 4.31. | |
Dubovsky JA, Beckwith KA, Natarajan G, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122(15):2539–2549. | |
Liu J, Fitzgerald ME, Berndt MC, Jackson CW, Gartner TK. Bruton tyrosine kinase is essential for botrocetin/VWF-induced signaling and GPIb-dependent thrombus formation in vivo. Blood. 2006;108(8):2596–2603. | |
Rushworth SA, MacEwan DJ, Bowles KM. Ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(13):1277–1278. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.