")
Back to Journals » Journal of Inflammation Research » Volume 16
AVE 0991 Suppresses Astrocyte-Mediated Neuroinflammation of Alzheimer’s Disease by Enhancing Autophagy
Authors Deng Y , Wang SY , Wang QG, Xu ZH , Peng Q, Chen SY, Zhu L, Zhang YD, Duan R
Received 19 October 2022
Accepted for publication 24 January 2023
Published 1 February 2023 Volume 2023:16 Pages 391—406
DOI https://doi.org/10.2147/JIR.S392599
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Yang Deng,1,* Si-Yu Wang,2,* Qing-Guang Wang,3,* Zhao-Han Xu,2 Qiang Peng,2 Shuai-Yu Chen,2 Lin Zhu,2 Ying-Dong Zhang,1,2 Rui Duan1,2
1Department of Neurology, Nanjing First Hospital, China Pharmaceutical University, Nanjing, People’s Republic of China; 2Department of Neurology, Nanjing First Hospital, Nanjing Medical University, Nanjing, People’s Republic of China; 3Department of Neurology, Jiangyin Hospital Affiliated to Nantong University, Jiangyin, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ying-Dong Zhang; Rui Duan, Department of Neurology, Nanjing First Hospital, China Pharmaceutical University, No.68, Changle Road, Nanjing, Jiangsu, People’s Republic of China, Email [email protected]; [email protected]
Purpose: Our previous study has shown that AVE 0991, a nonpeptide analogue of Ang-(1-7), ameliorates cognitive decline and inhibits NLRP3 inflammasome of astrocytes in Alzheimer’s disease model mice. Additionally, several studies have suggested that activation of autophagy appears to effectively inhibit the progression of neuroinflammation. However, it is unclear whether AVE 0991 can modulate astrocyte autophagy to suppress neuroinflammation in Alzheimer’s disease.
Materials and Methods: APP/PS1 mice and Aβ-treated primary astrocytes were used as the research objects in vivo and in vitro, respectively. Water maze test was used to evaluate cognitive function of mice, Nissl staining and immunofluorescence staining was used to assess neuronal damage. ELISA kits were used to detect the levels of Ang-(1-7) and Aβ in the cortex, and qRT-PCR was used to detect the expression of cortical inflammation-related mediators. The expression of autophagy-related proteins in cortex were detected by Western blot. The upstream molecular responses involved in inflammation inhibition by AVE 0991 were validated by means of using the Mas1 antagonist and autophagy inhibitor.
Results: We found that 30 days of intraperitoneal administration of AVE 0991 improved. Aβ deposition, neuronal death, and cognitive deficits in APP/PS1 Alzheimer’s disease model mice. Moreover, AVE 0991 treatment greatly suppressed astrocyte-mediated inflammation and up-regulated the expression of autophagy. Furthermore, the inhibitory effect of AVE 0991 on the expression of inflammatory factors was reversed by 3-MA, an autophagy inhibitor.
Conclusion: These findings suggest that regulation of autophagy is critical for inhibiting astrocyte neuroinflammatory responses and demonstrate a potential neuroprotective mechanism by which AVE 0991 could suppress neuroinflammatory responses by enhancing autophagy.
Keywords: AVE 0991, neuroinflammation, Alzheimer, autophagy, astrocytes
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized by impairments in learning, memory and cognition, as well as changes in mood.1 Previously, mainstream research focused on the pathogenic effects of the two main AD drivers, Aβ peptide (Aβ) and hyperphosphorylated tau (hp-Tau) protein.2,3 Recently, more and more evidence indicated that neuroinflammation plays a central role in the pathogenesis of AD.4,5 Emerging studies have suggested that controlling neuroinflammation mediated by glial cell activation is an important therapeutic strategy for AD6,7 and previous reports have also found that the glial responses activated by Aβ can promote the release of pro-inflammatory cytokines to cause neuronal dysfunction and memory impairment.8–10 Therefore, targeting neuroinflammation induced by glial activation may be an effective way to slow or prevent the progression of AD.
Astrocytes are the most abundant glial cells in the brain11 and play a vital role in maintaining brain homeostasis.12–14 When AD occurs, the deposition of Aβ will induce astrocytes to transform from a resting state to a reactive state, which is manifested by increased levels of glial fibrillary acidic protein (GFAP),15 which in turn produce more pro-inflammatory cytokines and mediators, such as interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α) and interleukin- 6 (IL-6), which exacerbates the progression of inflammation.16 Additionally, recent data found a specific accumulation of astrocytes associated with pro-inflammatory diseases in AD.17 Therefore, inhibiting astrocyte-mediated inflammation may be a potentially effective strategy for reducing cognitive impairment in AD patients.
Emerging data reveal that autophagy may play a critical role in the regulation of astrocyte-mediated inflammatory response during AD.18 Autophagy is one of the effective ways for the body to digest and degrade damaged or dysfunctional proteins and organelles in the cytoplasm and it plays an important role in maintaining cell survival, differentiation, development and homeostasis.19 Previous studies have found that autophagy dysfunction occurs in both AD animal models and clinical patients, leading to the pathological accumulation of tau aggregates and Aβ, which in turn aggravates the course of AD.20 Furthermore, another study demonstrated that Aβ-induced inflammatory cytokines release are related to astrocyte autophagy dysfunction, and activation of autophagy can inhibit Aβ-induced inflammatory responses.21 Therefore, we can speculate that in the inflammatory environment mediated by astrocytes, the activation of autophagy may delay the progression of AD.
AVE 0991 (5-Formyl-4-methoxy-2-phenyl-1-[[4-[2-(ethylaminocarbonylsulfonamido) 5-isobutyl-3-thienyl]-phenyl]-meth-yl]-imidazole) is a nonpeptide analogue of angiotensin-(1-7) [Ang-(1-7)], which can simulate the multiple functions of Ang-(1-7) in the nervous system. Our previous study proofed that intracerebroventricular injection of angiotensin-(1-7) can inhibit the excessive activation of autophagy in the brain of spontaneously hypertensive rats.22 In Parkinson’s disease, Ang-(1-7) reduced the aggregation of α-synuclein in rotenone-induced cell models by enhancing autophagy activity.23 In fact, our previous studies have directly confirmed that AVE 0991 inhibits the inflammatory response mediated by microglia in a Mas receptor-dependent manner, thereby reducing the aging‐related neuroinflammation.24 Notably, AVE 0991 also enhances the clearance of Nod-like receptor pyrin domain containing 3 (NLRP3) inflammasomes by promoting the process of autophagy, thereby protecting the brain from lipopolysaccharide (LPS)-induced excessive inflammation.25 However, it is not clear whether AVE 0991 regulates autophagy and its neuroprotective mechanisms in AD.
Therefore, we hypothesized that AVE 0991 exerts its neuroprotective effect by promoting autophagy of astrocytes and inhibiting neuroinflammation in AD. In the present study, we investigated the effects of AVE 0991 on behavior, inflammation and autophagy in amyloid precursor protein/presenilin 1 (APP/PS1) AD model mice. Meanwhile, we treated primary astrocytes with AVE 0991 to investigate whether AVE 0991 inhibited the neuroinflammatory response induced by Aβ1-42 in astrocytes by promoting autophagy. Our data indicated that AVE 0991 protects against astrocyte-mediated inflammation by promoting autophagy.
Materials and Methods
Animals and Treatments
Eight-month-old male APP/PS1 double-transgenic mice and age-matched male wild-type C57BL/6J mice (WT) were purchased from the Beijing Zhishan Co., Ltd. All experiments were performed in accordance with the guidelines for the care and use of laboratory animals, and were approved by the Animal Research and Ethics Committee of Nanjing First Hospital (protocol#: IACUC-1911032).
WT and APP/PS1 mice were randomly divided into six groups: WT group, APP/PS1 group, APP/PS1+ 1 mg/kg AVE 0991 group, APP/PS1+ 3 mg/kg AVE 0991 group, APP/PS1+ 10 mg/kg AVE 0991 group and APP/PS1+ 10 mg/kg AVE 0991+ 30 mg/kg 3-MA group. All mice were given intraperitoneal injection once daily for 30 consecutive days. Mice in WT group and APP/PS1 group were given equal volumes of normal saline. The Morris Water Maze (MWM) experiment was started on the 25th day. After the behavioral experiment was completed, all mice were quickly sacrificed. The brain cortex was promptly removed on ice, and then stored at −80°C for molecular detections such as gene and protein analysis.
Morris Water Maze (MWM) Test
The Morris Water Maze (MWM) was widely used to evaluate learning and memory of mice.26 In short, a hidden round platform was placed 1.00 cm below the water surface. A video tracking system (Ethovision 3.0, Noldus Information Technology B.V.) was used to automatically track mice. Data of learning and memory were collected for 5 days. The animals were randomly put into the water from four points every day until the platform was found and stayed for 10s within 60s. If the animals cannot find the platform within 60s, they then were guided to the platform. One day after the last trial, the platform was removed and the mice were placed into the water from the opposite quadrant of the platform. The path length to the merged platform was recorded, and the average value of four trials was calculated.
Nissl Staining
Nissl staining was performed as described.27 In brief, paraffin-embedded sections were deparaffinized and rehydrated according to standard protocols. The sections then were stained with tar violet (R23068, Saint-Bio, Shanghai) at 56 °C for 1 h. Next, the sections were dehydrated with gradient concentrations of ethanol, rinsed with xylene, and sealed with neutral gum after removing the dye by washing. Then a CX23 microscope (Olympus) was used to randomly select six fields of cortex on each coronal slice for quantitative analysis. Nuclei with round cell bodies, cytoplasmic Nissl material and nucleoli visible were identified as normal neurons, whereas nuclei with disorganized arrangement, Nissl material disappearance, nuclei surrounded by voids and unrecognizable nucleoli were identified as damaged neurons. Nissl positive neurons % = positive neurons/total neurons ×100%.
Oligomeric Aβ1-42 Preparation
The preparation of Aβ1-42 oligomer was performed as previously described.28 In short, Aβ1-42 monomer was prepared by evaporating 2 mg of Aβ1-42 dissolved in 1, 1, 1, 3, 3, 3, hexafluoro-2-propanol for 30 minutes under N2 gas at room temperature. Then, the monomer was diluted to 10 μM. Subsequently, the diluted solution was incubated to form oligomers. Next, The Aβ1-42 oligomer preparations were centrifuged to remove any insoluble fibrils.
Primary Astrocytes Culture and Treatment
Primary astrocytes were isolated from the brain cortex of newborn WT mice (< 24 h). Briefly, the minced brain cortex was digested, the digestion solution was filtered and centrifuged, and then the cells were seeded in Dulbecco’s Modified Eagle Medium/F12 (DMEM/F12) complete medium, and incubated in a 5% CO2 incubator at 37 °C. After 10 days of incubation, the cells were shaken to remove microglial contamination from adherent astrocytes. Then, the purified astrocytes were cultured in DMEM/F12 complete medium and placed in a humidified incubator at 37 °C and 5% CO2. The third generation of cells were used for subsequent experiments. The purity of astrocytes was evaluated by immunofluorescence staining with anti-GFAP antibody.
Astrocytes were treated for 24 h with 5 μM Aβ1-42 with or without 24 h incubation with AVE 0991 (1×10−8, 1×10−7 or 1×10−6 M) in the presence or absence of A-779 (1×10−6 M). Besides, 3-MA was added 1 h before Aβ1-42 treatment, while AVE 0991 was added for 24 h co-treatment upon Aβ1-42 exposure. DMEM (10% fetal bovine serum, FBS) was used as a control.
Immunofluorescence Staining for Brain Tissue and Cells
Brain tissue was paraffin-embedded and then de-paraffinized with xylene and dehydrated with graded concentrations of ethanol. Antigens were regenerated with sodium citrate buffer (pH 6.0) for 10 min. After blocking with 1% goat serum and phosphate-buffered saline (PBS) (containing 0.1% Triton X-100) for 30 min at room temperature, anti-MAP-2 (1:50, #4542, Cell Signaling Technology), anti-Mas1 (1:100, ab235914, abcam), anti-GFAP (1:200, #80788, Cell Signaling Technology) and anti-Iba-1 (1:1000, #17198, Cell Signaling Technology) were added and incubated overnight at 4°C. After 3 washes with PBS, the secondary antibody working solution was added and incubated at room temperature for 1 h. Sections were stained with 4’,6-diamidino-2-phenylindole (DAPI) for 5 min and observed using a confocal microscope (LSM780, Zeiss, Germany).
Astrocytes were washed three times with cold PBS, fixed at 4 °C in 4% paraformaldehyde for 15 min, penetrated with 0.5% Triton X-100 for 20 min, blocked in 5% serum at 37 °C for 60 min. Afterwards, astrocytes were incubated with GFAP primary antibody (1:300, #3670, Cell Signaling Technology) overnight at 4 °C. After washing with PBS three times, the cells were incubated with Alexa Fluor 488-labeled goat anti-mouse secondary antibody (1:500, ab150113, Abcam) at 37°C for 2 h. The nuclei were stained with DAPI, and a confocal microscope (LSM780, Zeiss, Germany) was used to observed the samples.
Cell Viability Assay
Cell viability was assessed using Cell Counting Kit-8 (CCK-8, Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer’s instructions. Astrocytes were seeded in 96-well plates at a density of 5×104 cells/well 24 h. The cells were then treated for 24 h with 5 μM Aβ1-42 with or without 24 h incubation with AVE 0991 (1×10−8, 1×10−7 or 1×10−6 M). Then, 10 μL CCK-8 was added to the cells and incubated at 37 °C for 2 h. The absorbance at 450 nm was measured using a microplate reader (Thermo Scientific, San Jose, CA, USA).
Enzyme-Linked Immunosorbent Assay (ELISA)
Freeze cortex was homogenized in ice-cold RIPA lysis buffer and centrifuged at 12,000 g for 10 min at 4 °C. Enzyme-linked immunosorbent assay (ELISA) assay kits (KMB3441, Thermo Fisher Scientific, Waltham, MA, USA) were used to quantify soluble Aβ1-42 levels in supernatants.
The concentrations of Ang-(1-7) and inflammatory cytokines including IL-1β, TNF-α and IL-6 in mouse brain cortex and the conditioned medium of each group were measured by commercial ELISA kits (R&D Systems, Inc.). The absorbance was detected and quantified with a microplate reader (Molecular Device Corporation, Sunnyvale, CA, USA).
Real-Time Quantitative Reverse-Transcription PCR (qRT-PCR)
Total RNA from mice brain cortex or astrocytes were isolated and extracted using Trizol reagent (Invitrogen, USA) according to the manufacturer’s protocol. Reverse transcription was performed using Exscript RT Reagent Kit (Takara Bio Inc., China). qRT-PCR was performed on a fluorescent thermocycler iQ5 (Bio-Rad) using SYBR® Premix Ex TaqTM II. β-actin was used as an endogenous control. Relative mRNA levels were analyzed using the 2−ΔΔCt method. The primers used in this study were listed in Supplementary Table S1.
Western Blot Analysis
Western blot was performed as previously described.29 The brain cortex or astrocytes were lysed by RIPA lysis buffer to extract protein. A BCA protein assay kit (Thermo, Hercules, CA) was used to determine the protein concentrations. Equal amounts of protein samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membrane (Merck Group, Darmstadt, Germany). The membrane was then blocked with 5% non-fat milk for 1 h at room temperature, and incubated with primary antibodies against Mas1 (1:200, PA5-77282, Thermo Fisher Scientific), Synaptophysin (1:1500, #SAB4502906, Sigma-Aldrich), GFAP (1:1000, #3670, Cell Signaling Technology), LC3B (1:1000, #43566, Cell Signaling Technology), Beclin-1 (1:1000, #3495, Cell Signaling Technology), p62 (1:1000, #23214, Cell Signaling Technology) and β-actin (1:1000, #4970, Cell Signaling Technology) overnight at 4 °C. After washing with TBST buffer, the membranes were incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibody. Next, the membranes were visualized with a chemiluminescent HRP substrate (#32132, Thermo Fisher Scientific, Waltham, MA, USA). Protein bands were quantified by Image J software.
Statistical Analysis
All experiments were performed at least three times. Prism 8.0 software (GraphPad, San Diego, CA, USA) was used for all analysis data. The results of the escape latency in MWM were analyzed by two-way analysis of variance with Tukey’s multiple comparison test. All other data were analyzed using one-way analysis of variance (ANOVA) and Tukey’s test for post hoc analysis. Data were presented as the mean ± standard error of the mean (SEM). A value of P < 0.05 was considered to be statistically significant.
Results
AVE 0991 Activates the Ang-(1-7)/Mas1 Axis in the Brain Cortex of APP/PS1 Mice
As a non-peptide analogue of Ang-(1-7), AVE 0991 acts on the Ang-(1-7)/Mas1 axis and exerts various functions in neurological diseases. As shown in Figure 1A, Ang (1–7) levels were significantly decreased in APP/PS1 mice compared with WT group, while AVE 0991 treatment (1, 3 and 10 mg/kg) have no influence on this level. Furthermore, AVE 0991 injections at 3 and 10 mg/kg dramatically increased Mas1 mRNA expression (Figure 1B). In coincide with the qRT-PCR results, Western blot analysis indicated that AVE 0991 administration (3 and 10 mg/kg) remarkably increased the protein expression level of Mas1 (Figure 1C and D).
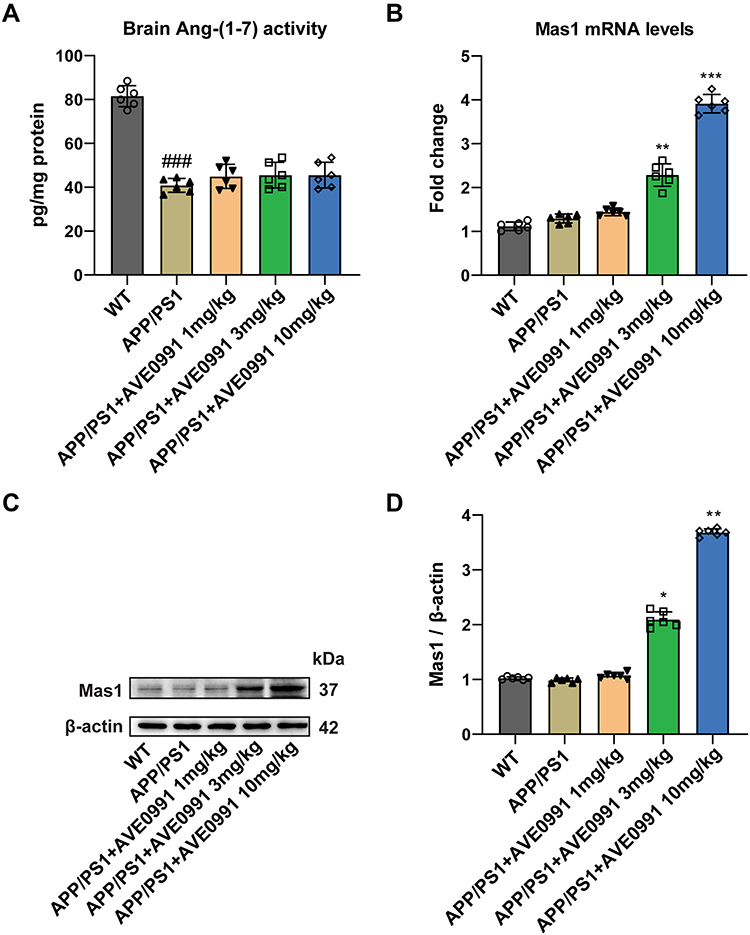 |
Figure 1 AVE 0991 activates the Ang-(1-7)/Mas1 axis in the brain cortex of APP/PS1 mice. (A) The protein expression of Ang- (1–7) in the brain cortex of mice injected with AVE 0991 was measured by ELISA assay (n = 6). (B) The mRNA expression of Mas1 in the brain cortex of mice injected with AVE 0991 was detected by qRT-PCR (n = 6). (C) The protein expression of Mas1 in the brain cortex of mice injected with AVE 0991 was evaluated by Western blot analysis. β-actin was used as the loading control (n = 6). (D) Quantitative analysis of Mas1 protein level was shown as bar chart (n = 6). All data are presented as mean ± SEM. ###P < 0.001 vs the WT group; *P < 0.05, **P < 0.01 and ***P < 0.001 vs the APP/PS1 group. |
AVE 0991 Attenuates Cognitive Deficits and Neuron Death in AD Mice
To study the effect of AVE 0991 treatment on AD model mice, 8-month-old APP/PS1 mice were intraperitoneally injected with AVE 0991 daily for 30 days. The MWM test was conducted for 5 days to assess their spatial learning and memory abilities before execution. As shown in Figure 2A, there was no difference in swimming speed between APP/PS1 mice and their age-matched WT controls, whereas injection of different doses of AVE 0991 did not significantly affect the swimming speed of APP/PS1 mice. Meanwhile, there was no significant difference in cognitive function among the five groups on 1–2 days during the MWM test. On day 3, 4 and 5, APP/PS1 mice exhibit significant spatial cognitive impairment, while AVE 0991 (3 and 10 mg/kg) rescues this impairment (Figure 2B). Nissl staining was used to observe the injury of mice neurons. The results indicated that there was significant neuronal death in the cortex of APP/PS1 mice, which was notably rescued by AVE 0991 treatment (3 and 10 mg/kg) (Figure 2C and D). Similar to the Nissl staining results, immunofluorescence staining data indicated that MAP-2 was strongly upregulated in the cortex of APP/PS1 mice after administration of AVE 0991 (3 and 10 mg/kg) (Figure 2E). As displayed in Figure 2F and G, compared with the WT group, APP/PS1 mice had significant synapse loss, while AVE 0991 treatment (3 and 10 mg/kg) strikingly increased synaptophysin protein levels. Taken together, these results demonstrated that AVE 0991 can alleviate memory and cognitive impairment in the development of AD. In this part, we selected three doses of AVE 0991 (1, 3, and 10 mg/kg) for in vivo assay, and the 1 mg/kg AVE 0991 group did not obtain a positive effect.
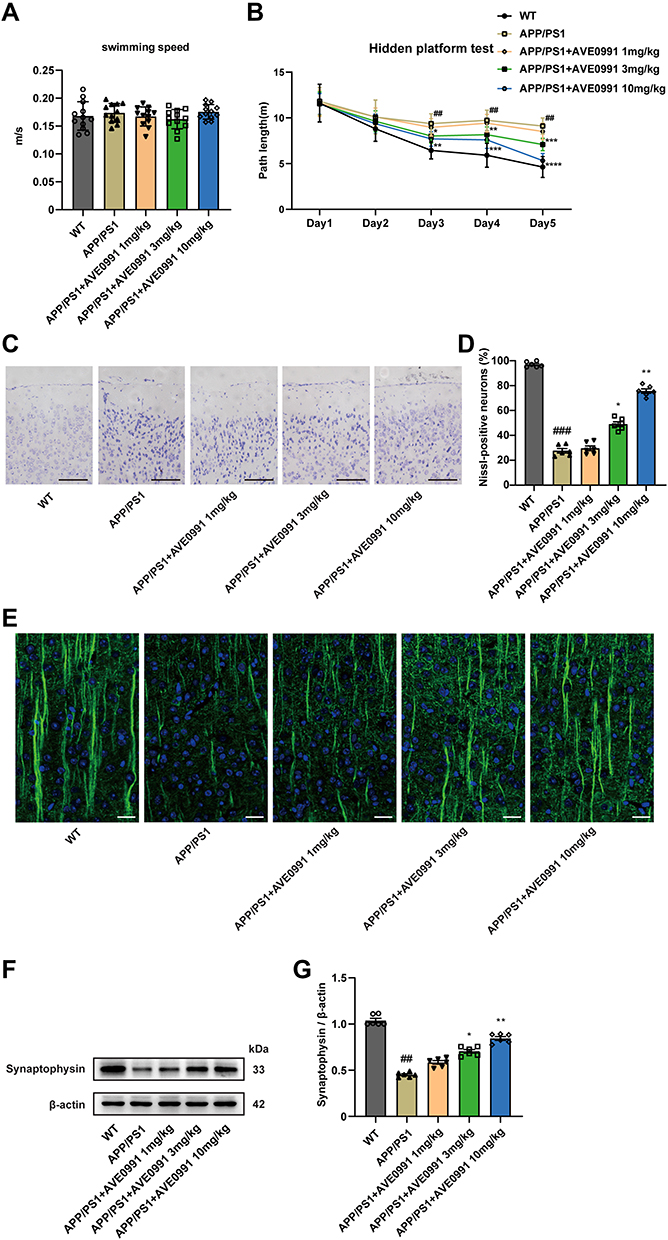 |
Figure 2 AVE 0991 attenuates cognitive deficits and neuron death in AD mice. (A) The swimming speed and (B) the path length were evaluated by MWM test for 5 days in mice injected with AVE 0991 (n = 12). (C) Neurons injuries in the cortex of mice injected with AVE 0991 were detected via Nissl staining. Scale bar, 100 μm (n = 6). (D) Quantitative analysis of Nissl-positive neurons was shown as bar chart (n = 6). (E) The protein expression of MAP-2 (green) in the brain cortex of mice injected with AVE 0991 were detected via Immunofluorescence staining. Nuclei subjected to DAPI-staining. Scale bar, 20 μm (n = 6). (F) The protein expression of synaptophysin in the brain cortex of mice injected with AVE 0991 was evaluated by Western blot analysis. β-actin was used as the loading control (n = 6). (G) Quantitative analysis of synaptophysin protein level was shown as bar chart (n = 6). All data are presented as mean ± SEM. ##P < 0.01 and ###P < 0.001 vs the WT group; *P < 0.05, **P < 0.01, ***P < 0.001 and ***P < 0.0001 vs the APP/PS1 group. |
AVE 0991 Reduces Soluble Aβ1-42 Concentrations in the Brain Cortex of APP/PS1 Mice
Next, we assessed soluble Aβ1-42 levels in the cortex, an important pathological feature of AD, using ELISA. As shown in Supplementary Figure 1, APP/PS1 mice exhibited significantly higher levels of soluble Aβ1-42 compared to WT mice, and AVE 0991 administration (3 and 10 mg/kg) significantly reduced soluble Aβ1-42 concentrations.
AVE 0991 Restrains Astrocyte Activation and Inflammation in AD Mice
In light of the fact that the deposition of Aβ in AD lesions can promote the proliferation of glial cells and trigger inflammatory response, and astrocytes are one of the most intensively studied cell types in neuroinflammation and AD.30 Therefore, we further tested the effects of AVE 0991 treatment on the activation of astrocytes and related pro-inflammatory factors in APP/PS1 mice, including IL-1β, TNF-α and IL-6. To begin with, the results of double immunofluorescence showed that Mas1 was not only expressed in microglia, but also remarkably co-localized with astrocytes (Supplementary Figure 2). Then, Western blot results suggested that the GFAP expression in the brain cortex of APP/PS1 mice was higher than that of the WT group, while was, however, inhibited by AVE 0991 treatment in a dose-dependent manner (Figure 3A and B). Next, to evaluate whether AVE 0991 can decrease the pro-inflammatory cytokines levels in the brain cortex of APP/PS1 mice, we measured the levels of IL-1β, IL-6 and TNF-α in mice brain cortex. As shown in Figure 3C–E, the mRNA expression of IL-1β, IL-6 and TNF-α was remarkably increased in the brain cortex of the APP/PS1 mice compared with the WT mice. Intriguingly, AVE 0991 treatment (3 and 10 mg/kg) markedly decreased the expression of these cytokines. In addition, in order to further verify the above results, we then tested the effect of AVE 0991 on IL-1β, IL-6 and TNF-α by ELISA. Results indicated that the concentrations of IL-1β, IL-6 and TNF-α in the brain cortex was in line with qRT-PCR results (Figure 3F–H). Predictably, AVE 0991 had no influence on the levels of pro-inflammatory cytokines in the cortex of WT mice (Supplementary Figure 3A–F). Taken together, these results showed that AVE 0991 could effectively inhibit the activation of astrocytes and pro-inflammatory cytokines transcription and secretion in APP/PS1 mice.
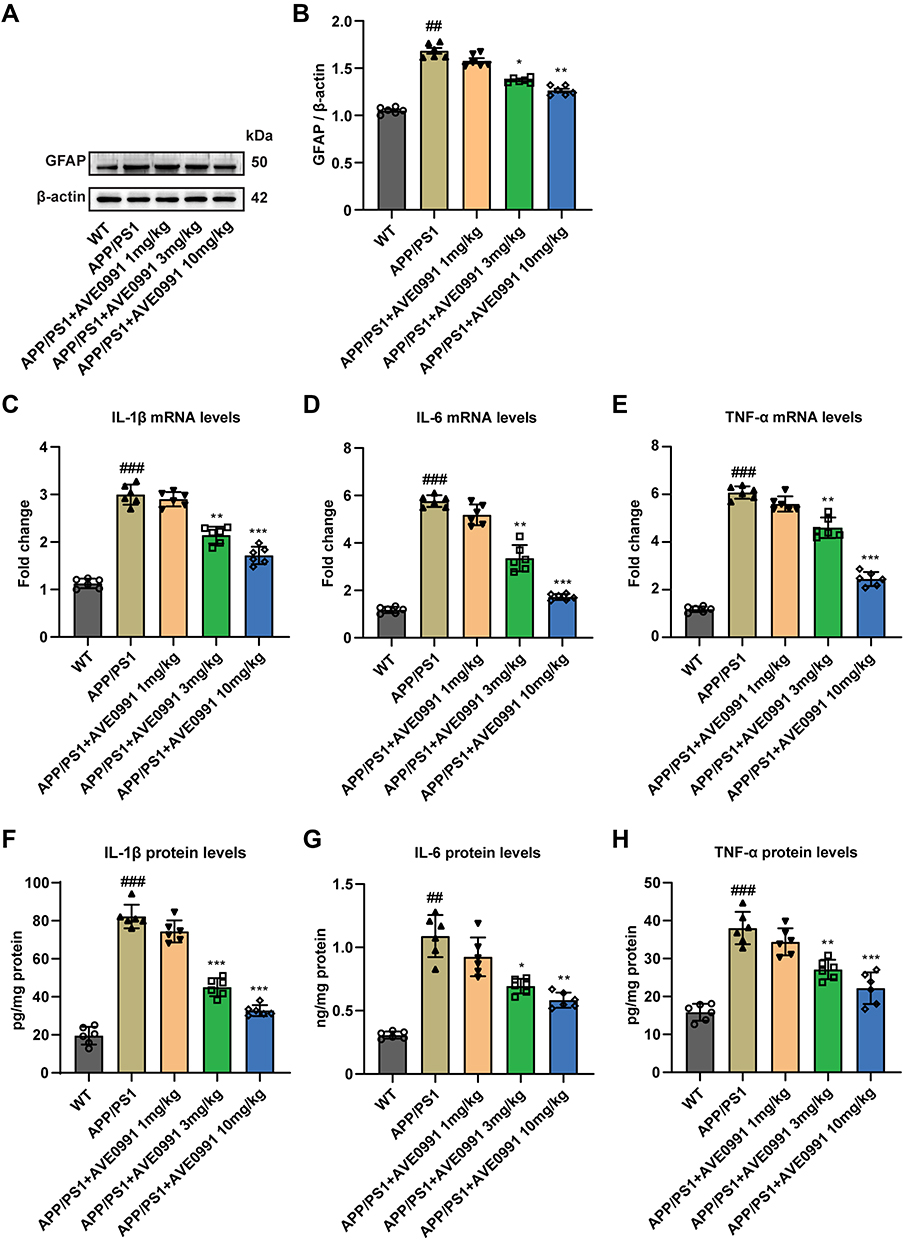 |
Figure 3 AVE 0991 restrains astrocyte activation and inflammation in AD mice. (A) The protein expression of GFAP in the brain cortex of mice injected with AVE 0991 was evaluated by Western blot analysis. β-actin was used as the loading control (n = 6). (B) Quantitative analysis of GFAP protein level was shown as bar chart (n = 6). The mRNA expressions of IL-1β (C), IL-6 (D) and TNF-α (E) in the brain cortex of mice injected with AVE 0991 were detected by qRT-PCR (n = 6). The protein expressions of IL-1β (F), IL-6 (G) and TNF-α (H) in the brain cortex of mice injected with AVE 0991 were measured by ELISA assay (n = 6). All data are presented as mean ± SEM. ##P < 0.01 and ###P < 0.001 vs the WT group; *P < 0.05, **P < 0.01 and ***P < 0.001 vs the APP/PS1 group. |
AVE 0991 Alleviates Inflammation and Promotes Autophagy in Aβ-Treated Astrocytes
In order to explore the effect of AVE 0991 on astrocyte-mediated inflammation, astrocytes were induced by Aβ1-42 in vitro experiments. First, immunofluorescence identification with GFAP proved that the primary astrocytes isolated in this study met the requirements (Figure 4A). Then we incubated astrocytes with different doses of AVE 0991 (1×10−8, 1×10−7和1×10−6 M) with or without Aβ1-42 for 24 h. CCK-8 results indicated that Aβ1-42 stimulation resulted in a prominent decrease in astrocyte viability, whereas AVE 0991 increased cell viability in a dose-dependent manner (Figure 4B). qRT-PCR results revealed that 1×10−7 and 1×10−6 M of AVE 0991 prominently increased Mas1 mRNA expression (Figure 4C). Consistent with the qRT-PCR results, Western blot analysis demonstrated that AVE 0991 administration (1×10−7 and 1×10−6 M) greatly increased the protein expression level of Mas1 (Figure 4D and E). Meanwhile, Western blot analysis showed that the expression of GFAP in astrocytes stimulated by Aβ1-42 was strongly increased compared with the normal group, while was inhibited by AVE 0991 in a dose-dependent manner (Figure 4F and G). Besides, compared with the normal group, Aβ1-42 considerably increased the mRNA expression of IL-1β, IL-6 and TNF-α in astrocytes. However, these increases were remarkably cancelled by AVE 0991 (Figure 4H–J). In addition, the protein levels of IL-1β, IL-6 and TNF-α in the culture medium also showed a similar trend by ELISA (Figure 4K–M). Remarkably, AVE 0991 treatment did not affect the expression of inflammatory cytokines in the absence of Aβ1-42 (Supplementary Figure 4A–F). These data suggest that the neuroprotective effect of AVE 0991 on Aβ 1–42-induced neurotoxicity is associated with reduced release of inflammatory cytokines in astrocytes.
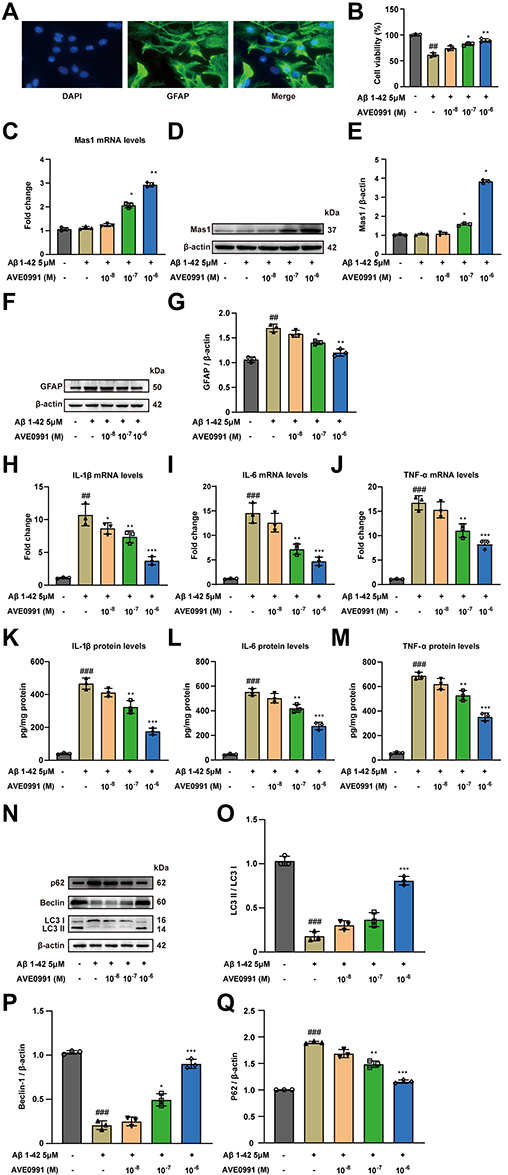 |
Figure 4 AVE 0991 alleviates inflammation and promotes autophagy in Aβ-treated astrocytes. (A) Images of immunofluorescence of primary astrocytes. Scale bar, 20 μm. (B) The cell viability was detected in Aβ-induced astrocytes treated with AVE 0991 using the CCK-8 assay (n = 3). (C) The mRNA expression of Mas1 in Aβ-induced astrocytes treated with AVE 0991 was detected by qRT-PCR (n = 3). (D) The protein expression of Mas1 in Aβ-induced astrocytes treated with AVE 0991 was evaluated by Western blot analysis. β-actin was used as the loading control (n = 3). (E) Quantitative analysis of Mas1 protein level was shown as bar chart (n = 3). (F) The protein expression of GFAP in Aβ-induced astrocytes treated with AVE 0991 was evaluated by Western blot analysis. β-actin was used as the loading control (n = 3). (G) Quantitative analysis of GFAP protein level was shown as bar chart (n = 3). The mRNA expressions of IL-1β (H), IL-6 (I) and TNF-α (J) in Aβ-induced astrocytes treated with AVE 0991 were detected by qRT-PCR (n = 3). The protein expressions of IL-1β (K), IL-6 (L) and TNF-α (M) in Aβ-induced astrocytes treated with AVE 0991 were measured by ELISA assay (n = 3). (N) The protein expressions of LC3, Beclin-1 and P62 in Aβ-induced astrocytes treated with AVE 0991 were evaluated by Western blot analysis. β-actin was used as the loading control (n = 3). Quantitative analysis of LC3 (O), Beclin-1 (P) and P62 (Q) protein levels was shown as bar chart (n = 3). All data are presented as mean ± SEM. ##P < 0.01 and ###P < 0.001 vs untreated astrocytes; *P < 0.05, **P < 0.01 and ***P < 0.001 vs Aβ-treated astrocytes. |
To further verify whether AVE 0991 can promote autophagy in Aβ1-42-induced astrocytes, the expression of autophagy-related proteins LC3, Beclin-1 and P62 in astrocytes was determined by Western blot. Aβ1-42 stimulation significantly reduced LC3 processing and Beclin-1 expression, while increased the protein abundance of P62 compared with the normal group, indicating that the autophagy function of astrocytes induced by Aβ1-42 was impaired (Figure 4N–Q). Different concentrations of AVE 0991 upregulated autophagy activity in Aβ1-42-induced astrocytes via increasing LC3 processing and Beclin-1 expression, whereas decreasing P62 level (Figure 4N–Q). Similarly, AVE 0991 had little effect on the expression of autophagy-related proteins in astrocytes stimulated without Aβ1-42 (Supplementary Figure 5A–D). These results indicate that AVE 0991 triggers autophagy in Aβ-treated astrocytes.
AVE 0991 Promotes Aβ-Induced Astrocytic Autophagy and Suppresses Inflammation Through a Mas1 Dependent Manner
Previous studies have identified that Mas1 is the functional receptor of Ang-(1-7) and is responsible for the beneficial physiological effects of Ang-(1-7).31 In order to determine whether the effect of AVE 0991, a selective agonist of Mas1, in regulating inflammation and autophagy was associated with the Mas1, astrocytes treated with AVE 0991 were co-incubated with Mas1 antagonist A-779 for 24 h concurrently. As expected, qRT-PCR results showed that AVE 0991 (1×10−6 M) substantially decreased the mRNA expression of cytokine in astrocytes, while A-779 abolished this inhibitory effect of AVE 0991 (Figure 5A–C). Similarly, ELISA results indicated that A-779 partially reversed the inhibitory effect of AVE 0991 on proinflammatory cytokines IL-1β, IL-6 and TNF-α (Figure 5D–F). In addition, Western blot analysis showed that AVE 0991 (1×10−6 M) notably increased the expression of LC3 processing and Beclin-1, while decreased the expression of P62. However, the effects of AVE 0991 on protein levels of LC3, Beclin-1 and P62 were partially eliminated by A-779 (Figure 5G–J). This observation indicated that AVE 0991 promoted astrocytic autophagy and inhibit astrocytic inflammation via a Mas1-dependent manner.
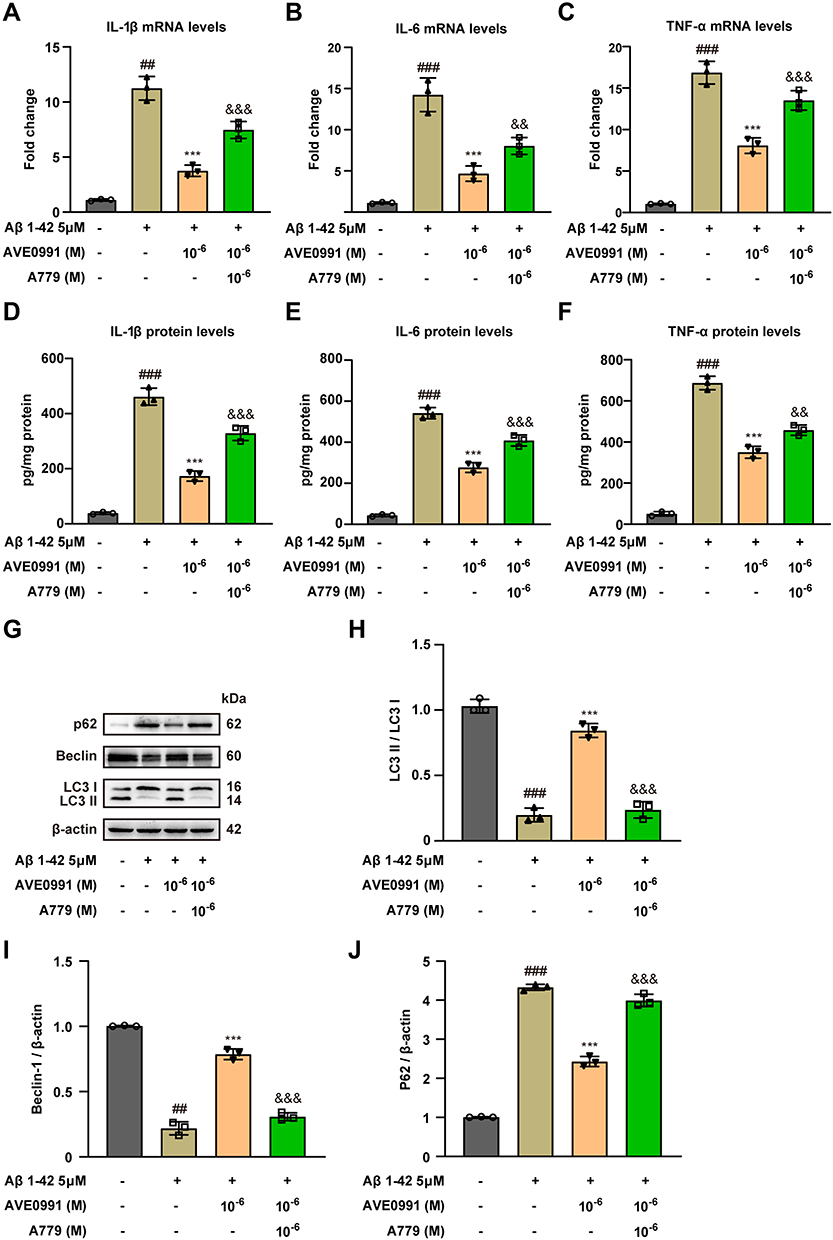 |
Figure 5 AVE 0991 promotes Aβ-induced astrocytic autophagy and suppresses inflammation through a Mas1 dependent manner. The mRNA expressions of IL-1β (A), IL-6 (B) and TNF-α (C) in Aβ-induced astrocytes treated with AVE 0991 with or without A-779 were detected by qRT-PCR (n = 3). The protein expressions of IL-1β (D), IL-6 (E) and TNF-α (F) in Aβ-induced astrocytes treated with AVE 0991 with or without A-779 were measured by ELISA assay (n = 3). (G) The protein expressions of LC3, Beclin-1 and P62 in Aβ-induced astrocytes treated with AVE 0991 with or without A-779 were evaluated by Western blot analysis. β-actin was used as the loading control (n = 3). Quantitative analysis of LC3 (H), Beclin-1 (I) and P62 (J) protein levels was shown as bar chart (n = 3). All data are presented as mean ± SEM. ##P < 0.01 and ###P < 0.001 vs untreated astrocytes; ***P < 0.001 vs Aβ-treated astrocytes; &&P < 0.01 and &&&P < 0.001 vs Aβ-treated astrocytes treated with AVE 0991 (1×10−6 M). |
AVE 0991 Suppresses Aβ-Induced Inflammatory Responses by Activating Astrocyte Autophagy
Recent studies have shown that autophagy can limit harmful and uncontrolled inflammation by removing pro-inflammatory cytokines. Considering the aforementioned neuroprotective ability of AVE 0991 in regulating autophagy and inflammation, we speculate that AVE 0991 may reduce neuroinflammation by promoting autophagy in Aβ1-42-induced astrocytes. 3-MA, the autophagy inhibitor, was used to block autophagy activation of astrocytes. 3-MA (5 μM) was added at 1 h before Aβ1-42 treatment for 24 h, and AVE 0991 was added at the same time as Aβ1-42 exposure. The protein abundance of autophagy markers in astrocytes was determined by Western blot. As expected, compared with Aβ1-42+AVE 0991 group, 3-MA treatment robustly decreased the expression of LC3 processing and Beclin-1, whereas the expression of P62 was enhanced by 3-MA (Figure 6A–D). This indicated that 3-MA prevents the promoting effect of AVE 0991 on astrocytes autophagy. Besides, we further evaluated the relationship between the inhibition of neuroinflammation and autophagy activation by AVE 0991 in Aβ1-42 treated astrocytes. ELISA results showed that AVE 0991 greatly reduced the increase of cytokines levels in astrocytes induced by Aβ1-42, while 3-MA eliminated the inhibitory effect of AVE 0991 on the pro-inflammatory cytokines IL-1β, IL-6 and TNF-α (Figure 6E–G). Consistent with ELISA results, qRT-PCR results proved that 3-MA significantly reduced the inhibitory effect of AVE 0991 on pro-inflammatory cytokines mRNA expression (Figure 6H–J). These findings suggested that AVE 0991 inhibits the inflammatory responses induced by Aβ1-42 by promoting autophagy in astrocytes.
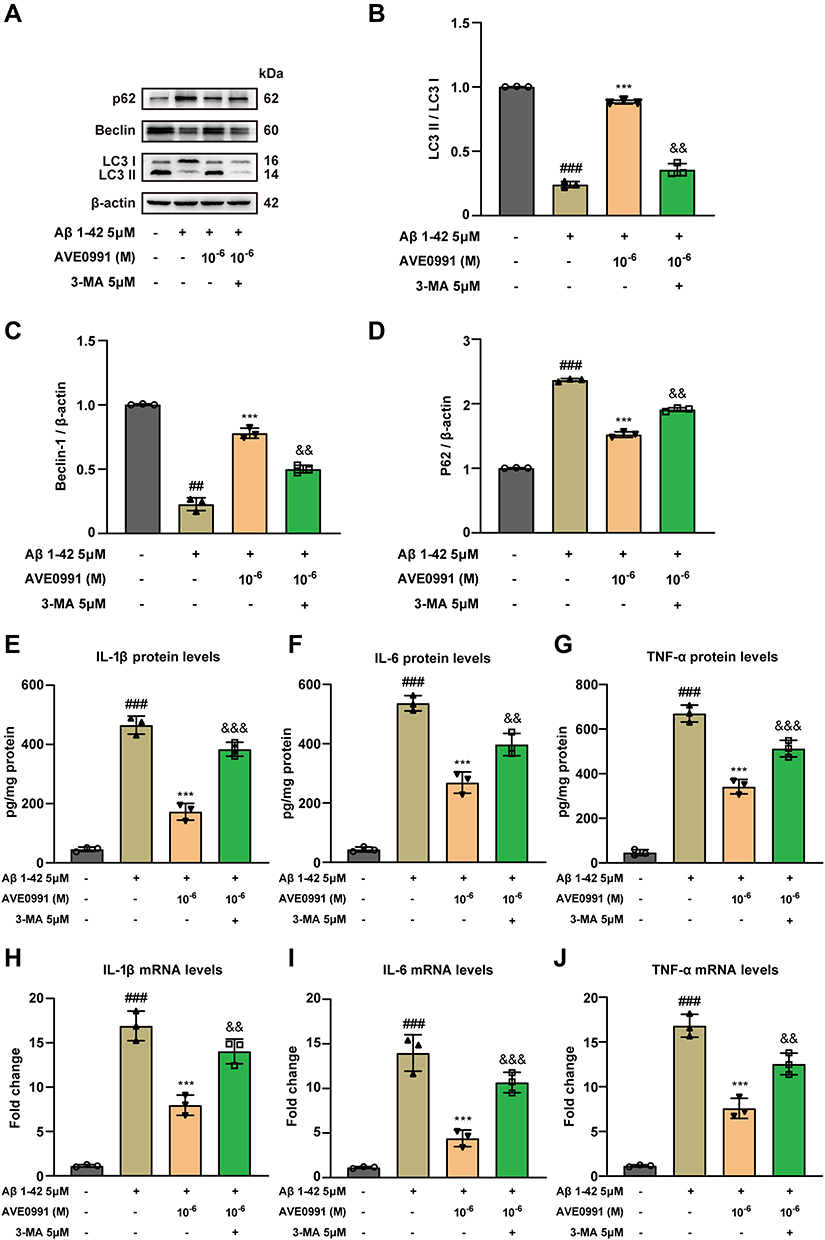 |
Figure 6 AVE 0991 suppresses Aβ-induced inflammatory responses by activating astrocyte autophagy. (A) The protein expressions of LC3, Beclin-1 and P62 in Aβ-induced astrocytes treated with AVE 0991 with or without 3-MA were evaluated by Western blot analysis. β-actin was used as the loading control (n = 3). Quantitative analysis of LC3 (B), Beclin-1 (C) and P62 (D) protein levels was shown as bar chart (n = 3). The protein expressions of IL-1β (E), IL-6 (F) and TNF-α (G) in Aβ-induced astrocytes treated with AVE 0991 with or without 3-MA were measured by ELISA assay (n = 3). The mRNA expressions of IL-1β (H), IL-6 (I) and TNF-α (J) in Aβ-induced astrocytes treated with AVE 0991 with or without 3-MA were detected by qRT-PCR (n = 3). All data are presented as mean ± SEM. ##P < 0.01 and ###P < 0.001 vs untreated astrocytes; ***P < 0.001 vs Aβ-treated astrocytes; &&P < 0.01 and &&&P < 0.001 vs Aβ-treated astrocytes treated with AVE 0991 (1x10−6 M). |
Discussion
AVE 0991 has been studied in different central nervous system diseases, including subarachnoid hemorrhage (SAH), chronic cerebral hypoperfusion (CCH) and ischemic stroke. For example, the expression of Ang-(1-7) and Mas1 observed was markedly increased in during cerebral ischemia, and AVE 0991 can protect neurons from ischemic damage when applied early during the ischemic period.32 Moreover, Mas expression was obviously decreased after SAH, while AVE 0991 reduced neuronal apoptosis and oxidative stress, and improved short-term and long-term neurological function.33 Furthermore, our previous study found that AVE 0991 significantly reduced hippocampal synaptic degeneration in CCH rats by promoting CBF recovery, reducing hippocampal Aβ levels and inhibiting neuroinflammatory responses.34 However, whether AVE 0991 has a neuroprotective effect in AD and its potential mechanisms are still largely unknown. In the current study, we demonstrated AVE 0991 plays a protective role in APP/PS1 mice, and illustrated its anti-inflammatory and pro-autophagy mechanisms in vivo and in vitro. In APP/PS1 mice, AVE 0991 treatment improved memory and cognitive impairment, neuronal loss and synaptic damage. Subsequently, we aimed to clarify whether the expression of pro-inflammatory cytokines and autophagy was regulated by AVE 0991. Our results indicated that AVE 0991 suppressed astrocyte-mediated neuroinflammation of Alzheimer’s disease by enhancing autophagy.
Neuroinflammation is currently considered to be widely involved in the occurrence and development of AD pathology.35 This inflammatory response can often promote tissue repair and remove cellular debris to produce beneficial effects. However, sustained inflammation is detrimental.36,37 In fact, elevated inflammatory markers such as cytokines IL-1β, IL-6 and TNF-α have been detected in AD patients or in different animal models with AD-like pathology.38 Therefore, maintaining the balance of inflammation in AD may be very important. Recently, the relationship between AVE 0991 and inflammation has been reported, suggesting that AVE 0991 may inhibit inflammation in response to stressful conditions. For instance, AVE 0991 alleviated pyrolysis and liver damage by inhibiting the reactive oxygen species (ROS)-NLRP3 inflammatory signaling pathway after heatstroke.39 In a mouse model of acute kidney injury (AKI) induced by bilateral ischemia/reperfusion (I/R) injury, AVE 0991 treatment attenuated local and systemic inflammatory responses and reduce renal functional impairment.40 Wang et al observed that AVE 0991 alleviated inflammation-induced arthritis by attenuating the nuclear transcription factor-kappaB (NF-κB) and mitogen-activated protein kinases (MAPK) pathways.41 In addition, our previous study confirmed that AVE 0991 inhibits microglia-mediated inflammatory response in a Mas receptor-dependent manner, thereby attenuating aging‐related neuroinflammation.24 However, whether AVE 0991 plays a protective role in AD by improving inflammation has not been confirmed. In the present study, our data found that AVE 0991 improved cognitive and memory impairment in mice. Furthermore, the inflammation levels of APP/PS1 mice were up-regulated, whereas AVE 0991 could down-regulated the expression of IL-1β, IL-6 and TNF-α in the brain cortex by a dose-dependent manner, indicating that AVE 0991 may play a neuroprotective effect in AD by suppressing inflammation.
Astrocytes and microglia have important activities in homeostasis and brain function, and are the major factors involved in the inflammatory process of AD.42 When inflammation occurs, both activated astrocytes and microglia produce and secrete several pro-inflammatory cytokines, including interleukin-1β (IL-1β), interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α), all of which could result in neurotoxicity.43 In fact, astrocytes are the most widely distributed cell type in brain and the time scale of pro-inflammatory signaling of astrocytes lasts longer than that of microglia.44,45 Besides, it is reported that GFAP expression increased in reactive astrocytes during AD, and this increase was associated with the progression of the disease.46 Moreover, among the different GFAP subtypes, GFAPα and GFAPδ immunoreactive astrocytes are often abundantly distributed near Aβ plaques.47 Thus, some authors proposed that Aβ-induced reactive astrocytes were the main driver of AD′s neuroinflammation.48 A previous study demonstrated that safflower leaf powder improved inflammatory responses and cognitive function in APP/PS1 mice by suppressing excessive astrocyte activation.49 In addition, Silibinin-loaded exosomes prevented astrocyte activation and attenuated astrocyte inflammation-mediated neuronal damage.50 Similar to these findings, our data proved that the elevation of GFAP in the brain cortex of APP/PS1 mice can be effectively inhibited by AVE 0991. Meanwhile, AVE 0991 treatment suppresses Aβ-stimulated astrocyte activation and decreases the production and secretion of pro-inflammatory cytokines in vitro, which was keeping with the results in vivo. These results indicated that the neuroprotective effect of AVE 0991 appears to be related to the reduced release of inflammatory cytokines after astrocyte activation.
Autophagy is one of the more effective ways by which the body removes misfolded proteins and damaged organelles, which is responsible for maintaining cell survival and normal function.19 It is reported that dysfunction in the process of autophagy is closely related to the pathology of AD.51 When AD occurs, it will lead to the accumulation of misfolded proteins (mainly composed of Aβ). Under physiological conditions, autophagy usually maintains Aβ homeostasis in healthy brains by clearing Aβ.52 Autophagy stimulation, moreover, can inhibit tau hyperphosphorylation.53 However, when autophagy is impaired, the clearance of these harmful proteins is delayed.54 Therefore, activating autophagy to enhance the elimination of harmful proteins was considered to be a promising target for discovery of anti-AD drugs. Unsurprisingly, our study found that soluble Aβ1-42 were significantly accumulated in the cerebral cortex of APP/PS1 mice, and treatment with AVE 0991 could partially eliminate this pathological change. Meanwhile, the association between AVE 0991 and autophagy has recently been studied. AVE 0991 treatment aggravated palmitic acid-induced autophagy and endoplasmic reticulum (ER) stress in human renal tubular epithelial cells.55 In LPS-stimulated microglia and mouse brain tissues, activation of Mas receptor by AVE 0991 can significantly trigger Forkhead box protein O1 (FoxO1) signaling and promote autophagy.25 Here, we investigated the potential ability of AVE 0991 to activate autophagy in AD. Our results showed that Aβ1-42 stimulation reduced the autophagy of astrocytes, as assessed by down-regulating LC3 processing, Beclin-1 expression and up-regulating p62 expression, while AVE 0991 treatment promoted the autophagy activity of astrocytes. This effect, nevertheless, can be partially eliminated by the Mas receptor antagonist A-779. This observation indicated that AVE 0991 promoted astrocyte autophagy through a Mas1 dependent manner. Current evidence suggested that astrocyte autophagy could limit harmful and uncontrolled inflammation, thereby acting as a central fulcrum to balance inflammatory responses.56 A recent study has confirmed that AVE 0991 enhanced the clearance of NLRP3 inflammasomes by promoting autophagy, thereby protecting the brain from excessive inflammation caused by LPS exposure.25 Therefore, we used 3-MA to study whether the anti-inflammatory effect of AVE 0991 was mediated by autophagy. Our results indicated that blockage of autophagy activation in astrocyte with 3-MA remarkably increased the release of pro-inflammatory cytokines induced by Aβ1-42 following AVE 0991 treatment. Similarly, the anti-inflammatory effect of AVE 0991 in vivo was remarkably eliminated by 3-MA. These data strongly indicated that AVE 0991 inhibited neuroinflammatory injury and brain damage in AD by promoting autophagy activation.
Notably, there are some limitations in this study. Firstly, in our present study, we did not explore in depth whether the increase of autophagy was due to the upregulation of autophagosome formation or the blockade of autophagic degradation and the specific period of action. Therefore, lysosomal protease inhibition or autophagic flux assays should be performed to distinguish these in the future. Secondly, in our present study, we only examined the effects of AVE 0991 on Aβ and autophagy, and focused on exploring the effects of autophagy on astrocyte inflammation after AD. Further research is necessary to investigate the correlation between autophagy and AD pathology.
In conclusion, our data demonstrated that AVE 0991 alleviated the activation of astrocytes, attenuated neuroinflammation and improved Aβ pathological deposition and memory cognitive function in AD. This effect may be regulated by astrocyte autophagy following AVE 0991 administration. Therefore, promoting autophagy to resist astrocyte-mediated inflammation may be a neuroprotective mechanism of AVE 0991 in AD. Our study proved that astrocyte autophagy may be a potential therapeutic target for AD, and provided a new insight into the neuroprotective mechanism of AVE 0991.
Funding
This work was supported by the National Science and Technology Innovation 2030 - Major program of “Brain Science and Brain-Inspired Intelligence Research” (2021ZD0201807), the Natural Science Foundation of Jiangsu Province (BK20201117), the International Joint Research and Development Project of Nanjing (202201030), the Natural Science Foundation of Jiangsu Province (BK20220196), China Postdoctoral Science Foundation (2022M711666) and Xinghuo Talent Program of Nanjing First Hospital.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Matej R, Tesar A, Rusina R. Alzheimer’s disease and other neurodegenerative dementias in comorbidity: a clinical and neuropathological overview. Clin Biochem. 2019;73:26–31. doi:10.1016/j.clinbiochem.2019.08.005
2. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi:10.1126/science.1072994
3. Braak H, Del Tredici K. The preclinical phase of the pathological process underlying sporadic Alzheimer’s disease. Brain. 2015;138(Pt 10):2814–2833. doi:10.1093/brain/awv236
4. Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16(6):358–372. doi:10.1038/nrn3880
5. Sala Frigerio C, Wolfs L, Fattorelli N, et al. The major risk factors for Alzheimer’s disease: age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep. 2019;27(4):1293–1306.e6. doi:10.1016/j.celrep.2019.03.099
6. Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405. doi:10.1016/S1474-4422(15)70016-5
7. Rossi D. Astrocyte physiopathology: at the crossroads of intercellular networking, inflammation and cell death. Prog Neurobiol. 2015;130. doi:10.1016/j.pneurobio.2015.04.003
8. Ledo JH, Azevedo EP, Beckman D, et al. Cross talk between brain innate immunity and serotonin signaling underlies depressive-like behavior induced by Alzheimer’s Amyloid-β oligomers in mice. J Neurosci. 2016;36(48):12106–12116. doi:10.1523/JNEUROSCI.1269-16.2016
9. Lourenco MV, Clarke JR, Frozza RL, et al. TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s β-amyloid oligomers in mice and monkeys. Cell Metab. 2013;18(6):831–843. doi:10.1016/j.cmet.2013.11.002
10. Bomfim TR, Forny-Germano L, Sathler LB, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Aβ oligomers. J Clin Invest. 2012;122(4):1339–1353. doi:10.1172/JCI57256
11. Colombo E, Farina C. Astrocytes: key regulators of neuroinflammation. Trends Immunol. 2016;37(9):608–620. doi:10.1016/j.it.2016.06.006
12. Clarke LE, Barres BA. Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci. 2013;14(5):311–321. doi:10.1038/nrn3484
13. Chung W-S, Clarke LE, Wang GX, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013;504(7480):394–400. doi:10.1038/nature12776
14. Liddelow S, Barres B. SnapShot: astrocytes in Health and Disease. Cell. 2015;162(5):1170–1170.e1. doi:10.1016/j.cell.2015.08.029
15. Choi M, Kim H, Yang E-J, Kim H-S. Inhibition of STAT3 phosphorylation attenuates impairments in learning and memory in 5XFAD mice, an animal model of Alzheimer’s disease. J Pharmacol Sci. 2020;143(4):290–299. doi:10.1016/j.jphs.2020.05.009
16. Liu L, Martin R, Chan C. Palmitate-activated astrocytes via serine palmitoyltransferase increase BACE1 in primary neurons by sphingomyelinases. Neurobiol Aging. 2013;34(2):540–550. doi:10.1016/j.neurobiolaging.2012.05.017
17. Habib N, McCabe C, Medina S, et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci. 2020;23(6):701–706. doi:10.1038/s41593-020-0624-8
18. Wang J-L, Xu C-J. Astrocytes autophagy in aging and neurodegenerative disorders. Biomed Pharmacother. 2020;122:109691. doi:10.1016/j.biopha.2019.109691
19. Ravanan P, Srikumar IF, Talwar P. Autophagy: the spotlight for cellular stress responses. Life Sci. 2017;188:53–67. doi:10.1016/j.lfs.2017.08.029
20. Wolfe DM, Lee J-H, Kumar A, Lee S, Orenstein SJ, Nixon RA. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur J Neurosci. 2013;37(12):1949–1961. doi:10.1111/ejn.12169
21. Hong Y, Liu Y, Zhang G, Wu H, Hou Y. Progesterone suppresses Aβ-induced neuroinflammation by enhancing autophagy in astrocytes. Int Immunopharmacol. 2018;54:336–343. doi:10.1016/j.intimp.2017.11.044
22. Jiang T, Gao L, Zhu X-C, et al. Angiotensin-(1-7) inhibits autophagy in the brain of spontaneously hypertensive rats. Pharmacol Res. 2013;71:61–68. doi:10.1016/j.phrs.2013.03.001
23. Gao Q, Chen R, Wu L, et al. Angiotensin-(1-7) reduces α-synuclein aggregation by enhancing autophagic activity in Parkinson’s disease. Neural Regen Res. 2022;17(5):1138–1145. doi:10.4103/1673-5374.324854
24. Jiang T, Xue L-J, Yang Y, et al. AVE0991, a nonpeptide analogue of Ang-(1-7), attenuates aging-related neuroinflammation. Aging. 2018;10(4):645–657. doi:10.18632/aging.101419
25. Dang R, Yang M, Cui C, et al. Activation of angiotensin-converting enzyme 2/angiotensin (1-7)/mas receptor axis triggers autophagy and suppresses microglia proinflammatory polarization via forkhead box class O1 signaling. Aging Cell. 2021;20(10):e13480. doi:10.1111/acel.13480
26. Yu J-Z, Li Y-H, Liu C-Y, et al. Multitarget therapeutic effect of fasudil in APP/PS1transgenic mice. CNS Neurol Disord Drug Targets. 2017;16(2):199–209. doi:10.2174/1871527315666160711104719
27. Jiang T, Yu J-T, Zhu X-C, et al. Triggering receptor expressed on myeloid cells 2 knockdown exacerbates aging-related neuroinflammation and cognitive deficiency in senescence-accelerated mouse prone 8 mice. Neurobiol Aging. 2014;35(6):1243–1251. doi:10.1016/j.neurobiolaging.2013.11.026
28. Klein WL. Abeta toxicity in Alzheimer’s disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem Int. 2002;41(5):345–352. doi:10.1016/S0197-0186(02)00050-5
29. Liu B, Liu J, Wang J, et al. Adiponectin protects against cerebral ischemic injury through AdipoR1/AMPK pathways. Front Pharmacol. 2019;10:597. doi:10.3389/fphar.2019.00597
30. Bandyopadhyay S. Role of neuron and glia in Alzheimer’s disease and associated vascular dysfunction. Front Aging Neurosci. 2021;13:653334. doi:10.3389/fnagi.2021.653334
31. Santos RAS, Simoes e Silva AC, Maric C, et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A. 2003;100(14):8258–8263. doi:10.1073/pnas.1432869100
32. Lee S, Evans MA, Chu HX, et al. Effect of a selective mas receptor agonist in cerebral ischemia in vitro and in vivo. PLoS One. 2015;10(11):e0142087. doi:10.1371/journal.pone.0142087
33. Mo J, Enkhjargal B, Travis ZD, et al. AVE 0991 attenuates oxidative stress and neuronal apoptosis via Mas/PKA/CREB/UCP-2 pathway after subarachnoid hemorrhage in rats. Redox Biol. 2019;20:75–86. doi:10.1016/j.redox.2018.09.022
34. Xue X, Duan R, Zhang -Q-Q, et al. A Non-Peptidic MAS1 agonist AVE0991 alleviates hippocampal synaptic degeneration in rats with chronic cerebral hypoperfusion. Curr Neurovasc Res. 2021;18(3):343–350. doi:10.2174/1567202618666211012095210
35. Saito T, Saido TC. Neuroinflammation in mouse models of Alzheimer’s disease. Clin Exp Neuroimmunol. 2018;9(4):211–218. doi:10.1111/cen3.12475
36. Kempuraj D, Thangavel R, Natteru PA, et al. Neuroinflammation induces neurodegeneration. J Neurol Neurosurg Spine. 2016;1(1):1003.
37. Russo MV, McGavern DB. Inflammatory neuroprotection following traumatic brain injury. Science. 2016;353(6301):783–785. doi:10.1126/science.aaf6260
38. Krabbe G, Halle A, Matyash V, et al. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One. 2013;8(4):e60921. doi:10.1371/journal.pone.0060921
39. Zhang M, Zhu X, Tong H, et al. AVE 0991 attenuates pyroptosis and liver damage after heatstroke by inhibiting the ROS-NLRP3 inflammatory signalling pathway. Biomed Res Int. 2019;2019:1806234. doi:10.1155/2019/1806234
40. Barroso LC, Silveira KD, Lima CX, et al. Renoprotective effects of AVE0991, a nonpeptide mas receptor agonist, in experimental acute renal injury. Int J Hypertens. 2012;2012:808726. doi:10.1155/2012/808726
41. Wang Z, Huang W, Ren F, et al. Characteristics of Ang-(1-7)/Mas-mediated amelioration of joint inflammation and cardiac complications in mice with collagen-induced arthritis. Front Immunol. 2021;12:655614. doi:10.3389/fimmu.2021.655614
42. Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21(3):383–421. doi:10.1016/S0197-4580(00)00124-X
43. Spangenberg EE, Green KN. Inflammation in Alzheimer’s disease: lessons learned from microglia-depletion models. Brain Behav Immun. 2017;61. doi:10.1016/j.bbi.2016.07.003
44. Rodríguez-Arellano JJ, Parpura V, Zorec R, Verkhratsky A. Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience. 2016;323:170–182. doi:10.1016/j.neuroscience.2015.01.007
45. Kato S, Gondo T, Hoshii Y, Takahashi M, Yamada M, Ishihara T. Confocal observation of senile plaques in Alzheimer’s disease: senile plaque morphology and relationship between senile plaques and astrocytes. Pathol Int. 1998;48(5):332–340. doi:10.1111/j.1440-1827.1998.tb03915.x
46. Simpson JE, Ince PG, Lace G, et al. Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol Aging. 2010;31(4):578–590. doi:10.1016/j.neurobiolaging.2008.05.015
47. Kamphuis W, Middeldorp J, Kooijman L, et al. Glial fibrillary acidic protein isoform expression in plaque related astrogliosis in Alzheimer’s disease. Neurobiol Aging. 2014;35(3):492–510. doi:10.1016/j.neurobiolaging.2013.09.035
48. Medeiros R, LaFerla FM. Astrocytes: conductors of the Alzheimer disease neuroinflammatory symphony. Exp Neurol. 2013;239:133–138. doi:10.1016/j.expneurol.2012.10.007
49. Zhang T, Zhang S, Peng Y, et al. Safflower leaf ameliorates cognitive impairment through moderating excessive astrocyte activation in APP/PS1 mice. Food Funct. 2021;12(22):11704–11716. doi:10.1039/D1FO01755A
50. Huo Q, Shi Y, Qi Y, Huang L, Sui H, Zhao L. Biomimetic silibinin-loaded macrophage-derived exosomes induce dual inhibition of Aβ aggregation and astrocyte activation to alleviate cognitive impairment in a model of Alzheimer’s disease. Mater Sci Eng C Mater Biol Appl. 2021;129:112365. doi:10.1016/j.msec.2021.112365
51. Carroll B, Hewitt G, Korolchuk VI, Lane JD. Autophagy and ageing: implications for age-related neurodegenerative diseases. Essays Biochem. 2013;55:119–131. doi:10.1042/bse0550119
52. Nilsson P, Loganathan K, Sekiguchi M, et al. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013;5(1):61–69. doi:10.1016/j.celrep.2013.08.042
53. Wang Y, Mandelkow E. Degradation of tau protein by autophagy and proteasomal pathways. Biochem Soc Trans. 2012;40(4):644–652. doi:10.1042/BST20120071
54. Ghavami S, Shojaei S, Yeganeh B, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24–49. doi:10.1016/j.pneurobio.2013.10.004
55. Kong Y, Zhao X, Qiu M, et al. Tubular Mas receptor mediates lipid-induced kidney injury. Cell Death Dis. 2021;12(1):110. doi:10.1038/s41419-020-03375-z
56. Cadwell K. Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat Rev Immunol. 2016;16(11):661–675. doi:10.1038/nri.2016.100
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.