")
Back to Journals » Lung Cancer: Targets and Therapy » Volume 6
AXL receptor tyrosine kinase as a therapeutic target in NSCLC
Received 13 February 2015
Accepted for publication 25 March 2015
Published 30 April 2015 Volume 2015:6 Pages 27—34
DOI https://doi.org/10.2147/LCTT.S60438
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Pan-Chyr Yang
Ross A Okimoto,1 Trever G Bivona1,2
1Division of Hematology and Medical Oncology, University of California San Francisco, San Francisco, CA, USA; 2Helen Diller Family Comprehensive Cancer Center, University of California San Francisco, San Francisco, CA, USA
Abstract: The AXL receptor tyrosine kinase and its ligand, Gas6, regulate key processes in lung cancer growth, metastasis, and epithelial–mesenchymal transition-associated drug resistance. Gas6 and AXL expression have been correlated with poor prognosis and advanced clinical stage in patients with lung cancer, and targeting the Gas6/AXL pathway demonstrates antitumor activity, decreases cellular invasion, and restores sensitivity in de novo and acquired drug resistance models. These findings implicate AXL as a promising therapeutic target in lung cancer. In this review, we explore the role of AXL in lung cancer progression, from tumor development to disseminated disease, and highlight the current clinical landscape of anti-AXL therapeutics.
Keywords: Gas6, lung cancer, targeted therapy, drug resistance
Introduction
The TAM family of receptor tyrosine kinases (RTKs) modulate a myriad of physiological processes, including cellular proliferation, survival, cell adhesion, and migration.1–3 AXL is a member of the TAM family of RTKs, which also includes TYRO-3 (also known as Sky) and MER (also known as Eyk, Nym, and Tyro12).4 In adult tissue, AXL is ubiquitously expressed in epithelial, mesenchymal, and normal and malignant hematopoietic cells.5 AXL has demonstrated oncogenic potential in NIH3T3 fibroblasts and has been found to be frequently overexpressed in many human cancers, including breast, pancreatic, and lung, where it has been correlated with poor prognosis, invasion, and metastasis.6 In lung cancer, AXL has been observed to be overexpressed in non-small-cell lung cancer (NSCLC) cell lines as well as in a significant proportion of patient-derived primary lung tissue specimens.7,8
Structurally, the AXL RTK is characterized by an extracellular domain that closely resembles cell adhesion molecules, containing two N-terminal immunoglobulin (Ig)-like domains and two fibronectin type III (FNIII) repeats that have been implicated in growth arrest-specific 6 (Gas6) ligand binding.9 The intracellular domain consists of a prototypical tyrosine kinase domain that selectively modulates downstream effector signaling pathways (Figure 1).
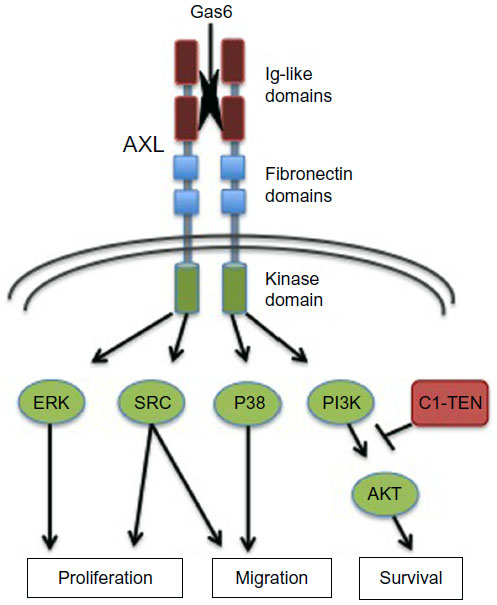 | Figure 1 AXL structure and effector pathways. |
AXL activation and downstream signaling pathways
The vitamin K-dependent protein Gas6 was initially identified in 1995 as the first endogenous ligand of AXL.10 Nearly 10 years later, the crystal structure of the Gas6/AXL complex revealed that Gas6 initiated signaling through receptor crosslinks, proving that dimerization occurred in the absence of extracellular receptor–receptor or ligand–ligand contacts.9
As an RTK, AXL is activated upon paracrine or autocrine binding of Gas6, facilitating trans-autophosphorylation of the intracellular tyrosine kinase domains. Upon phosphorylation at specific tyrosine residues (Y779, Y821, Y866), AXL mediates intracellular signaling predominantly through multi-substrate docking sites. Competitive inhibition of substrate binding revealed that Y821 modulated the interaction between AXL and multiple effector molecules including PLCγ, p85 proteins, Grb2, c-Src, and Lck. Additionally, Y779 demonstrated affinity for the p85 proteins and binding of PLCγ occurred through Y866.11 Once activated, AXL engages its downstream substrates to stimulate diverse cellular functions including cellular proliferation through the MEK–ERK pathway, survival and anti-apoptosis through the PI3K–AKT pathway, and migration through the Src and p38-MAPK signaling pathways.12–15 Negative regulators of AXL signaling have also been identified; in particular, the C1 domain-containing phosphatase and TENsin homologue (C1-TEN) has recently been shown to inhibit AXL-mediated PI3K–AKT pathway activation leading to decreased cellular survival and migration (Figure 1).16,17 Additional binding proteins, including the p55γ subunit of PI3K, SOCS-1, Nck2, and RanBPM, have since been uncovered through a yeast two-hybrid screen, which utilized the cytoplasmic domain of AXL as bait against a heart cDNA library.16 All together, through context-and tissue-specific interactions, AXL regulates a diverse network of signaling cascades, emphasizing the importance of identifying the critical components that drive the disease-specific phenotype.18
AXL overexpression and activation in lung cancer
While the initial transforming capacity of AXL in chronic myelogenous leukemia alluded to its importance in hematopoiesis, subsequent investigations have revealed that AXL is ubiquitously expressed and upregulated in a multitude of solid tumor malignancies including breast, gastric, prostate, ovarian, and lung. Importantly, the diverse biologic effects that arise in response to AXL activation are cell- and tissue type-specific. In lung cancer, for example, AXL overexpression was initially observed by immunohistochemistry (IHC) in 58% (7/12) of NSCLC cell lines.7 To further explore whether AXL expression correlated with clinical disease features of NSCLC, Shinh et al compared AXL expression in 58 patient-derived lung adenocarcinoma tissue specimens and identified ~48% (28/58) as positive and ~52% (30/58) with negative expression. AXL protein expression was retrospectively evaluated with clinicopathologic features and found to be significantly correlated with lymph node involvement (P<0.0001) and advanced clinical stage (P<0.0001).8 Further clinical outcome data in early-stage, resected lung adenocarcinomas demonstrated the prognostic impact of AXL expression on patient survival with a significant difference in 5-year overall survival rates comparing high and low AXL protein levels (38.6% and 77.5%, respectively; P<0.0001).19
The mechanism of AXL overexpression and activation in lung cancer is not well defined. Potential transcriptional regulators of AXL include specific mutant forms of p53. In a search for mutant p53 target genes, Vaughan et al have shown that H1299 lung cancer cells expressing various mutant p53 isoforms (R175H, R273H, and D281G) lead to upregulation of AXL, which is accompanied in part by gain-of-function phenotypes that are dependent on AXL expression.20 Another possible mechanistic link to upregulation of AXL in lung adenocarcinoma implicates the transcriptional regulator, YES-associated protein 1 (YAP1), where it is highly coexpressed in lung adenocarcinomas compared to normal lung tissue.21 Rankin et al have recently established a molecular link between the hypoxia-inducible transcription factor (HIF) pathway and AXL expression in metastatic renal cell carcinoma. AXL expression was directly activated by HIF-1 and HIF-2 in von Hippel–Lindau-deficient or hypoxic RCC cells and correlated with cellular invasion and metastasis.15 A more recent large-scale transcriptome sequencing effort to characterize 87 primary lung adenocarcinomas identified 45 gene fusions, with eight chimeric tyrosine kinases that were postulated to contribute to tumorigenesis. Among these, a novel AXL-MBIP fusion gene, which retained the kinase domain and dimerization units essential for catalytic activity, was identified. Further studies into the functional consequences of this rare novel AXL fusion and its contribution to lung cancer development are warranted.22 AXL induction by potent transcriptional regulators of tumorigenesis coupled with the relative absence of activating mutations suggests either the predominant role of AXL is to enhance or maintain the malignant phenotype or that AXL expression is regulated by epigenetic or posttranscriptional repression by noncoding RNAs. Utilizing integrative gene expression and promoter CpG profiling, Lin et al observed differential methylation patterns among a subset of epithelial–mesenchymal transition (EMT)-related genes, including AXL, in NSCLC.23 Additionally, utilizing a panel of NSCLC cell lines, Mudduluru et al identified an inverse correlation between AXL protein expression and miR-34a. Subsequent correlative studies revealed that miR-34a and miR-199a/b, two AXL-targeting noncoding RNAs, were significantly downregulated by promoter methylation in NSCLC tissue specimens.24 These findings highlight the dynamic interplay and complexities of AXL regulation at the genetic, epigenetic, and posttranscriptional levels in lung cancer.
Role of AXL in EMT, invasion, and metastasis
EMT is an embryonic program that is essential for normal morphogenesis that involves dissolution of intercellular junctions and loss of cell polarity. Carcinoma cells co-opt this developmental program to enhance migration, invasion, and metastasis.25 Accumulating experimental evidence indicates that AXL overexpression plays a critical role in cellular migration and strongly correlates with clinical metastasis in patients with NSCLC.8 Despite these findings, the effector pathways that mechanistically link AXL to a more migratory and invasive phenotype remain elusive. Specifically, our functional understanding of the role of AXL as a potent inducer or effector of the EMT process is currently unknown.
To date, a limited number of in vitro studies have attempted to address this critical question in lung cancer evolution and current evidence supports the role of AXL as a crucial activator of EMT. Notably, AXL signaling promotes an EMT gene signature including the upregulation of known EMT transcriptional regulators, snai1, snai2, and vim, with a corresponding decrease in epithelial markers, such as cdh1, resulting in an increased propensity for cellular migration.26 Additional functional evidence in NSCLC suggests that AXL may be promoting a morphologic transformation from a highly polarized epithelial state to a more migratory mesenchymal phenotype. Lay et al, for instance, exogenously overexpressed AXL in a panel of NSCLC cell lines and observed cytoskeletal rearrangements with increased filopodia formation and enhanced migratory capacity in Transwell chamber assays. Conversely, silencing of endogenous AXL resulted in the loss of characteristic spindle-like morphology and decreased migratory potential.27 Further molecular insight into the pathways by which AXL promotes dynamic cytoskeletal rearrangements implicates the Elmo scaffold proteins as substrates of AXL signaling through direct phosphorylation of a carboxyl-terminal tyrosine residue. Elmo proteins are known to directly interact with the Dock family of guanine nucleotide exchange factors (GEFs) to activate Rac-mediated cytoskeletal changes that drive cellular migration.28 Additional evidence that AXL-mediated cellular migration is a Rac1-dependent process comes from cellular redox regulation studies that identify AXL phosphorylation upon excessive reactive oxygen species (ROS) exposure. ROS-induced AXL phosphorylation leads to enhanced cellular migration via a Rac 1-dependent pathway.29
The role of AXL as an effector of EMT that promotes cellular migration and invasion is predicated upon its dynamic regulation by potent transcriptional programs regulated by YAP1 and the myeloid zinc finger 1 (MZF1). As previously noted, YAP1 is a nuclear effector of the Hippo pathway that has a demonstrated role in lung cancer tumorigenesis and EMT. YAP1 and AXL are highly expressed in lung adenocarcinomas as compared to normal lung tissue. Interestingly, knockdown of YAP1 dramatically reduced cellular invasion through downregulation of the AXL pathway, suggesting that AXL is a downstream target and effector of YAP1 oncogenic function.21 The MZF1 protein has also been shown to regulate cellular migration, invasion, and metastasis at least in part through the transcriptional upregulation of AXL.30 Overexpression of MZF1 leads to the transactivation of the AXL promoter and increased gene transcription, resulting in increased migration, invasion, and metastatic capacity both in vitro and in vivo. Conversely, genetic knockdown of AXL reduced MZF1-mediated migration and invasion, again suggesting that AXL is an effector of an MZF1 transcriptional program.
While it is becoming increasingly clear that AXL may have an intricate role in cellular migration, its precise role in EMT remains unknown. Intriguingly, analysis of either AXL or Gas6 germline knockout mice reveals that the Gas6/AXL cascade is not necessary for normal embryogenesis, a developmental program that requires EMT.31 Thus, despite mounting evidence that AXL is intimately associated with the EMT program, a direct mechanistic link is yet to be identified.
AXL expression correlates with drug response in lung cancer
It is well appreciated that greater than 90% of lung cancer mortality is related to the emergence of drug-resistant metastatic disease, yet the underlying mechanisms that drive this process remain unknown. EMT has been associated with metastasis and observed in ~25% of patient-derived tumor specimens that are resistant to EGFR-targeted therapies.32 A limited number of studies have explored the underlying molecular mechanisms that link EMT and drug resistance. Byers et al, for instance, have developed an EMT gene expression signature that predicts therapeutic resistance to EGFR and P13K inhibitors in vitro and identifies patients with relapsed or metastatic NSCLC. Additionally, the identification of AXL as a highly expressed EMT marker in mesenchymal NSCLC cell lines led these investigators to explore the therapeutic efficacy of AXL inhibition in erlotinib-refractory cell lines expressing high levels of AXL.33 AXL inhibition with the pharmacologic inhibitor, SGI-7079, sensitized otherwise refractory mesenchymal NSCLC cell lines to the EGFR inhibitor erlotinib, both in vitro and in mouse xenograft models. These findings suggested a synergistic role for combination therapy to overcome de novo resistance to EGFR blockade in AXL high mesenchymal NSCLC. In order to more broadly identify correlative expression patterns that accompany AXL expression, Wilson et al recently performed RNAseq transcriptome profiling of 643 human cancer cell lines and identified a strong correlation with the mesenchymal marker, vimentin.34 Among NSCLC cell lines, AXL tended to be overexpressed in erlotinib-insensitive cells that coexpressed high vimentin levels, while cells with lower vimentin and AXL expression correlated with greater erlotinib sensitivity.34 These findings corroborate the study of Byers et al33 and demonstrate the therapeutic efficacy of co-targeting AXL in mesenchymal NSCLC.
Over the past decade, the overwhelming majority of studies focused on elucidating the role of AXL in lung cancer have focused on the development of acquired resistance to TKI therapy. Recently, our laboratory along with two independent groups have shown that AXL inhibition can overcome acquired resistance to EGFR-targeted therapies in EGFR mutant lung cancer. In our study, we established multiple in vitro and in vivo erlotinib-resistant EGFR mutant lung cancer models and identified upregulation of AXL and an EMT phenotype to be associated with acquired resistance. Genetic and pharmacological inhibition of AXL restored sensitivity to erlotinib in TKI-resistant HCC827 lung adenocarcinoma cells. Consistent with our preclinical data, we revealed that 20% of EGFR mutant patient-derived tumors expressed higher levels of AXL upon acquiring resistance to erlotinib as compared to paired pretreatment samples.35 Recently, independent evaluation of 26 additional paired pretreatment EGFR mutant tumors that initially responded to gefitinib and subsequently acquired resistance in the Korean population revealed an increase in AXL expression in 19% (5/26) of patients.36
Consistent with these findings, AXL was observed to be upregulated in the setting of EMT in rociletinib (CO-1686)-resistant H1975 lung adenocarcinoma cell lines. In this study, however, only a modest decrease in growth inhibition was observed with effective genetic inhibition of AXL using RNAi knockdown. Combination therapies with rociletinib and two independent multikinase inhibitors that target AXL in two independent drug-resistant clones partially restored sensitivity to EGFR-targeting agents, suggesting a synergistic effect.37 Additional evidence for the dynamic interplay between AXL and EGFR signaling in lung cancer comes from the previously mentioned study by Byers et al33 that demonstrated enhanced therapeutic responses with combination AXL and EGFR targeted therapies. To further identify the precise relationship between AXL and EGFR, a recent computational modeling study proposed that activation of EGFR leads to transactivation of AXL in a ligand-independent manner that results in diversification of downstream signaling outputs beyond those triggered by either receptor alone.38 While adding an additional layer of complexity to AXL biology, the development of drugs that counteract this interaction may achieve clinical efficacy.
It has become increasingly clear that AXL plays a cooperative role in the development and maintenance of drug-resistant lung cancer. The functional consequences of enhanced AXL signaling are cell type-specific and seem to involve a dynamic interplay between existing parallel pathways and effector molecules. Thus it is unclear whether AXL inhibition will achieve clinical success in drug-resistant lung cancer, although upfront AXL-targeted therapy to forestall the emergence of drug resistance and EMT conversion may be a promising strategy to explore.
AXL as a clinical biomarker and potential therapeutic target in lung cancer
The Gas6/AXL pathway has recently emerged as a critical player in tumor progression, metastasis, and drug resistance. These findings make AXL inhibition a viable and attractive target in human lung cancer. Unfortunately, one of the major limitations of assessing the clinical efficacy of AXL-targeted agents is the absence of a validated prospective biomarker for patient enrichment. Current clinical assessment of AXL activity remains predominantly through IHC expression of the protein in tissue specimens. In order to improve upfront patient stratification and more rapidly identify subsets of patients who may benefit from AXL-targeted therapies, a recent study engineered a near-infrared fluorescence humanized anti-AXL antibody (h173) imaging probe that was validated in lung cancer xenograft models. Tumor uptake of the labeled probe in vivo was significantly higher in established AXL-high versus AXL-low cell lines, offering a novel methodology for real-time monitoring of AXL-expressing lung tumors.39 This is obviously a highly desirable noninvasive method for assessing AXL expression, which has been shown to temporally change with the acquisition of more invasive, drug-resistant phenotypes. Another promising avenue that might enhance noninvasive monitoring of AXL expression involves isolation of a soluble fragment of AXL (sAXL) from NSCLC patients that acquire resistance to EGFR or MET targeted therapies. Ise et al demonstrate proof-of-concept in vitro isolation of sAXL from TKI-resistant NSCLC cell lines that, if clinically validated, could potentially improve real-time therapeutic monitoring to small molecule inhibitors in lung cancer patients.40 As imaging modalities and serological biomarkers of response continue to evolve and achieve clinical validation, we postulate that they could better identify and direct patients into AXL-targeting therapeutic trials. One major obstacle with systemic tracking of AXL expression, however, is the ubiquitous expression of this protein. Investigators will need to optimize and overcome nonspecific labeling to more precisely monitor disease response.
Despite our significant progress in identifying the effectors of AXL signaling and the mechanisms that regulate AXL activation in lung cancer, there remains a paucity of small molecule inhibitors that specifically target this receptor. The majority of current preclinical and early-phase trials designed to target AXL utilize compounds that have been developed to target other kinases, yet demonstrate additional activity against AXL. Here, we will summarize the current clinical landscape of AXL TKIs in early-phase lung cancer clinical trials and highlight the most promising preclinical developments in alternative anti-AXL targeting strategies (Table 1).
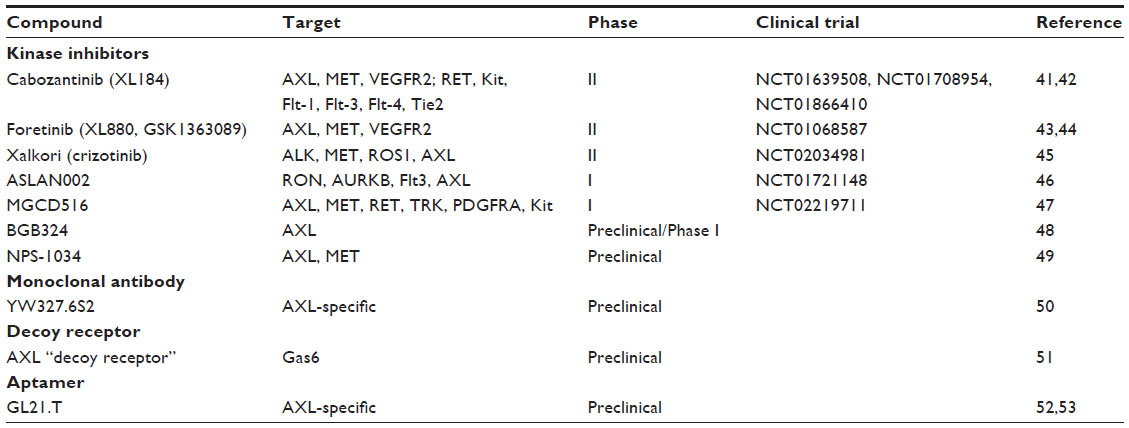 | Table 1 AXL inhibitors in clinical and preclinical development for lung cancer |
Cabozantinib (XL184) is a multikinase inhibitor that has activity against VEGFR, RET, MET, Flt-3, Kit, and AXL.41,42 It was approved by the US Food and Drug Administration (FDA) in 2012 for the treatment of medullary thyroid cancer and is currently being investigated in Phase II trials in patients with advanced or metastatic NSCLC with high AXL activity as assessed by IHC overexpression. Additionally, two Phase II trials are currently evaluating the efficacy of cabozantinib combination therapy with erlotinib in both wild-type and mutant EGFR NSCLC patients who progressed on standard systemic or targeted therapies. Another agent, foretinib (XL880, GSK1363089), is a multikinase inhibitor that targets MET, VEGFR-2, ROS1, and AXL and is currently being evaluated in Phase II trials in combination with erlotinib for advanced or metastatic NSCLC refractory to standard platinum-doublet chemotherapy.43,44 Crizotinib is an oral small-molecule TKI that targets ALK, MET, ROS1, and AXL and has demonstrated antitumor activity in patients with ALK-positive NSCLC.45 A Phase II trial is currently assessing the efficacy across multiple tumor types including NSCLC with a defined activating molecular alteration in a crizotinib target. In a Phase I study of ASLAN002, a RON, AURKB, FLT3, and AXL TKI is currently being assessed for tolerability and antitumor efficacy in patients with advanced or metastatic solid tumors.46 In another ongoing Phase I/Ib study of MGCD516, a TKI with nanomolar activity against Eph receptors, MET, VEGFR, and AXL, the clinical potential of targeting rare AXL gene rearrangements in a broad spectrum of advanced solid-tumor malignancies including NSCLC is being assessed.47 A new class of selective small molecule inhibitors that are designed to specifically target AXL are in preclinical and Phase I trials. BGB324, an oral AXL-specific inhibitor, was well tolerated in healthy patients and blocked the emergence of EMT-associated acquired resistance to EGFR-targeted agents in NSCLC preclinical xenografts.48 Lastly, newer TKIs that specifically address acquired resistance to EGFR-targeted therapies via bypass signaling of MET or AXL are currently in preclinical development for EGFR mutant NSCLC. Most notably, NPS-1034, a TKI that targets multiple kinases including MET and AXL, has demonstrated synergistic effects in EGFR mutant cell line-based models that acquired resistance to EGFR TKIs.49
While we eagerly await the early-phase results of AXL-specific inhibitors in NSCLC patients, ongoing clinical trials in lung cancer continue to utilize broad-spectrum multikinase inhibitors that target various oncogenic pathways. As a result, these nonspecific AXL TKIs have demonstrated modest antitumor efficacy with significant off-target effects that obscure the therapeutic response to direct AXL inhibition. As our understanding of what activates AXL and what AXL regulates at the cellular level improves, alternative strategies to target the Gas6/AXL pathway can be developed. Among these approaches, an anti-AXL monoclonal antibody, YW327.6S2, has been shown to bind to AXL with high affinity. This interaction abrogates ligand Gas6 binding to the receptor and downregulates receptor expression, activation, and downstream signaling in NSCLC cell lines. Amazingly, YW327.6S2 also inhibited tumor xenograft growth and decreased disseminated disease in experimental metastasis assays.50 Another recent innovative and promising approach has exploited our fundamental understanding of AXL biology, specifically the high picomolar affinity of AXL for its ligand Gas6. Kariolis et al engineered an AXL “decoy receptor” that binds Gas6 with femtomolar affinity, an 80-fold improvement over the wild-type receptor. Gas6 sequestration with this novel decoy receptor significantly reduced tumor burden and experimental metastasis in vivo, further validating AXL as a therapeutic target in tumor progression and metastasis.51 Another emerging class of therapeutic inhibitors that target the AXL pathway includes short nucleic acid aptamers. GL21.T is a selective RNA-based aptamer that binds the extracellular domain of AXL and inhibits its catalytic activity. GL21.T inhibited AXL-dependent signaling, cellular migration, and invasion, and demonstrated antitumor efficacy in lung xenograft models.52,53
In order to achieve clinical benefit from AXL inhibition, clinical trials will need to prospectively incorporate more predictive and reliable biomarkers of AXL dependency. It remains unclear if AXL inhibition in lung cancer can truly improve clinical outcomes, largely due to the broad context-dependent roles of AXL in lung cancer development, metastasis, and drug resistance. Furthermore, due to the relative dearth of direct and highly selective AXL antagonists, the translational potential of targeting AXL in lung cancer remains poorly defined. We postulate that the results of early-phase clinical trials with AXL-driven therapeutic interventions coupled with the implementation of novel non-TKI agents with high, direct affinity for the Gas6/AXL pathway will enhance our understanding of which patients will benefit most from an AXL-targeted strategy.
Conclusion
In the current landscape of clinically validated oncogenic drivers, AXL remains a relatively unexplored target despite its significant role in lung cancer tumorigenesis, metastasis, and drug resistance. This may in part be the result of preclinical data suggesting a cooperative role for AXL in lung cancer progression. We predict that as early-phase clinical trials come to fruition, AXL will emerge as a validated therapeutic target in human lung cancer. The specific context in which AXL inhibition may benefit patients, however, will only be revealed through biomarker-driven clinical trials that utilize compounds that directly target the AXL receptor. Lastly, as novel biomarker-based imaging methodologies are optimized and implemented into the clinical arena, real-time monitoring of AXL expression in response to systemic or targeted therapies will markedly enhance our clinical understanding of the role of AXL in lung cancer development and metastasis. Coupled with advanced biomarker development and assessment, we envision that as more specific AXL-targeting agents emerge, we can better define the true translational potential of targeting AXL in the clinic to improve patient outcomes.
Acknowledgments
The authors thank members of the Bivona lab for critical review of the manuscript and acknowledge funding support from the following sources: NIH Director’s New Innovator Award, NIH R01 CA169338, Howard Hughes Medical Institute, Doris Duke Charitable Foundation, American Lung Association, Sidney Kimmel Foundation for Cancer Research, Searle Scholars Program, California Institute for Quantitative Biosciences, and Li-Ka Shing Foundation (to TGB).
Disclosure
TGB is a consultant to Driver Group and to Novartis, Clovis Oncology, and Cleave Biosciences, and a recipient of a research grant from Servier. The authors report no other conflicts of interest in this work.
References
Sainaghi PP, Castello L, Bergamasco L, Galletti M, Bellosta P, Avanzi GC. Gas6 induces proliferation in prostate carcinoma cell lines expressing the Axl receptor. J Cell Physiol. 2005;204(1):36–44. | |
McCloskey P, Fridell YW, Attar E, et al. GAS6 mediates adhesion of cells expressing the receptor tyrosine kinase Axl. J Biol Chem. 1997;272(37):23285–23291. | |
Tai KY, Shieh YS, Lee CS, Shiah SG, Wu CW. Axl promotes cell invasion by inducing MMP-9 activity through activation of NF-kappaB and Brg-1. Oncogene. 2008;27(29):4044–4055. | |
O’Bryan JP, Frye RA, Cogswell PC, et al. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell Biol. 1991;11(10):5016–5031. | |
Neubauer A, Fiebeler A, Graham DK, et al. Expression of axl, a transforming receptor tyrosine kinase, in normal and malignant hematopoiesis. Blood. 1994;84(6):1931–1941. | |
Goruppi S, Ruaro E, Schneider C. Gas6, the ligand of Axl tyrosine kinase receptor, has mitogenic and survival activities for serum starved NIH3T3 fibroblasts. Oncogene. 1996;12(3):471–480. | |
Wimmel A, Glitz D, Kraus A, Roeder J, Schuermann M. Axl receptor tyrosine kinase expression in human lung cancer cell lines correlates with cellular adhesion. Eur J Cancer. 2001;37(17):2264–2274. | |
Shinh YS, Lai CY, Kao YR, et al. Expression of axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia. 2005;7(12):1058–1064. | |
Sasaki T, Knyazev PG, Clout NJ, et al. Structural basis for Gas6-Axl signalling. EMBO J. 2006;25(1):80–87. | |
Varnum BC, Young C, Elliot G, et al. Axl receptor tyrosine kinase stimulated by the vitamin K-dependent protein encoded by growth-arrest-specific gene 6. Nature. 1995;373:623–626. | |
Braunger J, Schleitfhoff L, Schulz AS, et al. Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is mediated mainly by a multi-substrate docking-site. Oncogene. 1997;14:2619–2631. | |
Goruppi S, Ruaro E, Varnum B, Schneider C. Requirement of phosphatidylinositol 3-kinase-dependent pathway and Src for Gas6-Axl mitogenic and survival activities in NIH 3T3 fibroblasts. Mol Cell Biol. 1997;17(8):4442–4453. | |
Hasanbasic I, Cuerquis J, Varnum B, Blostein MD. Intracellular signaling pathways involved in Gas6-Axl-mediated survival of endothelial cells. Am J Physiol Heart Circ Physiol. 2004;287(3):H1207–H1213. | |
Paccez JD, Vogelsang M, Parker MI, Zerbini LF. The receptor tyrosine kinase Axl in cancer: biological functions and therapeutic implications. Int J Cancer. 2014;134(5):1024–1033. doi:10.1002/ijc.28246. | |
Rankin EB, Fuh KC, Castellini L, et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc Natl Acad Sci U S A. 2014;111(37):13373–13378. | |
Hafizi S, Alindri F, Karlsson R, Dahlbäck B. Interaction of Axl receptor tyrosine kinase with C1-TEN, a novel C1 domain-containing protein with homology to tensin. Biochem Biophys Res Commun. 2002;299(5):793–800. | |
Hafizi S, Ibraimi F, Dahlbäck B. C1-TEN is a negative regulator of the Akt/PKB signal transduction pathway and inhibits cell survival, proliferation, and migration. FASEB J. 2005;19(8):971–973. | |
Axelrod H, Pienta KJ. Axl as a mediator of cellular growth and survival. Oncotarget. 2014;5(19):8818–8852. | |
Ishikawa M, Sonobe M, Nakayama E, et al. Higher expression of receptor tyrosine kinase Axl, and differential expression of its ligand, Gas6, predict poor survival in lung adenocarcinoma patients. Ann Surg Oncol. 2013;20 Suppl 3:S467–S476. | |
Vaughan CA, Singh S, Windle B, et al. Gain-of-function activity of mutant p53 in lung cancer through up-regulation of receptor protein tyrosine kinase Axl. Genes Cancer. 2012;3(7–8):491–502. | |
Cui ZL, Han FF, Peng XH, et al. YES-associated protein 1 promotes adenocarcinoma growth and metastasis through activation of the receptor tyrosine kinase Axl. Int J Immunopathol Pharmacol. 2012;25(4):989–1001. | |
Seo JS, Ju YS, Lee WC, et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012;22(11):2109–2119. | |
Lin SH, Wang J, Saintigny P, et al. Genes suppressed by DNA methylation in non-small cell lung cancer reveal the epigenetics of epithelial-mesenchymal transition. BMC Genomics. 2014;15:1079. | |
Mudduluru G, Ceppi P, Kumarswamy R, Scagliotti GV, Papotti M, Allgayer H. Regulation of Axl receptor tyrosine kinase expression by miR-34a and miR-199a/b in solid cancer. Oncogene. 2011;30(25):2888–2899. | |
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. | |
Gjerdrum C, Tiron C, Høiby T, et al. Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc Natl Acad Sci U S A. 2010;107(3):1124–1129. | |
Lay JD, Hong CC, Huang JS, et al. Sulfasalazine suppresses drug resistance and invasiveness of lung adenocarcinoma cells expressing AXL. Cancer Res. 2007;67(8):3878–3887. | |
Abu-Thuraia A, Gauthier R, Chidiac R, et al. Axl phosphorylates Elmo scaffold proteins to promote Rac activation and cell invasion. Mol Cell Biol. 2015;35(1):76–87. | |
Huang JS, Cho CY, Hong CC, et al. Oxidative stress enhances Axl-mediated cell migration through an Akt1/Rac1-dependent mechanism. Free Radic Biol Med. 2013;65:1246–1256. | |
Mudduluru G, Vajkoczy P, Allgayer H. Myeloid zinc finger 1 induces migration, invasion, and in vivo metastasis through Axl gene expression in solid cancer. Mol Cancer Res. 2010;8(2):159–169. | |
Lu Q, Gore M, Zhang Q, et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature. 1999;398(6729):723–728. | |
Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. | |
Byers LA, Diao L, Wang J, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19(1):279–290. | |
Wilson C, Ye X, Pham T, et al. AXL inhibition sensitizes mesenchymal cancer cells to antimitotic drugs. Cancer Res. 2014;74(20):5878–5890. | |
Zhang Z, Lee JC, Lin L, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012; 44(8):852–860. | |
Ji W, Choi CM, Rho JK, et al. Mechanisms of acquired resistance to EGFR-tyrosine kinase inhibitor in Korean patients with lung cancer. BMC Cancer. 2013;13:606. | |
Walter AO, Sjin RT, Haringsma HJ, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013;3(12):1404–1415. | |
Meyer AS, Miller MA, Gertler FB, Lauffenburger DA. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci Signal. 2013;6(287):ra66. | |
Li D, Liu S, Liu R, et al. Axl-targeted cancer imaging with humanized antibody h173. Mol Imaging Biol. 2014;16(4):511–518. | |
Ise N, Takemori N, Omi K, Goishi K, Higashiyama S. Abstract B46: Soluble AXL is a candidate circulating biomarker for predicting acquired and intrinsic resistance to molecular-targeted agents in NSCLC cells. Mol Cancer Ther. 2013;12(11 Suppl):B46. | |
Yakes FM, Chen J, Tan J, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10(12):2298–2308. | |
Drilon A, Wang L, Hasanovic A, et al. Response to Cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov. 2013;3(6):630–635. | |
Qian F, Engst S, Yamaguchi K, et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 2009;69(20):8009–8016. | |
Davare MA, Saborowski A, Eide CA, et al. Foretinib is a potent inhibitor of oncogenic ROS1 fusion proteins. Proc Natl Acad Sci U S A. 2013; 110(48):19519–19524. | |
Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–2394. | |
Schroeder GM, An Y, Cai ZW, et al. Discovery of N-(4-(2-amino-3-chloropyridin-4-yloxy)-3-fluorophenyl)-4-ethoxy-1-(4-fluorophenyl )- 2-oxo-1,2-dihydropyridine-3-carboxamide (BMS-777607), a selective and orally efficacious inhibitor of the Met kinase superfamily. J Med Chem. 2009;52(5):1251–1254. | |
Beaulieu N, Sainte-Croix H, Bonfils C, et al. Abstract 930: Preclinical characterization of MG516, a novel inhibitor of receptor tyrosine kinases involved in resistance to targeted therapies. Cancer Res. 2013; 73(8 Suppl):930. | |
Wnuk-Lipinska K, Tiron C, Gausdal G, et al. Abstract 1747: BGB324, a selective small molecule Axl kinase inhibitor to overcome EMT-associated drug resistance in carcinomas: therapeutic rationale and early clinical studies. Cancer Res. 2014;74(19 Suppl):1747. | |
Rho JK, Choi YJ, Kim SY, et al. MET and AXL inhibitor NPS-1034 exerts efficacy against lung cancer cells resistant to EGFR kinase inhibitors because of MET or AXL activation. Cancer Res. 2014;74(1):253–262. | |
Ye X, Li Y, Stawicki S, et al. An anti-Axl monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene. 2010;29(38):5254–5264. | |
Kariolis MS, Miao YR, Jones DS 2nd, et al. An engineered Axl ‘decoy receptor’ effectively silences the Gas6-Axl signaling axis. Nat Chem Biol. 2014;10(11):977–983. | |
Cerchia L, Esposito CL, Camorani S, et al. Targeting Axl with an high-affinity inhibitory aptamer. Mol Ther. 2012;20(12):2291–2303. | |
Esposito CL, Cerchia L, Catuogno S, et al. Multifunctional aptamer-miRNA conjugates for targeted cancer therapy. Mol Ther. 2014;22(6):1151–1163. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.