")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 16
Autophagy Inhibits Inflammation via Down-Regulation of p38 MAPK/mTOR Signaling Cascade in Endothelial Cells
Authors Zhou L, Wang J, Hou H, Li J, Li J, Liang J, Li J, Niu X, Hou R, Zhang K
Received 17 January 2023
Accepted for publication 5 March 2023
Published 14 March 2023 Volume 2023:16 Pages 659—669
DOI https://doi.org/10.2147/CCID.S405068
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jeffrey Weinberg
Ling Zhou,1 Juanjuan Wang,1 Hui Hou,1 Jiao Li,1 Juan Li,1 Jiannan Liang,1 Junqin Li,1 Xuping Niu,1 Ruixia Hou,1 Kaiming Zhang2
1Shanxi Key Laboratory of Stem Cells for Immunological Dermatosis, Institute of Dermatology, Taiyuan Central Hospital of Shanxi Medical University, Taiyuan, People’s Republic of China; 2Shanxi Key Laboratory of Stem Cells for Immunological Dermatosis, Institute of Dermatology, Taiyuan Central Hospital, Taiyuan, People’s Republic of China
Correspondence: Kaiming Zhang, Taiyuan Central Hospital, Taiyuan, People’s Republic of China, Tel/Fax +86-351-5656080, Email [email protected]; [email protected]
Objective: Autophagy, an intracellular process of self-digestion, has been shown to modulate inflammatory responses. In the present study, we determined the effects of autophagy on inflammatory response induced by M5 cytokines.
Methods: Human umbilical vein endothelial cells (HUVECs) were treated with M5 cytokines to induce inflammation. Expression levels of mRNA for inflammatory cytokines and BIRC2 were compared in HUVECs with vs without induction of autophagy with rapamycin (RAPA) by PCR, while cell apoptosis was assessed by flow cytometry and caspase-3 activity assay kit. Expression levels of LC3, p62, p-p38 MAPK (Thr180/Tyr182), p-mTOR (Ser2445) and p-ULK1 (Ser555) proteins were measured by Western blotting. The nitric oxide (NO) content, NO synthase (NOS) activity and cell angiogenesis were also evaluated.
Results: Induction of autophagy with RAPA decreased expression levels of IL6, IL8 and CCL20, in addition to reduction in inflammation-induced apoptosis in HUVECs. Moreover, RAPA increased LC3II, while decreasing p62 expression. Likewise, expression levels of p-p38 MAPK and p-mTOR proteins were markedly decreased by the treatment with RAPA. Finally, RAPA treatment increased the NO content and the NOS activity, and inhibited angiogenesis.
Conclusion: Induced autophagy can improve the function of endothelial cells in psoriasis, suggesting approaches to induce autophagy can be used to ameliorate psoriasis.
Keywords: autophagy, inflammatory, HUVECs, p38 MAPK/mTOR
A Letter to the Editor has been published for this article.
A Response to Letter by Mr Maha has been published for this article.
Introduction
Autophagy is an evolutionarily conserved catabolic process that degrades cytoplasmic materials and provides a substrate for energy metabolism during nutrient deficiency and metabolic stress in order to maintain cellular homeostasis and adapt to adverse environments.1–5 Alterations of autophagy are associated with a number of inflammatory diseases, including psoriasis.6,7 A large number of studies have shown that autophagy and autophagy-related proteins are involved in immune regulation, such as intracellular bacterial clearance, secretion of inflammatory cytokines, antigen presentation and lymphocyte development.6,8 Autophagy is initiated by inducing autophagy genes for microtubule-associated protein light chain 3 (LC3), Beclin-1 and other autophagy-related proteins, which all play an important role in the maintenance cell homeostasis under physiological and pathological conditions.9–11
The regulation of autophagy is a very complex process. Mammalian target of rapamycin (mTOR), phosphoinositide 3-kinase (PI3K)/Akt, MAPK and other pathways are considered as major regulatory pathways of autophagy and have been widely studied.12,13 Mammalian rapamycin mechanistic target is a typical inhibitor of autophagy, which is related to growth factor nutrient and energy signals. Rapamycin can inhibit mTOR complex 1 (mTORC1), which effectively inhibits autophagy by phosphorylating ULK1. In addition to regulation of autophagy, the mTORC1 signaling pathway also regulates various processes in innate immune cells through various mechanisms such as metabolic protein translation and antigen presentation.14 In addition, mitogen-activated protein kinase (MAPK) signaling pathway regulated cell growth and differentiation. MAPK pathway is also considered to be the main regulation pathway of autophagy.15
Psoriasis is a chronic, multifactorial, immune-mediated skin disease.16 Psoriasis is considered as a systemic disease because psoriatic inflammation is involved both cutaneous and extracutaneous tissues.17 Previous studies demonstrated that abnormal autophagy contributes dermal angiogenesis,18 neovascularization, and extravasation of inflammatory cells into the lumen in psoriasis.19 Since autophagy deficiency can induce the production of proinflammatory cytokines by increasing the expression of p62,20 inflammation in psoriatic endothelial cells could also be linked to altered autophagy. Therefore, we studied here the regulatory role of autophagy in inflammation, apoptosis and endothelial cell function in vitro.
Materials and Methods
Materials
Materials and sources were as following: EBM-2 (Lonza, Germany, lot NO.: 9MB833), rapamycin (Solarbio, Beijing, Lot. No.: 1018N033), trypsin solution (Gibco Invitrogen, New York, USA, lot NO.: 1563418), eECL Western Blot Kit (CWBIO, Beijing, China, Lot No.: 20507), SB203580 (Med Chem Express, New Jersey, CAS No.: 152121-47-6), Chloroquine (CQ) (Med Chem Express, New Jersey, CAS No.: 54-05-7), BD matrigel (Corning, NewYork, USA, lot No.: 6172007), Annexin V-FITC/PI Apoptosis Assays Kit (KeyGenBio TECH, Nanjing, China, Cat. NO.: KGA107), capase-3 Kit (Beyotime, Shanghai, China, Lot NO.: 070320200803), NO Kit (JianCheng, Nanjing, China, NO.: A013-2-1), NOS Kit (JianCheng, Nanjing, China, NO.: A014-2-2), DAPI (Solarbio, China, lot-no. 20170412). Antibodies against β-actin, LC3, p62 were obtained from Abcam (Cambridge, England). p38 MAPK (8690), p-p38 MAPK (Thr180/Tyr182; 4511), ULK1 (8054), p-ULK1 (Ser555, 5869), mTOR (2983) and p-mTOR (Ser2445; 5536) were obtained from Cell Signaling Technology (Bossdun, USA).
Cell Culture and Treatment
Umbilical cord was provided by the department of obstetrics and gynecology of Taiyuan Central Hospital, Shanxi Medical University. Human umbilical vein endothelial cells (HUVECs) were cultured as described previously.21 HUVECs at about 70–80% confluency were treated with M5 cytokines (IL-1, IL-17, IL-22, TNF-α, Oncostatin M) for 4h (M5-HUVEC). Prior to the treatment with M5 cytokines, autophagy of HUVECs was induced by incubation of HUVECs with 200nM rapamycin (RAPA) in EBM-2 for 1h (R-M5-HUVEC).
Quantitative RT-PCR (qRT-PCR)
Quantitative RT-PCR was used to assess the expression levels of IL6, IL8, CCL20 and BIRC2 in HUVECs with and without RAPA treatment, as described previously.22 Total RNA was extract from control, M5-HUVEC and R-M5-HUVEC. RNA was reversely transcribed into cDNA. For the PCR assay, cDNA was mixed with QuantiTect SYBR Green PCR Master Mix, primers, and RNase-Free Water, and tested on Step OneTM. Primers information is shown in Table 1.
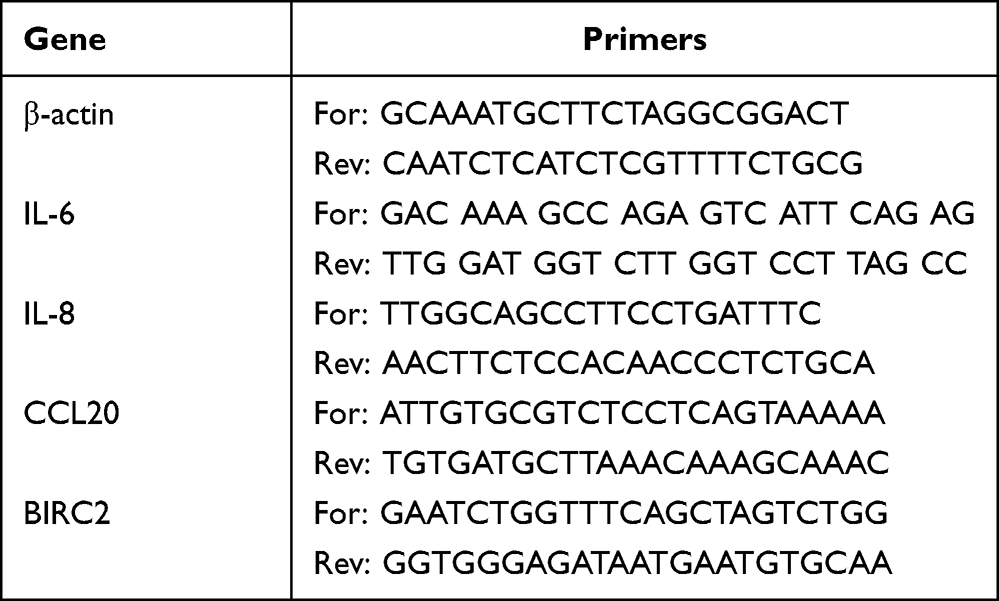 |
Table 1 Primers Used for RT-PCR |
Immunofluorescence
Cells on chamber slide were washed with PBS for 3 times, followed by fixation with 4% paraformaldehyde and permeation with 0.5%Triton x-100 for 20min at room temperature. After blocking with serum, cells were incubated with primary antibody LC3 (1:1000) overnight at 4°C. Afterward cells were incubation with secondary antibody for 1h. After DAPI staining, immunofluorescence staining was observed under an immunofluorescence microscope.
Western Blotting
Total protein was extracted from HUVECs for Western blot analysis. Cells were collected and lysed with ice-cold lysis buffer. Protein samples were bathed in metal bath for 10min at 95°C. LC3 was detected by traditional Western Blot. Briefly, a total of 20μg protein was loaded for Western blot assay. Electrophoresis was carried out using 12% separation glue and the transfer condition was 70V for 1.5h. Blotting was incubated overnight with LC3 rabbit primary antibody at 4°C, followed by washing with Tris-buffered saline containing 0.1% Tween for 3 times. The membrane was then incubated with a second antibody conjugated to horseradish peroxidase for 1h at room temperature. eECL Western Blotting reagent was used to detect the labeled proteins. Imaging was performed using the Protein Simple Fluor Chem Q imaging system (Protein Simple, USA).
Protein Simple was used to detect the expression levels of p62, p38 mitogen-activated protein kinase (p38 MAPK), phosphorylated p38 MAPK, unc-51 like kinase 1 (ULK1), phosphorylated ULK1, mammalian target of rapamycin (mTOR) and phosphorylated mTOR. Protein samples and monoclonal antibodies against p62, p38 MAPK, p-p38 MAPK, ULK1, p-ULK1, mTOR, p-mTOR (antibody ratio 1:100) were added according to manufacturer’s instructions, tested on WES system.
Apoptosis Was Detected by Flow Cytometry
After digestion and collection, cells were washed twice with PBS and centrifuged at 2000rpm for 5min. The cells were resuspended with 500μL Binding Buffer, followed by addition of 5μL Annexin V-FITC and 5μL Propidium Iodide. After incubation at room temperature for 5–15min in dark, apoptosis was detected by flow cytometry.
Measurement of Caspase-3 Activity
The cells were collected and the protein was extracted by adding 100μL lysate per 2*106 cells. Protein concentrations were measured by Bradford’s method and caspase-3 activity was detected with caspase-3 activity assay kit according to the manufacturer’s instructions.
NO/NOS
Expression of NO and NOS activity were detected with respective kits. Assay was performed according to the manufacturer’s protocol. The absorbance of OD value was measured at wavelength of 550nm with a microplate reader.
Angiogenesis Experiment
The angiogenesis experiment was performed as described previously.23 Precooled tip was used to add BD matrigel glue to 96-well plate, 50μL/well. Afterward the cells were digested and inoculated with 1*104cells/cm2 for 24h, and then cultured at 37°C and 5% CO2 for 6h. Under the microscope, five fields were randomly selected to count the numbers of junction and mesh. The data were expressed as percentages of control, and the control was set at 100%.
Statistical Analysis
One-way ANOVA with Tukey’ s multiple comparisons was used to determine significant differences when three or more groups were compared, while an unpaired t-test was used to determine significance between two groups. p < 0.05 was considered statistically significant. All analyses were performed using SPSS.
Results
Induction of Autophagy Alleviates Inflammation Induced by M5 Cytokines
Because activation of autophagy can inhibit inflammation in croakers,24 we first assessed here whether induction of autophagy can also inhibit inflammation in HUVECs. HUVEC inflammation was induced by incubation with M5 cytokines for 4h. As seen in Figure 1a, addition of M5 cytokines to HUVEC culture significantly increased expression levels of mRNA for IL-6, IL-8 and CCL20, whereas induction of autophagy with RAPA lowered the expression levels of IL-6, IL-8 and CCL20 mRNA to the levels comparable to that of the controls. To ascertain whether RAPA induces autophagy, we measured expression levels of autophagy-associated biomarkers in the HUVEC cultures. As expected, RAPA treatment markedly increased the ratio of LC3I/LC3II protein (Figure 1b and c), while decreasing p62 expression (Figure 1d and e). In parallel, RAPA treatment increased fluorescence intensity of LC3 (Figure 1f and g). The results show that RAPA induces autophagy, likely contributing to the alleviation of the M5 cytokines-induced inflammation in HUVECs.
 |
Figure 1 Induction of autophagy alleviates inflammation in HUVECs. Inflammation of HUVECs was induced by addition of M5 cytokines to the cultures, while autophagy was induced by the treatment of HUVECs with Rapamycin. (a) Expression levels of mRNA for IL6, IL8 and CCL20; (b and c) Expression levels of LC3 protein and ratio of LC3I/LC3II; (d and e) Expression levels of p62 protein; (f and g) Expression of LC3 assessed by immunofluorescence. n=5, **p<0.01. |
Autophagy Inhibits Inflammation-Induced Apoptosis
Autophagy and apoptosis, different forms of cell death, interact with each other. Autophagy can antagonize apoptosis by promoting cell survival.25 We next determined whether autophagy can inhibit apoptosis of HUVECs. Flow cytometry showed a significant increase in M5-HUVEC apoptosis (both early and late apoptosis) compared to the controls (Figure 2a–d). Induction of autophagy with RAPA (R-M5-HUVEC) significantly reduced apoptosis (both early and late apoptosis). Correspondingly, expression levels of BIRC2 mRNA, an inhibitor of apoptosis, were dramatically increased in R-M5-HUVEC compared with M5-HUVEC (Figure 2e). In contrast, caspase-3 activity was significantly decreased following the treatment of M5-HUVECs with RAPA (Figure 2f), indicating an inhibition of apoptosis. These results indicate that autophagy inhibits inflammation-induced apoptosis in HUVECs.
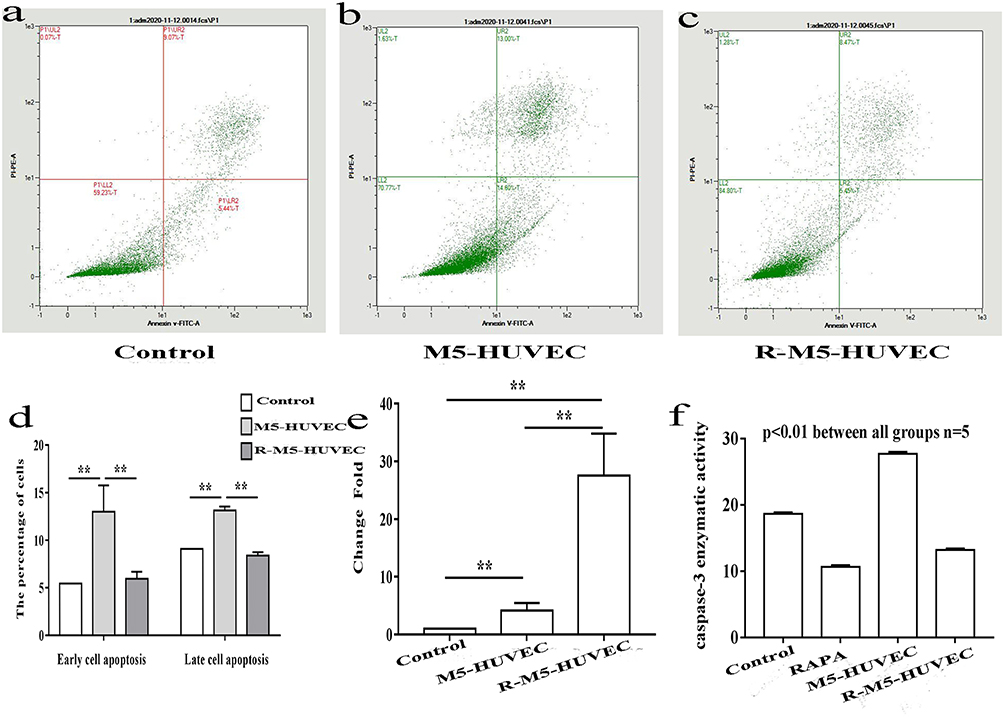 |
Figure 2 Induction of autophagy inhibits apoptosis in HUVECs. (a–c) Cell apoptosis assessed by flow cytometry; (d) Quantitative data of percentage of apoptotic cells; (e) Expression levels of BIRC2 mRNA, and (f) caspase-3 activity. n=5, **p<0.01. |
Autophagy Inhibits Inflammation Through the p38 MAPK/mTOR Pathway
p38 MAPK pathway plays an important role in autophagy.26,27 ULK is the only core protein with serine/threonine kinase activity in autophagy signaling pathway. ULK1 complex acts as a bridge between upstream nutrient or energy receptor mTOR and downstream autophagosome in vivo. Phosphorylated ULK1 has long been considered a key regulator of autophagy.28 To assess the involvement of p38 MAPK-mTOR-ULK1 signaling in the regulation of inflammation by autophagy, we measured expression levels of LC3, p62, p38 MAPK, p-p38 MAPK (Thr180/Tyr182), mTOR, p-mTOR (Ser2448), ULK1 and p-ULK1 (Ser555). M5-HUVEC displayed significantly higher expression levels of LC3II (Figure 3a and b) and p62 (Figure 3c) compared with controls. RAPA treatment increased the ratio of LC3II/LC3I in R-M5-HUVEC while decreasing p62 expression, indicating an induction of autophagy. Chloroquine (CQ) can inhibit autophagy by lysosomal acidification and subsequently blocks the fusion of the autophagosome with lysosome, leading to the accumulation of autophagosome. Inhibition of autophagy with CQ increased both the ratio of LC3II/LC3I and p62 expression (Figure 3a–c), indicating autophagy initiation is normal, but autophagy flow is disrupted. Inhibition of p38 MAPK with SB203580 increased LC3II protein expression while decreasing p62 expression, indicating enhanced autophagy (Figure 3a–c). In addition, the expression levels of both p-p38 MAPK and p-mTOR in R-M5-HUVEC and SB-R-M5-HUVEC were significantly decreased compared with M5-HUVEC (Figure 3d and f), and the expression of p-p38 MAPK in SB-R-M5-HUVEC was significantly decreased compared with R-M5-HUVEC (Figure 3d), indicating that p38 MAPK is a negative regulator of autophagy. In contrast, inhibition of autophagy with CQ increased expression levels of p-mTOR in RAPA-treated M5-HUVECs (Figure 3f). On the other hand, p38 MAPK inhibitor increased the expression of p-ULK1 in RAPA-treated M5-HUVEC while autophagy inhibitor, CQ, decreased p-ULK1 (Figure 3e). The results show that RAPA-induced autophagy negatively regulates inflammation through the p38 MAPK/mTOR pathway.
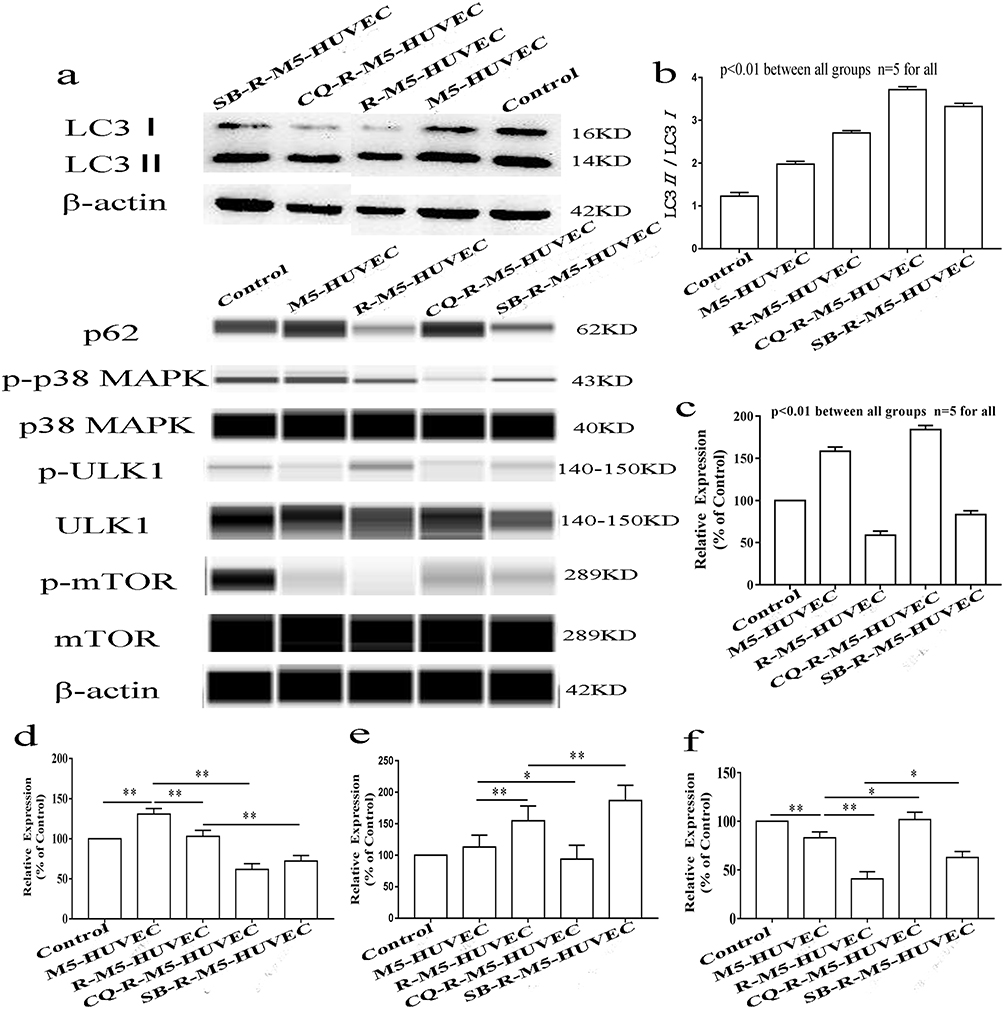 |
Figure 3 Autophagy-induced inhibition of inflammation is via p38 MAPK/mTOR signaling pathway. (a) is the representative images of Western blot; (b and c) are quantitative diagrams of expression levels of LC3 and p62; (d–f) are quantitative diagrams of expression levels of p-p38 MAPK, p-ULK1, and p-mTOR, respectively. n=5, *p<0.05, **p<0.01. |
Induction of Autophagy Improves the Function of Inflamed Endothelial Cells
Dysfunctions in psoriatic endothelial cells include increased pro-inflammatory response, decreased vasodilation, vasogenesis and thrombosis.29 The decreased vasodilation is attributable to the reduced production of nitric oxide (NO).30 Therefore, we detected NO content and NOS activity in HUVECs. As can be seen in Figure 4a and b, both NO content and NOS activity declined significantly in M5-HUVECs vs normal controls. Induction of autophagy with RAPA significantly increased both NO content and NOS activity in M5-HUVECs, while inhibition of autophagy reversed the effect of RAPA on both NO content and NOS activity, indicating that autophagy increases NO content and NOS activity in M5-HUVECs.
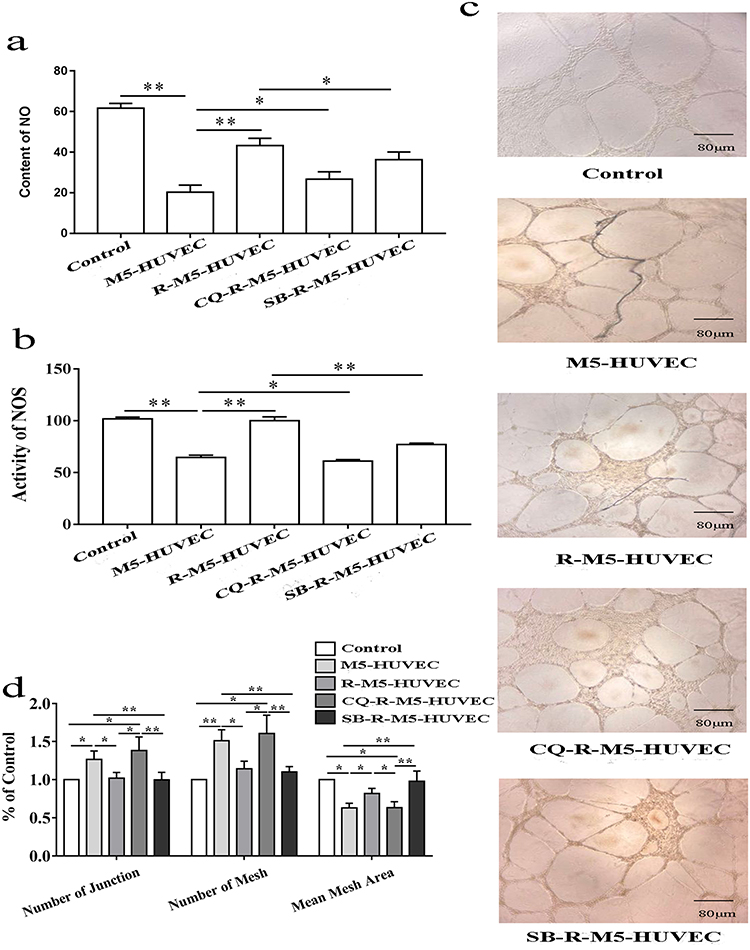 |
Figure 4 Autophagy improves endothelial cell function. (a and b) show NO levels and NOS activity, respectively; (c and d) display representative images of angiogenesis and quantitative diagrams of angiogenesis in HUVEC culture, respectively. n=5, *p<0.05, **p<0.01. |
Finally, we assessed whether the influence of autophagy on HUVEC function is reflected in angiogenesis, an abnormality in psoriasis. Our results showed that the numbers of both junction and mesh were increased and mean mesh areas were decreased in M5-HUVECs in comparison to normal controls (Figure 4c and d). Inhibition of autophagy overcame the effect of RAPA on the formations of junction and mesh, mean mesh areas in M5-HUVECs. In contrast, neither the numbers of junction nor mesh or mean areas of mesh differed significantly in R-M5-HUVECs treated with or without p38 MAPK inhibitor. Taken together, these results demonstrate that autophagy improves the function of inflamed endothelial cells via inhibition of p38 MAPK.
Discussion
The psoriasis-involved skin is characterized by increased blood vessels and angiogenesis, suggesting the pathogenic role of blood vessels and endothelial cells in psoriasis. So far, the studies on the role of endothelial cells in the pathogenesis of psoriasis are limited although regulation of CARD14+ ECs in production of cytokines and chemokines (IL-8 and CXCL1 etc.) has been documented.31 However, the pathomechanisms of endothelial cell inflammation in psoriasis have not been well defined. Since it is difficult to culture psoriatic endothelial cells from psoriatic skin, we established a psoriatic endothelial cell model by incubation of HUVECs with the M5 cytokines. This model exhibits some phenotypes of psoriatic endothelial cells such as increased expression levels of IL-6, IL-8 and CCL20. We show here that altered autophagy contributes, at least in part, to the development of inflammation in psoriatic endothelial cells.
Autophagy, also called programmed type II death, is a conserved degradation of the cells, removing unnecessary or dysfunctional components through a lysosome-dependent regulated mechanism, which is different from apoptotic programmed type I death.32,33 Autophagy and inflammation are highly intertwined cellular processes.10 Autophagy exerts anti-inflammatory property by regulating innate immune signaling pathway and inflammatory body activity.34,35 Moreover, autophagy can also be activated during the occurrence of inflammation. Autophagy not only affects the relief of infectious diseases and the pathological process of inflammatory diseases but also can inhibit inflammatory response to the damages of non-infectious tissues.36 Growing evidences indicate that autophagy dysfunction not only causes psoriasis but also aggravates the inflammation in the pathogenesis of psoriasis. Autophagy-related proteins regulate multiple immune functions, including secretion of inflammatory cytokines, bacterial clearance and lymphocyte development. Because of the possible pathogenic role of autophagy in psoriasis, it could be a target in the treatment of psoriasis.37 Correspondingly, in the present study, we demonstrate induction of autophagy inhibits inflammation and apoptosis in HUVECs.
The mechanism by which autophagy inhibits inflammation is not clear. But evidence points to the involvement of p38 MAPK/mTOR signaling pathway. MAPK is an important cellular transduction pathway, regulating cell growth and differentiation. Recent studies have shown that p38 MAPK, an intracellular signal transduction molecule, regulates a variety of inflammatory responses, including the expression of pro-inflammatory cytokines, leukocyte adhesion and chemotaxis. MAPK signaling pathways are involved in the regulation of autophagy.38 Autophagy is a catabolic pathway regulated by a complex signal network. p38 MAPK is a stress-activated protein kinase because it is frequently activated in response to inflammatory responses induced by various environmental stresses (eg, REDOX stress, UV irradiation of cytokines, heat shock and osmotic shock), which is a key process in the host defense system. Moreover, p38 MAPK regulates cell cycle, promoting apoptosis, differentiation and senescence39 and inhibits basic autophagy by blocking Atg9.40 Furthermore, ULK1 is a key upstream regulator of autophagy.27 p38α MAPK can directly phosphorylate ULK1 and inhibit ULK1 kinase activity, leading to destruction of ULK1 functional complex with ATG13, consequently resulting in reduced autophagy. In the present study, we showed that p38 MAPK was activated by inflammation, and induction of autophagy reduced the phosphorylation of p38 MAPK. Additionally, mTOR is a serine/threonine protein kinase and a key negative regulator of autophagy initiation.41 It phosphorylates an important autophagy protein ULK1 and inhibits its activity, thereby preventing the formation of ULK1-ATG13-FIP200 complex and inhibiting autophagy.42 Finally, mTOR signaling pathway inhibits autophagy by phosphorylating the transcription factor EB and preventing its nuclear translocation from expressing autophagy genes.43 Our results showed that either induction of autophagy or inhibition of p38 MAPK lowered expression levels of p-mTOR. Thus, autophagy-induced inhibition of inflammation is in part via inhibition of p38 MAPK-mTOR-ULK1 signaling pathways.
Considering the correlation between NO/NOS and endothelial cell function, we assessed the expression levels of NO and the activity of NOS. NO, synthesized by NOS, has a variety of physiological and pathological functions. NO is essential for maintaining microvascular endothelial cell function and vascular homeostasis by inducing vasodilation and inhibiting platelet adhesion and aggregation.44 Under inflammatory conditions, the physiological activity of eNOS may be impaired, leading to a so-called uncoupled state characterized by the production of superoxide O2− instead of NO.45 Therefore, the bioavailability of NO in patients with psoriasis is reduced, leading to systemic microvascular dysfunction. Reduced bioavailability of nitric oxide impaired endothelium-dependent vasodilation.46 The results of the present study showed that induction of autophagy increased NO expression and NOS activity and stimulated endothelial angiogenesis. In agreement with our findings, other studies showed that autophagy stimulates No production,47 but the specific mechanism remains to be elucidated.
Conclusion
In this study, we established a psoriatic endothelial cell model to investigate the effects of autophagy on inflammation and apoptosis of HUVEC. Induction of autophagy can inhibit inflammation and apoptosis, while improving endothelial cell functions in HUVECs treated with M5 cytokines, mediated in part by p38 MAPK/mTOR signaling pathway. Induction of autophagy can improve the function of endothelial cells in psoriasis, potentiating the utility of approaches to enhance autophagy in the treatment of psoriasis.
Abbreviations
HUVECs, Human umbilical vein endothelial cells; LC3, Light chain 3; p38 MAPK, p38 Mitogen-activated protein kinase; ULK1, unc-51 like kinase 1, mTOR, mammalian target of rapamycin; RAPA, Rapamycin; CQ, Chloroquine; NO, Nitric oxide; EGM-2, Endothelial cell growth medium-2.
Data Sharing Statement
All data are available upon request.
Ethics Approval and Consent to Participate
Ethical approval for the experiments was obtained from the Medical Ethics Committee of Taiyuan City Centre Hospital, and all subjects provided informed consent.
Funding
This project was supported by the National Natural Science Foundation of China (grant no.81773336, 81803146, 81472888).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R, Tang D. Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem Biol. 2020;27(4):420–435. doi:10.1016/j.chembiol.2020.02.005
2. Hazari Y, Bravo-San Pedro JM, Hetz C, Galluzzi L, Kroemer G. Autophagy in hepatic adaptation to stress. J Hepatol. 2020;72(1):183–196. doi:10.1016/j.jhep.2019.08.026
3. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi:10.1016/j.cell.2007.12.018
4. Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell. 2014;159(6):1263–1276. doi:10.1016/j.cell.2014.11.006
5. Netea-Maier RT, Plantinga TS, van de Veerdonk FL, Smit JW, Netea MG. Modulation of inflammation by autophagy: consequences for human disease. Autophagy. 2016;12(2):245–260. doi:10.1080/15548627.2015.1071759
6. Wu DJ, Adamopoulos IE. Autophagy and autoimmunity. Clin Immunol. 2017;176:55–62. doi:10.1016/j.clim.2017.01.007
7. Li XM, Jung KE, Yim SH, et al. Autophagy suppresses toll-like receptor 3-mediated inflammatory reaction in human epidermal keratinocytes. Biomed Res Int. 2020;2020:4584626. doi:10.1155/2020/4584626
8. Cao Y, Chen J, Ren G, Zhang Y, Tan X, Yang L. Punicalagin prevents inflammation in LPS-Induced RAW264.7 macrophages by Inhibiting FoxO3a/autophagy signaling pathway. Nutrients. 2019;11(11):2794. doi:10.3390/nu11112794
9. Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333(6046):1109–1112. doi:10.1126/science.1201940
10. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–335. doi:10.1038/nature09782
11. Zhang Q, Kang R, Zeh HJ, et al. DAMPs and autophagy: cellular adaptation to injury and unscheduled cell death. Autophagy. 2013;9(4):451–458. doi:10.4161/auto.23691
12. Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12(9):814–822. doi:10.1038/ncb0910-814
13. Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124–131. doi:10.1016/j.ceb.2009.11.014
14. Ko JH, Yoon S-O, Lee HJ, Oh JY. Rapamycin regulates macrophage activation by inhibiting NLRP3 inflammasome-p38 MAPK-NFκB pathways in autophagy- and p62-dependent manners. Oncotarget. 2017;8(25):40817–40831. doi:10.18632/oncotarget.17256
15. Sui X, Kong N, Ye L, et al. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic. Cancer Lett. 2014;344(2):174–179. doi:10.1016/j.canlet.2013.11.019
16. Vicic M, Kaštelan M, Brajac I, et al. Current concepts of psoriasis immunopathogenesis. Int J Mol Sci. 2021;22(21):11574. doi:10.3390/ijms222111574
17. Aryanian Z, Jafaripour I, Kohneshin E, et al. Echocardiographic and electrocardiographic assessments in patients with psoriasis. Caspian J Intern Med. 2021;12(2):162–166. doi:10.22088/cjim.12.2.162
18. Locker F, Vidali S, Holub BS, et al. Lack of galanin receptor 3 alleviates psoriasis by altering vascularization, immune cell infiltration, and cytokine expression. J Invest Dermatol. 2018;138(1):199–207. doi:10.1016/j.jid.2017.08.015
19. Rubina KA, Yu Sysoeva V, Zagorujko EI, et al. Increased expression of uPA, uPAR, and PAI-1 in psoriatic skin and in basal cell carcinomas. Arch Dermatol Res. 2017;309(6):433–442. doi:10.1007/s00403-017-1738-z
20. Barrera M-J, Aguilera S, Castro I, et al. Tofacitinib counteracts IL-6 overexpression induced by deficient autophagy: implications in Sjögren’s syndrome. Rheumatology. 2021;60(4):1951–1962. doi:10.1093/rheumatology/keaa670
21. Zhou L, Wang J, Liang J, et al. Psoriatic mesenchymal stem cells stimulate the angiogenesis of human umbilical vein endothelial cells in vitro. Microvasc Res. 2021;136:104151. doi:10.1016/j.mvr.2021.104151
22. Hou R, Yin G, Peng A, et al. DNA methylation of dermal MSCs in psoriasis: identification of epigenetically dysregulated genes. J Dermatol Sci. 2013;72(2):103–109. doi:10.1016/j.jdermsci.2013.07.002
23. Zhou L, Niu X, Liang J, et al. Efficient differentiation of vascular endothelial cells from dermal-derived mesenchymal stem cells induced by endothelial cell lines conditioned medium. Acta Histochem. 2018;120(8):734–740. doi:10.1016/j.acthis.2018.08.004
24. Yang B, Renlei J, Xueshan L, et al. Activation of autophagy relieves linoleic acid-induced inflammation in large yellow croaker (Larimichthys crocea). Front Immunol. 2021;12:649385. doi:10.3389/fimmu.2021.649385
25. Hamada M, Kameyama H, Iwai S, Yura Y. Induction of autophagy by sphingosine kinase 1 inhibitor PF-543 in head and neck squamous cell carcinoma cells. Cell Death Discov. 2017;3:17047. doi:10.1038/cddiscovery.2017.47
26. Liu Y, Fan D. Ginsenoside Rg5 induces G2/M phase arrest, apoptosis and autophagy via regulating ROS-mediated MAPK pathways against human gastric cancer. Biochem Pharmacol. 2019;68:285–304. doi:10.1016/j.bcp.2019.07.008
27. He Y, She H, Zhang T, et al. p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1. J Cell Biol. 2018;217(1):315–328. doi:10.1083/jcb.201701049
28. Wang C, Wang H, Zhang D, et al. Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat Commun. 2018;9(1):3492. doi:10.1038/s41467-018-05449-1
29. Mercurio L, Failla CM, Capriotti L, et al. Interleukin (IL)-17/IL-36 axis participates to the crosstalk between endothelial cells and keratinocytes during inflammatory skin responses. PLoS One. 2020;15(4):e0222969. doi:10.1371/journal.pone.0222969
30. Alba BK, Greaney JL, Ferguson SB, Alexander LM. Endothelial function is impaired in the cutaneous microcirculation of adults with psoriasis through reductions in nitric oxide-dependent vasodilation. Am J Physiol Heart Circ Physiol. 2018;314(2):H343–H349. doi:10.1152/ajpheart.00446.2017
31. Harden JL, Lewis SM, Pierson KC, et al. CARD14 expression in dermal endothelial cells in psoriasis. PLoS One. 2014;9(11):e111255. doi:10.1371/journal.pone.0111255
32. Yufeng X, Rong J, Wang Y. Roles of Annexin A protein family in autophagy regulation and therapy. Biomed Pharmacother. 2020;130:110591. doi:10.1016/j.biopha.2020.110591
33. Krysko DV, Berghe TV, Parthoens E, et al. Methods for distinguishing apoptotic from necrotic cells and measuring their clearance. Methods Enzymol. 2008;442:307–341.
34. Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13(10):722–737. doi:10.1038/nri3532
35. Harris J, Hartman M, Roche C, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286(11):9587–9597. doi:10.1074/jbc.M110.202911
36. Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy. 2018;14(2):243–251. doi:10.1080/15548627.2017.1402992
37. Lv H, Liu X, Chen W, et al. Yangxue jiedu fang ameliorates psoriasis by regulating vascular regression via Survivin/PI3K/Akt pathway. J Immunol Res. 2021;2021:4678087. doi:10.1155/2021/4678087
38. Pan S-T, Qin Y, Zhou Z-W, et al. Plumbagin induces G2/M arrest, apoptosis, and autophagy via p38 MAPK- and PI3K/Akt/mTOR-mediated pathways in human tongue squamous cell carcinoma cells. Drug Des Devel Ther. 2015;9:1601–1626. doi:10.2147/DDDT.S76057
39. Zhao Y, Huina W, Xing X, et al. CD13 induces autophagy to promote hepatocellular carcinoma cell chemoresistance through the P38/ Hsp27/CREB/ATG7 pathway. J Pharmacol Exp Ther. 2020;374(3):512–520. doi:10.1124/jpet.120.265637
40. Webber JL, Tooze SA. New insights into the function of Atg9. FEBS Lett. 2010;584(7):1319–1326. doi:10.1016/j.febslet.2010.01.020
41. Williams A, Sarkar S, Cuddon P, et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4(5):295–305. doi:10.1038/nchembio.79
42. Ganley IG, Lam DH, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284(18):12297–12305. doi:10.1074/jbc.M900573200
43. Unno R, Kawabata T, Taguchi K, et al. Deregulated MTOR (mechanistic target of rapamycin kinase) is responsible for autophagy defects exacerbating kidney stone development. Autophagy. 2020;16(4):709–723. doi:10.1080/15548627.2019.1635382
44. Saito H, Godo S, Sato S, et al. Important role of endothelial Caveolin-1 in the protective role of endothelium-dependent hyperpolarization against nitric oxide-mediated nitrative stress in microcirculation in mice. J Cardiovasc Pharmacol. 2018;71(2):113–126. doi:10.1097/FJC.0000000000000552
45. Karbach S, Wenzel P, Waisman A, Munzel T, Daiber A. eNOS uncoupling in cardiovascular diseases--The role of oxidative stress and inflammation. Curr Pharm Des. 2014;20(22):3579–3594. doi:10.2174/13816128113196660748
46. Karbach S, Croxford AL, Oelze M, et al. Interleukin 17 drives vascular inflammation, endothelial dysfunction, and arterial hypertension in psoriasis-like skin disease. Arterioscler Thromb Vasc Biol. 2014;34(12):2658–2668. doi:10.1161/ATVBAHA.114.304108
47. Pestana CR, Oishi JC, Salistre-Araújo HS, et al. Inhibition of autophagy by chloroquine stimulates nitric oxide production and protects endothelial function during serum deprivation. Cell Physiol Biochem. 2015;37(3):1168–1177. doi:10.1159/000430240
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.