")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Autophagy inhibition enhances RAD001-induced cytotoxicity in human bladder cancer cells
Authors Lin J , Lin Y, Yang S, Tsai T , Chen H, Chou K, Hwang TI
Received 7 September 2015
Accepted for publication 19 November 2015
Published 18 April 2016 Volume 2016:10 Pages 1501—1513
DOI https://doi.org/10.2147/DDDT.S95900
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Ji-Fan Lin,1 Yi-Chia Lin,2,3 Shan-Che Yang,1 Te-Fu Tsai,2,3 Hung-En Chen,2 Kuang-Yu Chou,2,3 Thomas I-Sheng Hwang2–4
1Central Laboratory, Shin-Kong Wu Ho-Su Memorial Hospital, Taipei, Taiwan; 2Division of Urology, Department of Surgery, Shin-Kong Wu Ho-Su Memorial Hospital, Taipei, Taiwan; 3Division of Urology, School of Medicine, Fu-Jen Catholic University, New Taipei City, Taiwan; 4Department of Urology, Taipei Medical University, Taipei, Taiwan
Background: Mammalian target of rapamycin (mTOR), involved in PI3K/AKT/mTOR pathway, is known to play a central role in regulating the growth of cancer cells. The PI3K/AKT/mTOR pathway enhances tumor survival and proliferation through suppressing autophagy, which sustains energy homeostasis by collecting and recycling cellular components under stress conditions. Conversely, inhibitors of the mTOR pathway such as RAD001 induce autophagy, leading to promotion of tumor survival and limited antitumor efficacy. We thus hypothesized that the use of autophagy inhibitor in combination with mTOR inhibition improves the cytotoxicity of mTOR inhibitors in bladder cancer.
Materials and methods: The cytotoxicity of RT4, 5637, HT1376, and T24 human bladder cancer cells treated with RAD001 alone or combined with autophagy inhibitors (3-methyladenine (3-MA), bafilomycin A1 (Baf A1), chloroquine, or hydroxychloroquine) was assessed using the WST-8 cell viability kit. The autophagy status in cells was analyzed by the detection of microtubule-associated light chain 3 form II (LC3-II), using immunofluorescent staining and Western blot. Acidic vesicular organelle (AVO) formation in treated cells was determined by acridine orange vital staining. Inhibition of mTOR pathway by RAD001 was monitored by using a homemade quantitative polymerase chain reaction gene array, while phospho-mTOR was detected using Western blot. Induced apoptosis was determined by measurement of caspase 3/7 activity and DNA fragmentation in cells after treatment.
Results: Advanced bladder cancer cells (5637, HT1376, and T24) were more resistant to RAD001 than RT4. Autophagy flux detected by the expression of LC3-II showed RAD001-induced autophagy. AVO formation was detected in cells treated with RAD001 and was inhibited by the addition of 3-MA or Baf A1. Cotreatment of RAD001 with autophagy inhibitors further reduced cell viability and induced apoptosis in bladder cancer cells.
Conclusion: Our results indicate that simultaneous inhibition of the mTOR and autophagy pathway significantly enhances apoptosis, and it is suggested to be a new therapeutic paradigm for the treatment of bladder cancer.
Keywords: autophagy, apoptosis, bladder cancer, chloroquine, RAD001
Introduction
Bladder cancer is one of the most common noncutaneous malignancies with high mortality in general population, particularly in men.1 Approximately, one-third of patients diagnosed with high-risk non–muscle-invasive bladder cancer are expected to have progression to advanced disease within 5 years.2 Radical cystectomy is a common clinical modality for advanced bladder cancer, but it fails to increase survival rates due to recurrences and metastases.3 To improve survival rates, a novel chemotherapeutic strategy against bladder cancer is in an urgent need, which prevents and/or delays progression of bladder cancer.
Activation of mammalian target of rapamycin (mTOR) signaling has been demonstrated in aggressive cancers such as gastric and cervical cancer.4–6 The effect of mTOR signaling has also been evaluated in bladder cancer.7 The expression of phosphor-S6 (a marker of mTOR activity) was found in 55% of muscle-invasive bladder cancers with evident lymph node metastases. mTOR activity was demonstrated to be associated with increased pathological stage and reduced patient survival. Inhibition of mTOR using rapamycin-reduced tumor volume in a T24-xenograft model was also observed.7 Therefore, targeting mTOR is a reasonable strategy to manage bladder cancer. RAD001 is an mTOR inhibitor that shares similar pharmacological activity with rapamycin as a chemotherapeutic agent.8 RAD001 has been demonstrated to exert antiproliferative activities against human breast cancer cell lines, gastric cancer, lymphoma cells, and prostate cancer cells.9–12 Its antitumor effects on different solid cancers such as renal and endometrial cancers are under clinical evaluation.13,14 It has a relatively short half-life and favorable bioavailability. RAD001 specifically binds with FKBP-12 after entering cells, forming a complex that binds to mTOR.15 RAD001 thus inhibits growth factor-induced transduction signaling that is involved in cellular division, cell cycle, and apoptosis.16
The aim of this study was to investigate the mechanism underlying the anticancer effects of RAD001 in different grades of bladder cancer cells. We found that RAD001 induced autophagy in bladder cancer cells and that sensitivity to RAD001 was associated with the grade of cancer cells. Furthermore, the inhibition of autophagy could lead to the enhancement of RAD001-mediated cytotoxicity.
Materials and methods
Chemicals
RAD001 was purchased from Sigma-Aldrich (St Louis, MO, USA). A 10 mM stock solution for cell culture was prepared in dimethyl sulfoxide (DMSO), and aliquots were stored at −20°C. All other chemicals, unless otherwise mentioned, were purchased from Sigma-Aldrich.
Cell culture
Human bladder cancer cell lines RT4 (American Type Culture Collection, ATCC, Manassas, VA, USA; #HTB-2), 5637 (ATCC#HTB-9), HT1376 (ATCC#CRL-1472), and T24 (ATCC#HTB-4) were obtained from Bioresource Collection and Research Center (Hsinchu, Taiwan) and maintained at 37°C under 5% CO2. RT4 and HT1376 cells were cultured in McCoy’s 5A and minimum essential medium, respectively. 5637 and T24 cells were cultured in Roswell Park Memorial Institute-1640 medium. Culture media were supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA), 2 mM GlutaMAX-1 (Thermo Fisher Scientific), 100 units/mL penicillin, and 100 μg/mL streptomycin (Thermo Fisher Scientific). Cells were treated with the indicated concentration of RAD001 with or without the treatment with autophagy inhibitors, including bafilomycin A1 (Baf A1), 3-methyladenine (3-MA), and chloroquine (CQ), while control cells received an equal volume of DMSO. The final concentration of DMSO was less than 0.1%. Ethical permission for the use of cell lines was not necessary for this study, as only patients’ samples or animal research requires such approval from the institutional review board or relevant ethical committee.
Cell viability assays
The effect of RAD001 on the growth of human bladder cancer cells was examined using the WST-8 reagent (Sigma-Aldrich) as previously described.17 In some experiments, cell viability was detected in RAD001-treated cells cotreated with autophagy inhibitors, including 3-MA, Baf A1, or CQ.
Detection of mTOR-related genes by quantitative polymerase chain reaction
Genes involved in mTOR signaling, including mTOR complexes (MLST8 and RPTOR (mTOR complex 1 [mTORC1]) and RICTOR (mTOR complex 2 [mTORC2])), mTORC1-positive regulation (EIF4B, RPS6, and RPS6KB1), and mTORC2-positive regulation (AKT1, GSK3B, and RPS6KB1) were detected using quantitative polymerase chain reaction (qPCR). Total RNA was extracted, using TRIZol reagent (Thermo Fisher Scientific), from T24 cells treated with 1 μM RAD001 for 24 hours. RNA yields were measured using Nanodrop 2000 (Thermo Fisher Scientific). Two micrograms of total RNA was subjected to reverse transcription using SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific). qPCR was performed as previously described.18 GAPDH was employed as internal control. Relative expression was calculated by the comparative Ct and expressed as fold of control. Sequences of primer pairs used in this study are listed in Table S1.
Detection of mTORC1 downstream effectors by Western blot
Cells were harvested and lysed using radioimmunoprecipitation assay buffer, followed by measurement of protein concentration using a BCA protein assay (Pierce, Rockford, IL, USA). Protein samples were subjected to a 14% sodium dodecyl sulfate polyacrylamide gel electrophoresis and subsequently transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). Antibodies against RPS6 and phospho-RPS6 (p-RPS6) were purchased from Cell Signaling Technology (Danvers, MA, USA); antibody against β-actin served as internal control and was obtained from Sigma-Aldrich. Immunoblotting procedures were performed using a chemiluminescence procedure (Millipore) per the manufacturer’s instructions. The intensity of immunoreactive bands were determined using GeneTools software (Syngene, Frederick, MD, USA) after scanning the chemiluminescence signals on the membrane by a Multigel-2 multifunction gel image system (TOP BIO CO., Taipei, Taiwan). Results are presented as mean ± standard deviation of three independent experiments.
Autophagy assays
Induction of autophagy was monitored in cells treated with indicated concentration of RAD001 with or without 2 hours pretreatment with 200 nM Baf A1 by the formation of acidic vesicular organelles using acridine orange staining as described;19 and the detection of microtubule-associated protein 1B-light chain 3 (LC3) processing using Western blot as described.17
Caspase-3/7 activity
Caspase-3 activity was assayed using (H-Asp-Glu-Val-Asp)2-Rhodamine 110 ((Z-DEVD)2-R110, Bachem, Torrance, CA, USA) substrate as previously described.17,18 In brief, cells were seeded in 96-well plates and incubated overnight. The cells were then treated with DMSO (control) or 1 μM RAD001 with or without the 2 hours pretreatment of inhibitors (1 mM 3-MA or 200 nM Baf A1) for 48 hours. The cells were subsequently lysed with caspase-3 assay buffer containing (Z-DEVD)2-R110 substrates and incubated at 37°C for 1 hour. The fluorescent intensity of proteolytically released fluorophore R110 was then measured using a plate reader (Victor X2; PerkinElmer, Waltham, MA, USA) with an excitation wavelength of 485 nm and emission wavelength of 535 nm.
Statistical analysis
All data are expressed as the mean ± standard deviation. The statistical significance of the differences in variables between groups was calculated using Student’s t-test, and P<0.05 was considered statistically significant. Each experiment was performed at least in triplicate.
Results
RAD001 inhibits the growth of bladder cancer cells
The effect of RAD001 on the survival of bladder cancer cell lines was determined using WST-8 assay. Treatment of bladder cancer cell lines with RAD001 resulted in increased cell death in a dose-dependent manner (Figure 1). Our data revealed that RAD001 exerted inhibitory effects on RT4 cells (Grade I with wild-type p53) at a concentration as low as 50 nM at 24 hours, whereas the other bladder cell lines had no response to the treatment (Figure 1). Advanced bladder cancer cells, HT1376, 5637, and T24, were found responding to the RAD001 treatment at concentrations of over 20 μM. Prolonged exposure to low concentrations of RAD001 had a greater effect on the survival of 5637 and HT1376 cells (Grade II transitional cell carcinoma [TCC] with mutant p53) compared with that of T24 cells (Figure 1B). Given the findings, the concentration of RAD001 at 1 and 5 μM was used for following experiments.
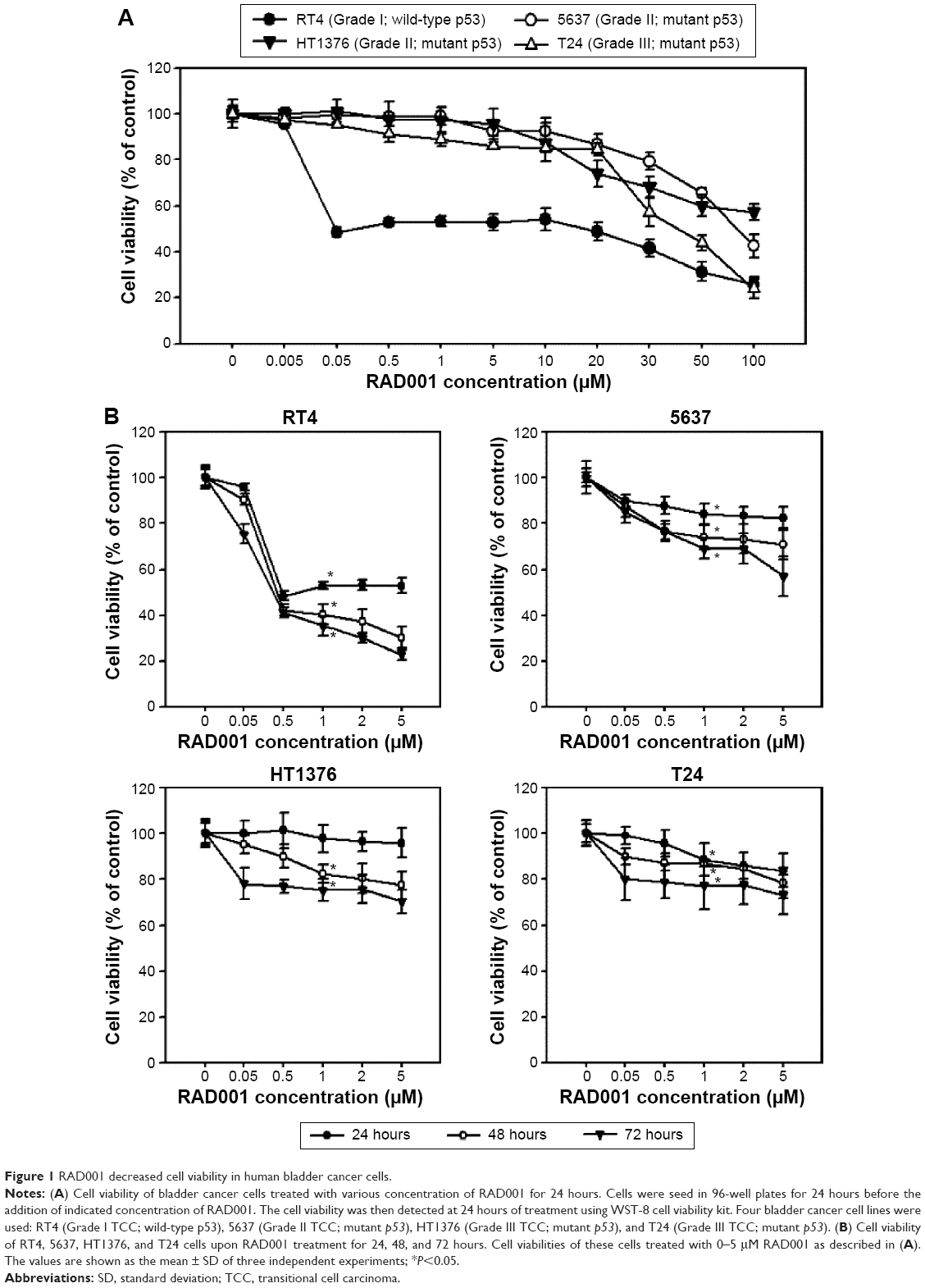 | Figure 1 RAD001 decreased cell viability in human bladder cancer cells. |
RAD001 induces autophagy in bladder cancer cells
We next examined the effects of RAD001 on mTOR signaling in advanced bladder cancer cells. q-PCR was performed to analyze the levels of mTOR-related genes after a single 24-hour exposure of T24 cells to 1 or 5 μM of RAD001. As shown in Figure 2, RAD001 treatment resulted in a downregulated expression of mTOR-related genes (Figure 2A), including RPS6 and RPS6KB1, which are positively regulated by mTORC1 (mTOR-positive regulation; Figure 2B). It is known that RAD001 has an effect on mTORC1 but not on mTORC2 protein complex signaling.20 Interestingly, the results revealed that RAD001 induced a downregulation of mTORC2-related gene expression, such as those of AKT1 and RPS6KB1 (mTORC2-positive regulation; Figure 2C). However, AKT1 expression was increased in cells treated with 5 μM RAD001 compared to those cells treated with 1 μM RAD001 at 24 hours posttreatment (Figure 2C). We further detected the expression of the downstream effector of mTORC1, RPS6; the results showed that RAD001 potently inhibited RPS6 activation throughout the duration of treatment (Figure 2D).
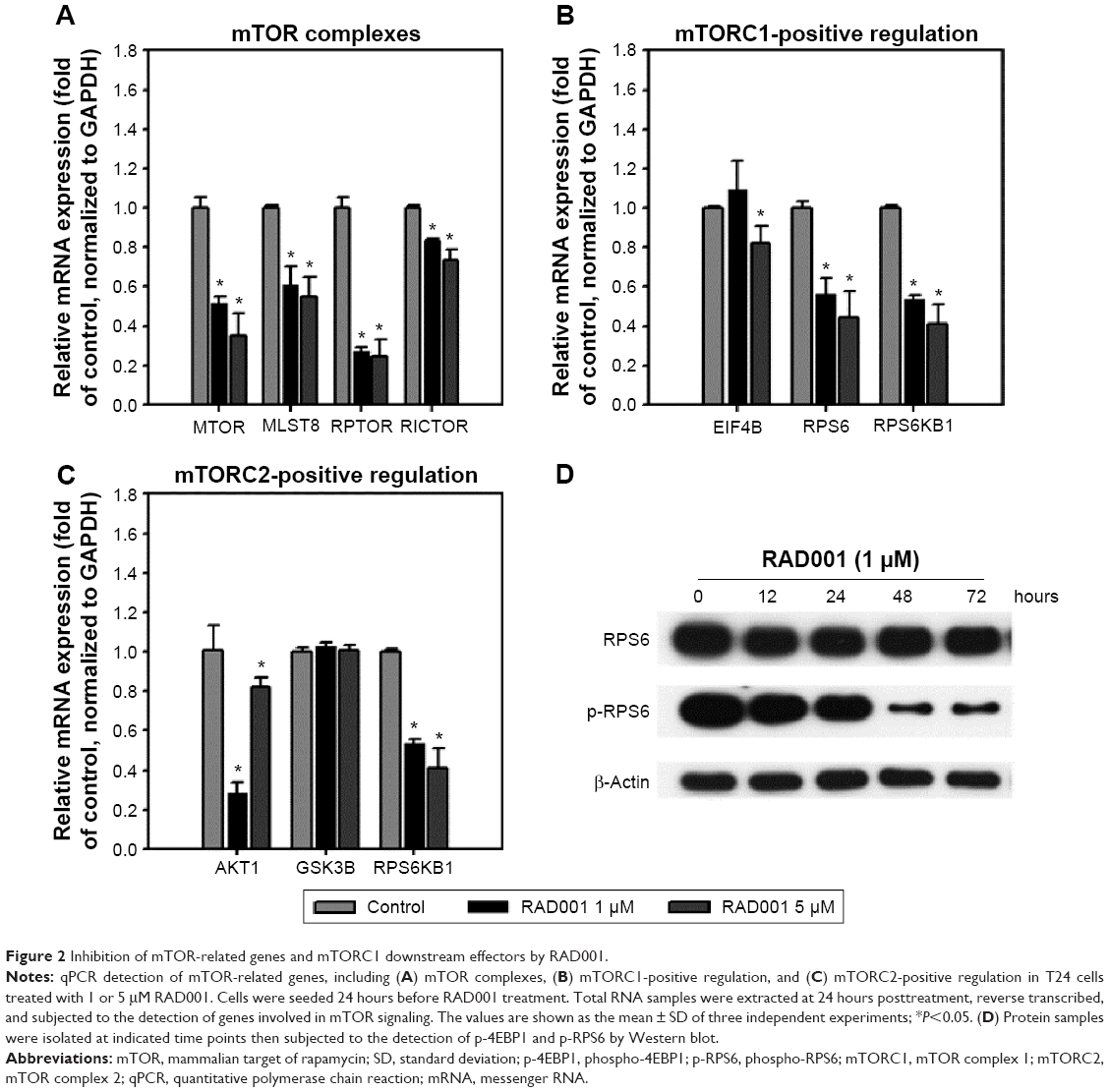 | Figure 2 Inhibition of mTOR-related genes and mTORC1 downstream effectors by RAD001. |
To investigate the role of autophagy in RAD001-induced cytotoxicity, fluorescent imaging was employed to morphologically assess the formation of autophagosome in bladder cancer cells. As shown in Figure 3A, treatment with 1 μM of RAD001 for 24 hours resulted in an accumulation of lysosomal vacuoles exhibiting autolysosomal characteristics, whereas the control cells exhibited few vacuoles in number. Western blotting showed that RAD001 treatment increased the breakdown of p62 (also known as SQSTM1, a polyubiquitin-binding protein that is degraded by autophagy)21 and LC3-I, with a corresponding increase in LC3-II level in a dose-dependent manner in T24 cells (Figure 3B). These results indicated that RAD001 induces autophagy in human bladder cancer cells. Administration of autophagy inhibitor, Baf A1, significantly increased the accumulation of p62 and LC3-II protein, suggesting that RAD001 induces a continuous autophagic flux in T24 cells (Figure 3C). The data of Western blotting confirmed the inhibition of autophagy in the presence of Baf A1 in bladder cancer cells by blocking autophagolysosome-mediated degradation of p62 and LC3-II protein.
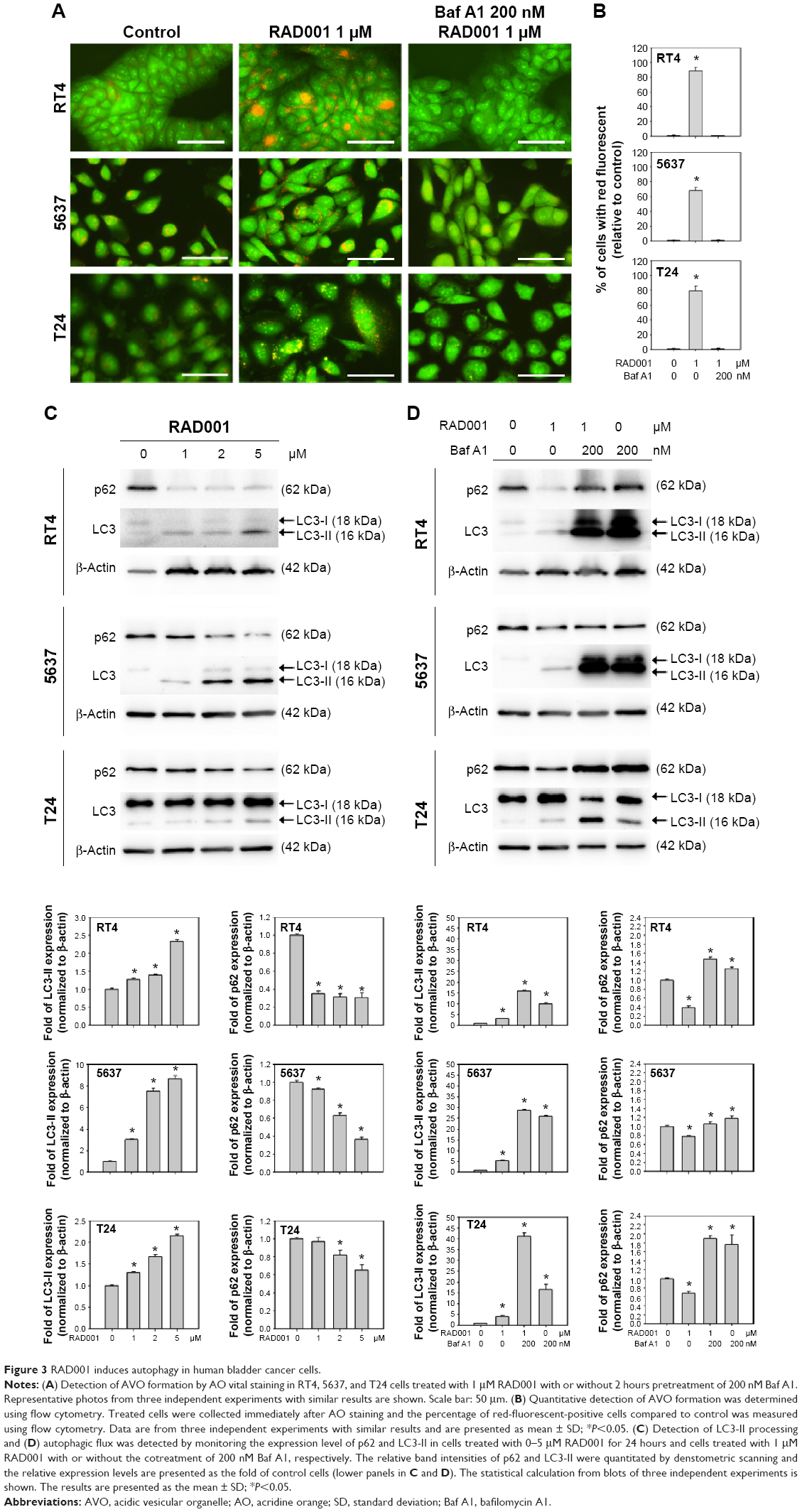 | Figure 3 RAD001 induces autophagy in human bladder cancer cells. |
Inhibition of autophagy enhances RAD001-induced cytotoxicity in bladder cancer cells
We next examined whether the inhibition of autophagy sensitizes bladder cancer cells to RAD001-induced cell death using WST-8 assay. As presented in the “RAD001 inhibits the growth of bladder cancer cells” section, sensitivity of bladder cancer cells to RAD001 is suggested to be related to the grade of the tumor. Therefore, three bladder cancer cell lines were exposed to RAD001 in the absence or presence of autophagy inhibitors (including 3MA, Baf A1, or CQ). The low-grade bladder cancer cell line, RT4, exhibited a significantly decreased viability in the presence of a combination of RAD001 and autophagy inhibitor compared with that of RAD001 only (Figure 4A). The data revealed that the viability of bladder cancer cells was significantly decreased in the presence of RAD001 and autophagy inhibitor compared with that exposed only to RAD001. In high-grade bladder cancer cell lines, a significant difference in cell viability between the absence and presence of late-stage autophagy inhibitor was observed at high concentrations (Figure 4B and C).
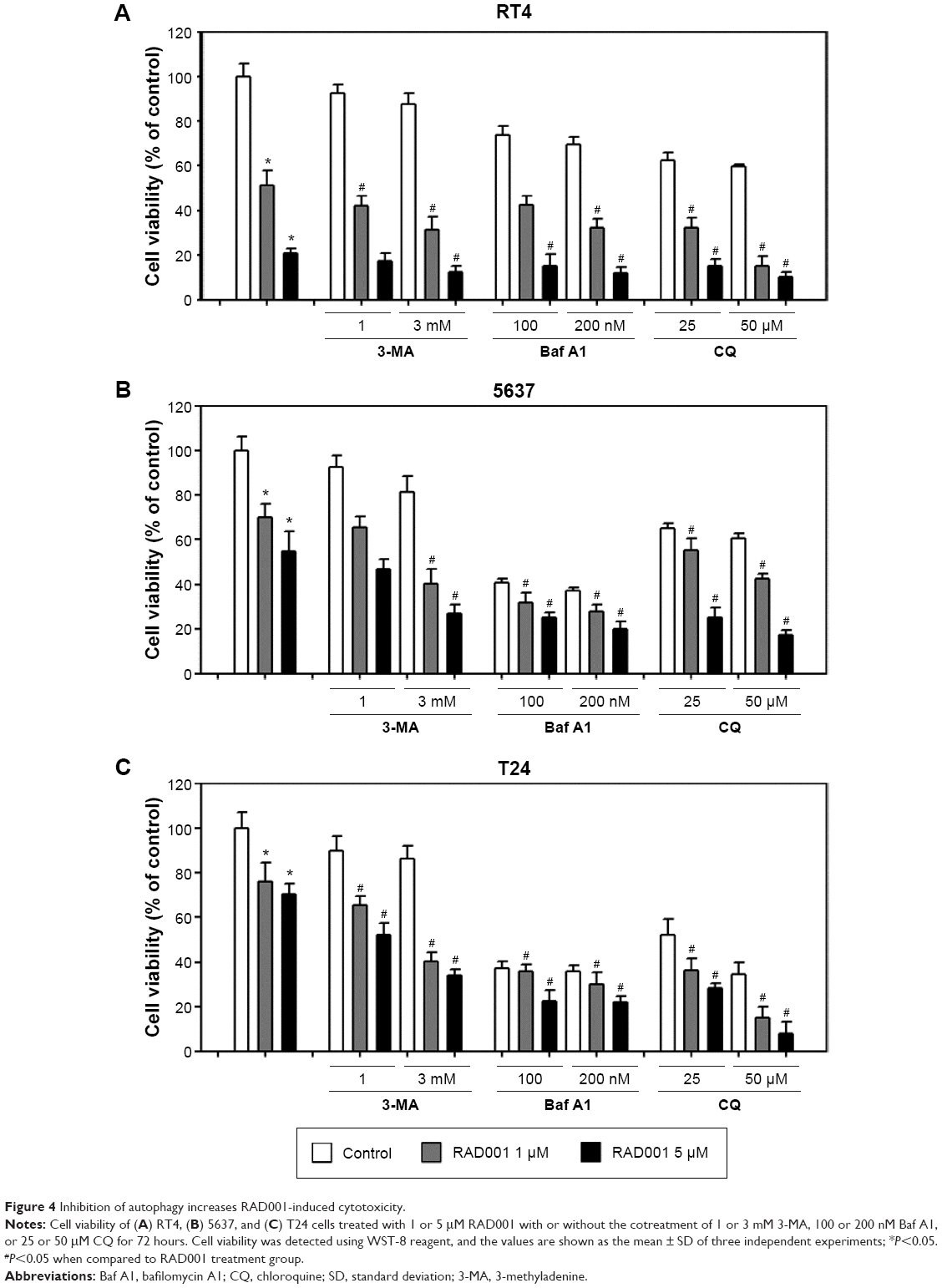 | Figure 4 Inhibition of autophagy increases RAD001-induced cytotoxicity. |
Autophagy inhibition triggers apoptosis in bladder cancer cells
We demonstrated that cytotoxicity of RAD001 in bladder cancer cells was enhanced in response to cotreatment with autophagy inhibitors. It is of interest to elucidate the mechanism by which the combination of mTOR inhibitor and autophagy inhibitor cause cell death in bladder cancer cells. Treatment of RT4 with 1 μM of RAD001 resulted in a significant increase in caspase 3/7 activity, while the induction of apoptosis was absent in 5637 and T24 cell lines. The combination of RAD001 and 3MA or Baf A1 led to a substantial elevation of caspase 3/7 activity in 5637 and T24 cell lines compared with that in the absence of autophagy inhibitors (Figure 5). These data suggested that inhibition of autophagy significantly enhanced RAD001-induced apoptosis in high-grade bladder cancer cells.
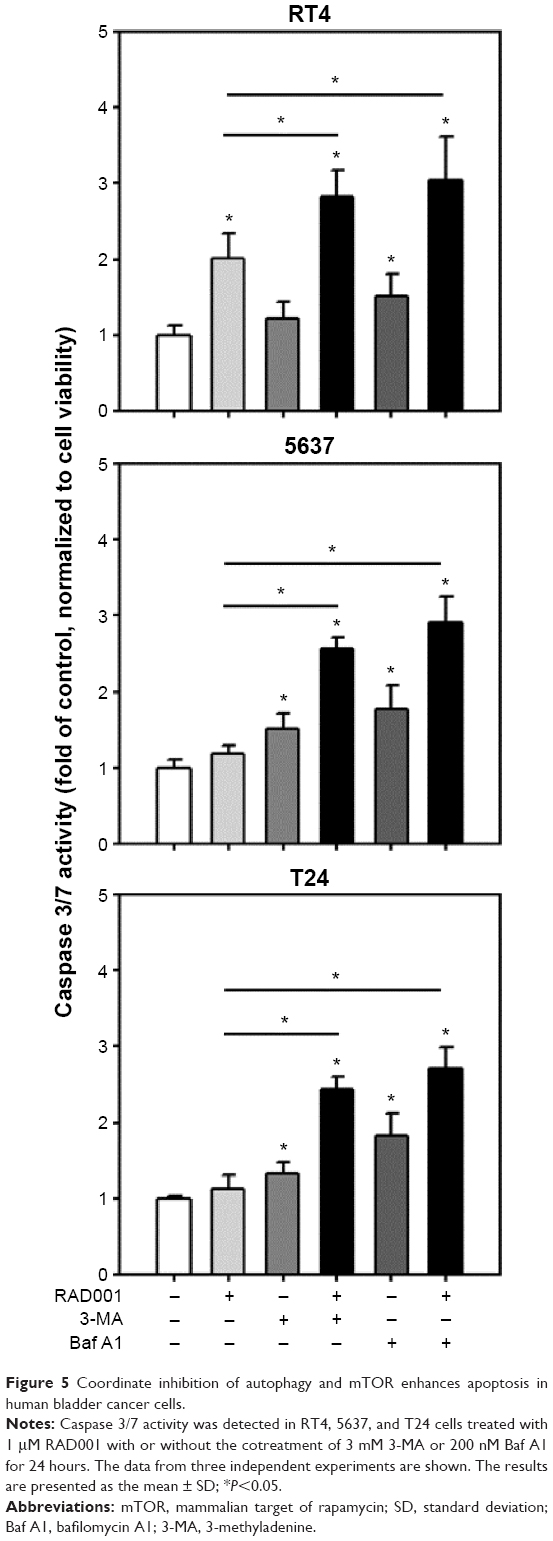 | Figure 5 Coordinate inhibition of autophagy and mTOR enhances apoptosis in human bladder cancer cells. |
Discussion
In this study, we reported that RAD001 treatment induced autophagy in bladder cancer cells. We found that a decreased sensitivity to RAD001 was associated with high-grade bladder cancer. Furthermore, coordinate inhibition of mTOR and autophagy led to enhanced cell death in high-grade bladder cancer cells.
The mTOR protein is known for its role in regulation of cell growth and metabolic processes. Deregulated mTOR signaling pathway has been shown to contribute to the development of many disorders including cancer.22 mTOR protein forms two distinct complexes, mTORC1 and mTORC2. The mTORC1 is the nutrient-sensitive complex consisting of the mTOR interacting protein raptor. It is involved in cell growth and cell size by regulating protein synthesis and autophagy.23 Upon activation, mTORC1 phosphorylates the translational regulator eukaryotic translation initiation factor 4E (eIF4E) binding protein 1 (4E-BP1) and S6 kinase (S6K). mTORC1 inhibition has been shown to impair protein synthesis and alter gene transcription.24 In this study, we demonstrated that the mTORC1 inhibitor RAD001 exerts anticancer activity in bladder cancer cells. It is suggested that mTOR inhibition by RAD001 is effective in disturbing the growth of bladder cancer cells. However, the sensitivity to RAD001 varied among the bladder cancer cell lines tested, with RT4 cells being the most responsive and T24 cells being the least responsive. It has been reported that cellular response to mTORC1 inhibition is associated with the p53 status in many types of tumor.25–27 Recent studies have suggested that rapamycin-mediated tumor suppression is dependent on p53, which reduces cellular proliferation after mTORC1 inhibition.25,28 Our findings that RT4 cells with wild-type p53 were more sensitive to RAD001 than the other ones with mutated p53 support the results of previous studies, implying a heterogeneous response to RAD001-mediated mTOR inhibition. Although inhibition of mTORC1 was expected to diminish cancer cell survival, the extent of cytotoxicity can be reduced by additional changes that occur. For example, the mTORC2 is suggested to be insensitive to the presence of rapamycin and its derivatives and was shown to regulate the prosurvival kinase AKT by phosphporylation on Ser473.29 Therefore, inhibition of mTORC1 results in inhibition of negative feedback loops and leads to rapamycin resistance in some cell types.30,31 Our results showed a decreased expression of mTOR complexes and AKT1 transcripts in RAD001-treated T24 cells, and only the expression of AKT1 was slightly increased upon 5 μM RAD001 treatment. Rapamycin and RAD001 are well recognized as specific inhibitors that act on mTORC1, and the inhibition usually accompanies with feedback activation of mTORC2 and AKT prosurvival pathway as already mentioned. Our results from the transcription level may also help to explain why prolonged inhibition of mTORC1 inhibits mTORC2 assembly and AKT activation.32 The differences in bladder cancer cells responding to mTOR inhibition may be attributed to other factors, such as variability of mTORC1 and mTORC2 response or activation/inhibition of other pathways upon mTOR inhibition. RAD001 has been shown to activate MAP kinase (MAPK) through S6K/PI3K/Ras signaling, which, in turn, enhances survival of cells.33 These complex interplays between mTOR and other pathways are postulated to account for differences in sensitivity to RAD001.
Autophagy is a fine-tuned catabolic process that is critical in organelle degradation and protein turnover. It is present at low levels in normal cell and upregulated in response to metabolic stresses. It is evident that mTOR (as a sensor of cellular nutritional status, stress, and growth factor signals), particularly mTORC1, plays a role in autophagy signaling pathway. A previous study has demonstrated that autophagy is induced by mTORC1 inhibition, whereas stimulation of mTORC1 inhibits this process.34 The mTORC1 was shown to control autophagy by regulation of a protein complex consisting of ULK1, ATG13, and FIP200. Inhibition of mTORC1 resulted in the decreased phosphorylation of ULK1 and ATG13, and therefore induction of autophagy.35 Although starvation or stress signaling is not always mediated by mTORC1, and it is possible that other mTORC1-independent pathways regulate ULK complexes,36 the current knowledge suggests that mTORC1 and ULK complexes constitute the main axis of the pathways that regulates growth and autophagy. In this study, we observed that RAD001, as an mTORC1 inhibitor, induces autophagy in bladder cancer cells corresponding with variable cytotoxicity. Autophagy was initially considered a process that suppressed malignant transformation.37 Activation of the PI3K/AKT pathway via activating PI3K mutations, AKT amplifications, or PTEN loss has been reported to attenuate autophagy in many settings largely through mTOR activation.38,39 The p53 protein has been shown to have opposing roles in autophagy, which is activated by nutrient deprivation or genotoxic stress leading to activation of autophagy as well as inhibition of mTOR. In contrast, p53 has been demonstrated to suppress autophagy in the basal state.40,41
Despite a role in tumor suppression, autophagy has been demonstrated to play a potentiating role in cancer development, promoting survival in cells under a number of stresses.40,42,43 In addition, autophagy has been shown to mediate therapeutic resistance in a variety of situations.44 For example, our previous study demonstrated that natural chemopreventive compound, benzyl isothiocyanate, induces protective autophagy in human prostate cancer cells.17 Furthermore, we reported that high-grade bladder cancer exhibits high basal level of autophagy.45 It is possible that inhibition of mTORC1 with RAD001 further enhances autophagy and accounts for the resistance of this drug in high-grade bladder cancer cells. Recent studies have demonstrated that autophagy is induced by restoration of p53 expression or by alkylating chemotherapy and that inhibition of autophagy enhances the antiproliferative activity of chemotherapy.46–48 In this study, we showed that combination of RAD001 with autophagy inhibitor leads to significantly increased apoptotic cell death in bladder cancer cells. It is suggested that the mTOR inhibition by RAD001 induces a protective autophagy blocking apoptosis pathway. Our results were consistent with the report that inhibition of mTOR and autophagy coordinately enhances cell death in melanoma.49 In addition, a phase I trial of temsirolimus (another mTORC1 inhibitor) and hydroxychloroquine (an autophagy inhibitor that disrupt autophagosomes fusion with lysosomes) demonstrated that the combination of temsirolimus and hydroxychloroquine is safe and tolerable in patients with advanced solid tumors and melanoma.50 Therefore, inhibition of autophagy is suggested to enhance RAD001-induced apoptotic cell death in bladder cancer.
Conclusion
The antiproliferation effect of RAD001 on bladder cancer cells is heterogeneous. Inhibition of autophagy significantly enhances cytotoxicity of RAD001 in bladder cancer cells. Combination of mTOR inhibitor and autophagy inhibitor represents a potential effective approach to manage bladder cancer. Further studies are needed to elucidate the mechanism of mTOR regulation and its clinical role in the treatment of bladder cancer.
Acknowledgments
This work was supported by Shin Kong Wu Ho-Su Memorial Hospital, Taipei, Taiwan (SKH-8302-101-DR-06 and SKH-8302-102-DR-10 to YCL and SKH-8302-101-0202 to JFL).
Disclosure
The authors report no conflicts of interest in this work.
References
Malats N, Real FX. Epidemiology of bladder cancer. Hematol Oncol Clin North Am. 2015;29:177–189, vii. | ||
van der Meijden AP. Optimal treatment for intermediate- and high-risk, nonmuscle-invasive bladder cancer. ScientificWorldJournal. 2006;6:2611–2616. | ||
Dovedi SJ, Davies BR. Emerging targeted therapies for bladder cancer: a disease waiting for a drug. Cancer Metastasis Rev. 2009;28:355–367. | ||
Hashimoto I, Koizumi K, Tatematsu M, et al. Blocking on the CXCR4/mTOR signalling pathway induces the anti-metastatic properties and autophagic cell death in peritoneal disseminated gastric cancer cells. Eur J Cancer. 2008;44:1022–1029. | ||
Lamouille S, Derynck R. Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol. 2007;178:437–451. | ||
Faried LS, Faried A, Kanuma T, et al. Predictive and prognostic role of activated mammalian target of rapamycin in cervical cancer treated with cisplatin-based neoadjuvant chemotherapy. Oncol Rep. 2006;16:57–63. | ||
Hansel DE, Platt E, Orloff M, et al. Mammalian target of rapamycin (mTOR) regulates cellular proliferation and tumor growth in urothelial carcinoma. Am J Pathol. 2010;176:3062–3072. | ||
Fasolo A, Sessa C. mTOR inhibitors in the treatment of cancer. Expert Opin Investig Drugs. 2008;17:1717–1734. | ||
Huang JJ, Li ZM, Huang Y, et al. Schedule-dependent inhibition of T-cell lymphoma cells by cotreatment with the mTOR inhibitor everolimus and anticancer drugs. Invest New Drugs. 2012;30:223–235. | ||
Hurvitz SA, Kalous O, Conklin D, et al. In vitro activity of the mTOR inhibitor everolimus, in a large panel of breast cancer cell lines and analysis for predictors of response. Breast Cancer Res Treat. 2015;149:669–680. | ||
Lee SJ, Lee J, Lee J, et al. Phase II trial of capecitabine and everolimus (RAD001) combination in refractory gastric cancer patients. Invest New Drugs. 2013;31:1580–1586. | ||
Wedel S, Hudak L, Seibel JM, et al. Inhibitory effects of the HDAC inhibitor valproic acid on prostate cancer growth are enhanced by simultaneous application of the mTOR inhibitor RAD001. Life Sci. 2011;88:418–424. | ||
Motzer RJ, Escudier B, Oudard S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–456. | ||
Erdemoglu E, Guney M, Take G, Giray SG, Mungan T. RAD001 (Everolimus) can prevent tamoxifen-related endometrial and stromal hyperplasia. Int J Gynecol Cancer. 2009;19:375–379. | ||
Liu E, Marincola P, Oberg K. Everolimus in the treatment of patients with advanced pancreatic neuroendocrine tumors: latest findings and interpretations. Therap Adv Gastroenterol. 2013;6:412–419. | ||
Vignot S, Faivre S, Aguirre D, Raymond E. mTOR-targeted therapy of cancer with rapamycin derivatives. Ann Oncol. 2005;16:525–537. | ||
Lin JF, Tsai TF, Liao PC, et al. Benzyl isothiocyanate induces protective autophagy in human prostate cancer cells via inhibition of mTOR signaling. Carcinogenesis. 2013;34:406–414. | ||
Chang CJ, Lin JF, Chang HH, Lee GA, Hung CF. Lutein protects against methotrexate-induced and reactive oxygen species-mediated apoptotic cell injury of IEC-6 cells. PLoS One. 2013;8:e72553. | ||
Lin JF, Lin YC, Lin YH, et al. Zoledronic acid induces autophagic cell death in human prostate cancer cells. J Urol. 2011;185:1490–1496. | ||
O’Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. | ||
Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. | ||
Huang S, Houghton PJ. Targeting mTOR signaling for cancer therapy. Curr Opin Pharmacol. 2003;3:371–377. | ||
Wengrod JC, Gardner LB. Cellular adaptation to nutrient deprivation: crosstalk between the mTORC1 and eIF2alpha signaling pathways and implications for autophagy. Cell Cycle. 2015;14:2571–2577. | ||
Tang L, Zirpoli GR, Guru K, et al. Intake of cruciferous vegetables modifies bladder cancer survival. Cancer Epidemiol Biomarkers Prev. 2010;19:1806–1811. | ||
Cam M, Bid HK, Xiao L, et al. p53/TAp63 and AKT regulate mammalian target of rapamycin complex 1 (mTORC1) signaling through two independent parallel pathways in the presence of DNA damage. J Biol Chem. 2014;289:4083–4094. | ||
Wall M, Poortinga G, Stanley KL, et al. The mTORC1 inhibitor everolimus prevents and treats Emu-Myc lymphoma by restoring oncogene-induced senescence. Cancer Discov. 2013;3:82–95. | ||
Zhang WB, Wang Z, Shu F, et al. Activation of AMP-activated protein kinase by temozolomide contributes to apoptosis in glioblastoma cells via p53 activation and mTORC1 inhibition. J Biol Chem. 2010;285:40461–40471. | ||
Akeno N, Miller AL, Ma X, Wikenheiser-Brokamp KA. p53 suppresses carcinoma progression by inhibiting mTOR pathway activation. Oncogene. 2015;34:589–599. | ||
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. | ||
Hsu PP, Kang SA, Rameseder J, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332:1317–1322. | ||
Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. | ||
Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. | ||
Carracedo A, Ma L, Teruya-Feldstein J, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–3074. | ||
Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 2005;12(Suppl 2):1509–1518. | ||
Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–1991. | ||
Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009;16:46–56. | ||
Guertin DA, Sabatini DM. An expanding role for mTOR in cancer. Trends Mol Med. 2005;11:353–361. | ||
Lamoureux F, Zoubeidi A. Dual inhibition of autophagy and the AKT pathway in prostate cancer. Autophagy. 2013;9:1119–1120. | ||
Cao C, Subhawong T, Albert JM, et al. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res. 2006;66:10040–10047. | ||
Huo Y, Cai H, Teplova I, et al. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discov. 2013;3:894–907. | ||
Lee HY, Chung KJ, Hwang IH, et al. Activation of p53 with ilimaquinone and ethylsmenoquinone, marine sponge metabolites, induces apoptosis and autophagy in colon cancer cells. Mar Drugs. 2015;13:543–557. | ||
Toshima T, Shirabe K, Matsumoto Y, et al. Autophagy enhances hepatocellular carcinoma progression by activation of mitochondrial beta-oxidation. J Gastroenterol. 2014;49:907–916. | ||
Shi Y, Han JJ, Tennakoon JB, et al. Androgens promote prostate cancer cell growth through induction of autophagy. Mol Endocrinol. 2013;27:280–295. | ||
Morselli E, Galluzzi L, Kepp O, et al. Anti- and pro-tumor functions of autophagy. Biochim Biophys Acta. 2009;1793:1524–1532. | ||
Lin YC, Lin JF, Wen SI, et al. Inhibition of high basal level of autophagy induces apoptosis in human bladder cancer cells. J Urol. Epub 2015 Oct 28. | ||
Pal I, Parida S, Prashanth Kumar BN, et al. Blockade of autophagy enhances proapoptotic potential of BI-69A11, a novel Akt inhibitor, in colon carcinoma. Eur J Pharmacol. Epub 2015 Oct 15. | ||
Li LQ, Xie WJ, Pan D, Chen H, Zhang L. Inhibition of autophagy by bafilomycin A1 promotes chemosensitivity of gastric cancer cells. Tumour Biol. Epub 2015 Aug 15. | ||
Ruan Y, Hu K, Chen H. Autophagy inhibition enhances isorhamnetininduced mitochondriadependent apoptosis in nonsmall cell lung cancer cells. Mol Med Rep. 2015;12(4):5796–5806. | ||
Xie X, White EP, Mehnert JM. Coordinate autophagy and mTOR pathway inhibition enhances cell death in melanoma. PLoS One. 2013;8:e55096. | ||
Rangwala R, Chang YC, Hu J, et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy. 2014;10:1391–1402. |
Supplementary material
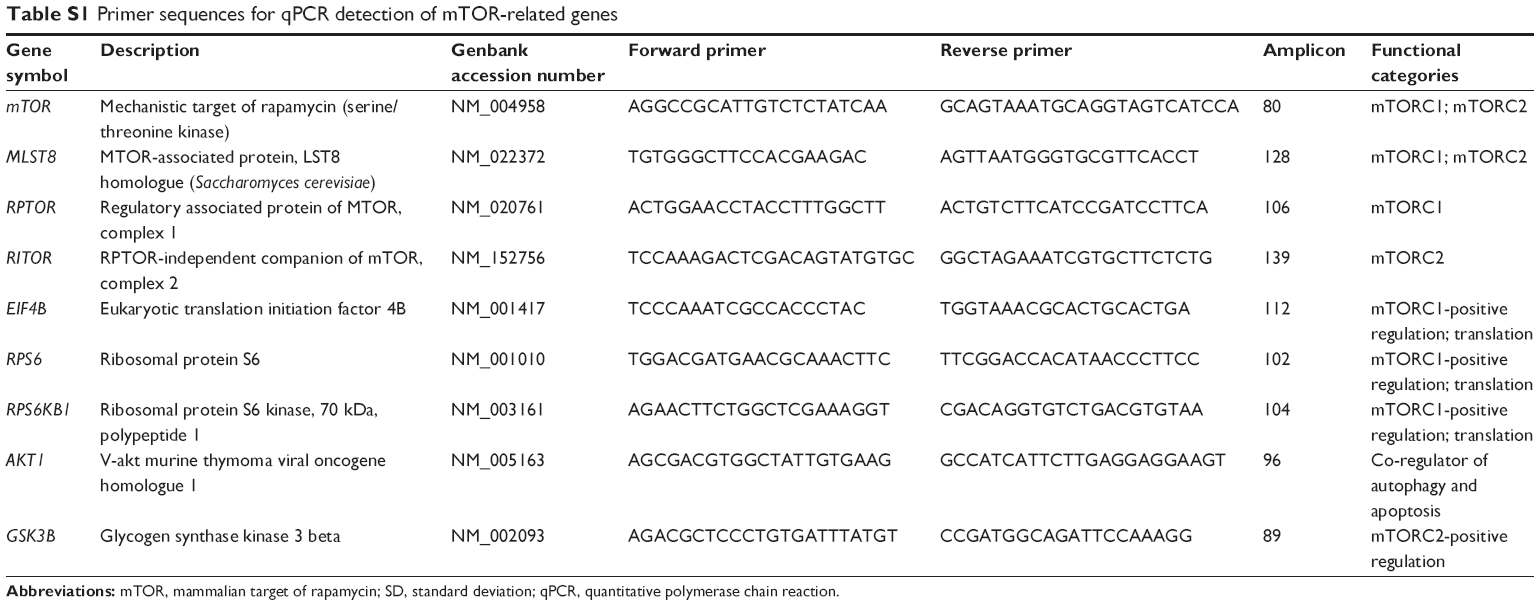 | Table S1 Primer sequences for qPCR detection of mTOR-related genes |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.