")
Back to Archived Journals » Reports in Medical Imaging » Volume 14
Aventriculy: A Rare Case Report
Authors Dandena A , Sisay S, Mekonnen A , Beza K
Received 12 September 2020
Accepted for publication 22 January 2021
Published 9 February 2021 Volume 2021:14 Pages 9—13
DOI https://doi.org/10.2147/RMI.S281603
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Tarik Massoud
Abdi Dandena,1 Samuel Sisay,1 Abebe Mekonnen,1 Kalkidan Beza2
1Department of Radiology, Addis Ababa University, Addis Ababa, Ethiopia; 2Department of Pediatrics and Child Health, Addis Ababa University, Addis Ababa, Ethiopia
Correspondence: Abdi Dibaba Tel +251 921566429
Email [email protected]
Abstract: In this paper, we will describe a week old neonate who was referred to Black Lion Referral Hospital with a diagnosis of ambiguous genitalia and was screened for associated congenital anomalies. The neonate was evaluated with Trans-fontanel Ultrasound and Brain MRI, which showed the absence of the lateral and third ventricles and associated Intracranial multiple anomalies.
Keywords: brain malformations, aventriculy, rhomboencephalosynapsis, holoprosencephaly
Introduction
Central nervous system (CNS) anomalies are rare among the other congenital disorders of various organ systems. Furthermore, each CNS anomaly offers a unique challenge to Radiologists, since they have their own unique and complicated imaging findings. Moreover, accurate identification of all the CNS anomalies is essential for determining the proper course of management and prediction of the patient’s outcome.1
CNS anomalies can be classified based on Magnetic Resonance Imaging (MRI) appearances and, based on the appearance, the timing of the gestational disorders can be estimated. MRI, because of its multiplanar, multi-sequential characteristics, and excellent soft-tissue contrast differentiation, can be an invaluable tool in the classification of congenital CNS anomalies.2
Disorders of diverticulation, cleavage, sulcation, and cellular migration are among the common congenital CNS anomalies, and, under this classification, we can have several disorders, including Holoprosenephally, Lisencephaly, cortical Dysplasia, Heterotrophic grey matter, schizencephaly, and Rhomboencephalosynapsis.1,2
Holoprocenephally (HPE) is the most common developmental disorder of the Human Forebrain, and it is estimated to occur in one out of 10,000 live and stillbirths. HPE is believed to be a result of a complex interaction of genetic and environmental factors.3
Holoprocenephally can be classified using various classification schemes; the most commonly used classification scheme is the DeMyers classification, which classifies HPE into Alobar, semi lobar, and lobar holoprocenephally from the most severe end of the spectrum to the least severe end of the spectrum. However, other variants of these conditions have been described in the literature since then.4
Rhomboencephalosynapsis (RES) is a midline brain malformation characterized by different levels of missing cerebellar vermis with an apparent fusion of cerebellar hemispheres. RES can be seen in isolation or associated other CNS and extra CNS anomalies. The severity of the RES usually correlates with the associated anomalies, which include the agenesis of the olfactory bulb, dysgenesis of the corpus callosum, absent septum pellucidum, and in rare cases, atypical forms of holoprocenephally.5
Case Presentation
A 3 days old male neonate born from a 29 years old para I Diabetic mother and 31-year-old father after eight months and two weeks of amenorrhea. The baby was delivered by spontaneous vaginal delivery after labor of 9 hours duration. The baby had an APGAR score of 4 and 5 at the 1st and 5th minutes, respectively and the baby was resuscitated with bag and mask ventilation for the first 5 minutes.
The mother was diagnosed with Type II diabetes for the past two years but failed to take her prescribed medications during her pregnancy. The father had no known chronic medical illness, and the parents belonged to different ethnic groups, and there was no chance of consanguinity between them.
The antenatal follow-up was uneventful. The mother had regular follow-up (3x) during the entire pregnancy as per the national guidelines and was taking iron and folate supplementation. The mother did not consume alcohol, cigarette, or any illicit drugs during the entire pregnancy.
After delivery, the neonate was resuscitated for 5 minutes. Afterward, the neonate had stable vitals. The baby weighed 2,900 grams and had a length of 49 cm; the head circumference was 30 cm, which was below the 3rd percentile for his age. On precordial examination, there was a grade II holo systolic murmur in the lower left sternal border. There was no anal opening visualized on perineal inspection, and meconium was seen to be passing out through a small perineal opening at the base of the scrotum. On musculoskeletal examination, there was a fusion of the 3rd and 4th digit in both hands (syndactyly). The rest of the physical examination findings were unremarkable.
Blood samples were taken, and complete blood count, serum electrolytes, renal and liver function tests were performed and were all within the normal range. Genetic testing was not performed on the child or the parents.
The neonate’s abdominal sonography showed an ectopic and malrotated right kidney, located in the pelvic region with a moderate degree of hydronephrosis.
Transthoracic echocardiography showed a small mid-ventricular muscular septal defect and a patent ductus arteriosus with a left to right shunt.
Transfontanell ultrasound examination showed the absence of a lateral and third ventricle with partially formed anterior interhemispheric fissure and falx cerebra. The cisterna magna was shown to be enlarged, measuring 1.2 cm in the anterior–posterior diameter. On color doppler evaluation of the intracranial vasculature, there was a fusion of the anterior cerebral arteries (azygous anterior cerebral arteries).
A subsequent multiplanar and multi-sequential MRI was performed using a 1.5T Philips scanner with T1, T2, FLAIR, and inversion recovery sequences on the 5th day of life and was repeated on the 7th day due to the poor quality of the initial images. The baby was not sedated during the acquisition of the images leading to motion degradation in some sequences. The MRI images showed the absence of the lateral and third ventricles with a fusion of the thalami at the midline. The corpus callosum was not visualized. There was abnormal sulcation of the cerebral hemispheres with multiple small shallow sulci (Polymicrogyria) in the midline and parasagittal cerebral hemispheres. There was a reduction in the number of gyri in the bilateral temporal lobes (pachygyria). There is a generalized thinning in the cortical thickness, which was especially prominent in the bilateral temporal regions where the cortex size is barely perceptible. A band like T2 hypointense ribbon like structure in the white matter of the cerebellar hemispheres likely represents a band heterotopia. The basal ganglia nuclei appear indistinct and dysmorphic with no clear separation between the different deep brain nuclei. The hippocampal region appears malformed with distortion of the internal architecture. There is an azygous anterior cerebral artery coursing between the interhemispheric fissures. The falx cerbri is not visualized. There is a fusion of the cerebellar hemispheres with non-visualization of the cerebellar vermis (rhomboencephalosynapsis). There is an enlargement of the cisterna magna, which measures 1.2 cm in the maximum anterior–posterior diameter. The cochlea has normal dimensions but no visible internal partitioning, the vestibule and semicircular canals are dysmorphic bilaterally, suggesting incomplete partition type I. There is a left high parietal T1 hyper-intense and T2 mixed hypo-intense scalp collection which likely represents a cephalohematoma (Table 1 and Figure 1, Figure 2, Figure 3).
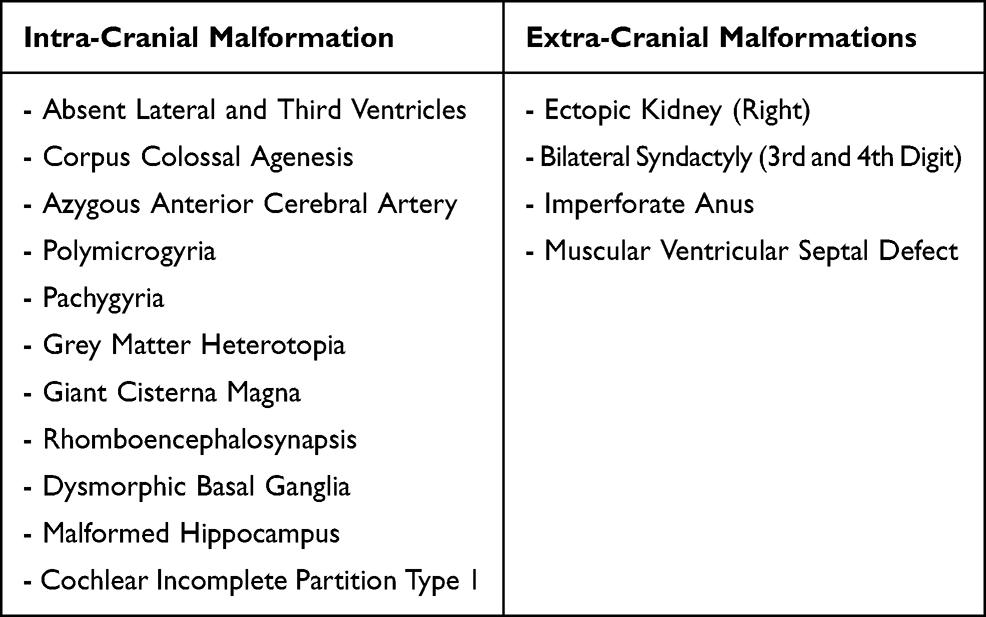 |
Table 1 Summary of the Intra-Cranial and Extra-Cranial Congenital Malformations Detected in the Child |
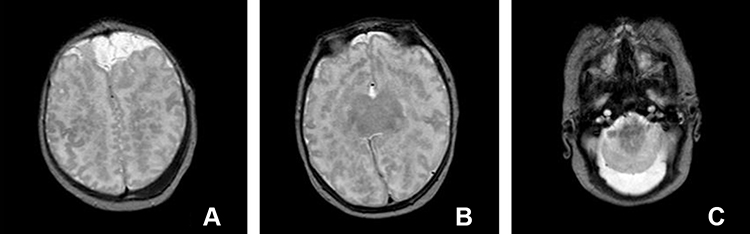 |
Figure 1 Axial T2 weighted images showing absence of the lateral and third ventricles with fusion of the thalami at the midline (B). Azygous ACA coursing in the interhemispheric fissure (A) and an enlarged cisterna magna with fusion of the cerebellar hemispheres (C). Abnormal sulcal patterns with areas of pachygyria and polymicrogyria is also seen (A and B). |
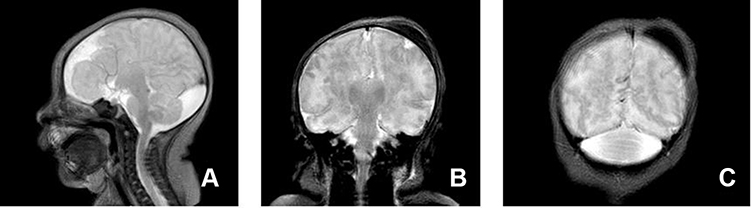 |
Figure 2 Sagittal and coronal T2 weighted images show the absence of the lateral and third ventricles with a prominent cisterna magna (A) with fusion of thalami (B) and the cerebellar hemispheres (C) and cephalohematoma (B and C). |
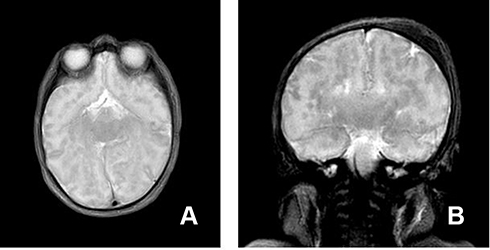 |
Figure 3 Axial and coronal T2 weighted images at the level of the basal ganglia (A) and the hippocampus (B) showing dysmorphic and ill-defined basal nuclei and malformed hippocampus. |
Discussion
Holoprocenephally represents a continuum of forebrain malformation that results from a disordered porecncephalic ventral induction. However, there may not be a well-defined, clear cut distinction between the different subcategories. The DeMyers classification has classified HPE into three types: alobar, semi-lobar, and lobar. Most recently, newer variants of holoprocenephally have been described, including the middle interhemispheric variant or syntelencephaly.6
Although HPE is primarily a disorder of pore cephalic (forebrain) cleavage, associated intra- and extracranial malformations have been described in the literature, such as craniofacial abnormalities, rhomboencephalosynapsis, and subcortical heterotopia being described as well as the associated extracranial malformations, the most common ones are genital defects (24%), postaxial polydactyly (8%), vertebral defects (5%), limb reduction defects (4%), and transposition of the great arteries (4%).4
The cause for holoprocenephally is believed to be a result of interaction between different genetic, environmental, and possible socio-cultural factors. Moreover, around eight candidate genes have been identified that have an association with HPE: SHH, SIX3, ZIC2, TGIF, PATCHED1, GL12, TDGF1, and FAST1. However, mutations in these genes were only identified in around 15–20% of the cases. Some of the environmental factors that have been incremented to have an association with HPE include maternal diabetes, alcohol consumption, cigarette smoking, and exposure to retinoic acid.7
Aventriculy was first described in the lterature by Garfunkel back in 1996 when he described the absence of the lateral, third, and fourth ventricles in an 11-year-old girl who presented with multiple aspiration episodes of pneumonia. In this case, the pregnancy was also complicated by maternal diabetes and multiple episodes of toxemia and colds. The child also had a head circumference below the 3rd percentile (microcephaly). On subsequent neuroimaging, the child had corpus callosal agenesis, Azygous ACA, Polymicrogyria, fused thalami, dysplastic cerebral hemispheres, and Rhomboencephalosynapsis, which were also presented in our case. The significant differences between the case by Garfunkel and our case include observable Sylvian fissures, the presence of the 4th ventricle, and the presence of an enlarged Cisterna Magna in our case.8
The second case of aventriculy was described in Turkey by Sener where he described the absence of the supratentorial ventricles, corpus callosum, and falx cerebri in a 1-month-old girl who presented seizures. The case also shared other similarities, such as a head circumference below the 3rd percentile and areas of pachygyria. Some of the notable differences between our cases are seen in the posterior fossa, where a Dandy-Walker variant malformation was described in the case of Sener with dilation of the 4th ventricle and hypogenetic inferior vermis.9
The most recently reported case of aventriculy was by Citfcioglu et al in 2015, where they described the absence of the supratentorial ventricular system in a 6-month-old infant suffering from motor and mental retardation. Other similar findings to our case include Microcephaly, a fusion of the Thalami in the midline, and the presence of corpus callosal agenesis. The case differs from ours due to the presence of a well-formed falx cerebri and absence of any posterior fossa anomalies.10
It can be concluded that aventriculy is a rare congenital CNS malformation which shares many similar features with holoprocenephally and commonly associates with other congenital intracranial and extracranial malformations. Although genetic testing and further work up was not performed in all described cases of aventriculy, the imaging features may suggest aventriculy may be a rare variant of holoprocenephally.
Ethical Consideration
Written informed consent was obtained from the patient's parents to publish this case report and any accompanying images. Institutional Approval was not required in the writing of the Case Report.
Author Contributions
All authors contributed to data analysis, drafting, or revising of the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Singh TG, Srivastav V, Singhania P, Devi SM. Congenital brain anomalies: neuroimaging findings. J Dr NTR Univ Health Sci. 2014;3(2):77–85. doi:10.4103/2277-8632.134827
2. Poe LB, Coleman LL, Mahmud F. Congenital central system anomalies nervous. Radiographics. 1989;9(5):801–826. doi:10.1148/radiographics.9.5.2678293
3. Kauvar EF, Muenke M. Holoprosencephaly: recommendations for diagnosis and management. Curr Opin Pediatr. 2010;22(6):687. doi:10.1097/MOP.0b013e32833f56d5
4. Winter TC, Kennedy AM, Woodward,PJ. Holoprosencephaly: a survey of the entity, with embryology and fetal imaging. Radiographics. 2015;35(1):275–290. doi:10.1148/rg.351140040
5. Ishak GE, Dempsey JC, Shaw DWW, et al. malformation associated with incomplete hydrocephalus and a broad spectrum of severity. Brain. 2013;(December).
6. Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V. Holoprosencephaly. Orphanet J Rare Dis. 2007;14(2):1–14.
7. Stashinko EE, Clegg NJ, Kammann HA, et al. A retrospective survey of perinatal risk factors of 104 living children with holoprosencephaly. Am J Med Genet A. 2004;128A(2):114–119. doi:10.1002/ajmg.a.30070
8. Garfinkle WB. Aventriculy: a new entity? Am J Neuroradiol. 1996;17(9):1649–1650.
9. Sener RN. Aventriculi associated with holoprosencephaly. Comput Med Imaging Graph. 1998;22(4):345–347. doi:10.1016/S0895-6111(98)00029-9
10. Report C. Absence of the lateral and third ventricles associated with holoprosencephaly. Anat Cell Biol. 2015;48(3):222–224. doi:10.5115/acb.2015.48.3.222
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.