")
Back to Journals » Journal of Inflammation Research » Volume 16
Autoimmune Hemolytic Anemia After Cord Blood Transplantation: A Retrospective Single-Center Experience
Authors Yuan J , Liang ZY, Dong YJ, Ren HY
Received 29 October 2022
Accepted for publication 21 December 2022
Published 4 January 2023 Volume 2023:16 Pages 1—6
DOI https://doi.org/10.2147/JIR.S395375
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Adam D Bachstetter
Jing Yuan,1 Ze-Yin Liang,2 Yu-Jun Dong,2 Han-Yun Ren2
1Department of Hematology, The Second Hospital of Hebei Medical University, Shijiazhuang, People’s Republic of China; 2Department of Hematology, Peking University First Hospital, Beijing, People’s Republic of China
Correspondence: Ze-Yin Liang; Han-Yun Ren, Department of Hematology, Peking University First Hospital, Beijing, People’s Republic of China, Email [email protected]; [email protected]
Objective: To describe the incidence, possible risk factors, and treatment options of autoimmune hemolytic anemia (AIHA) occurring after cord blood transplantation (CBT).
Methods: We retrospectively analyzed the patients who underwent CBT at Peking University First Hospital between January 2004 and July 2022.
Results: We totally identified thirty-six patients who received CBT. Median age was 27.5 years (range, 1.6– 52). With a median 6 (range 0.6– 10.0) years survivor follow-up, six patients developed AIHA (2 Evans syndrome included) at a median of 168 (range, 122– 264) days post-CBT for 8% cumulative incidence density 3 years. Its mortality was 50% and mainly associated with concomitant infections (CMV reactivation rate nearly 100%). The possible risk factors for developing AIHA are CMV reactivation, GvHD and HLA mismatch.
Conclusion: AIHA is a clinically significant common complication in recipients post-CBT. Corticosteroids combined with intravenous immunoglobulin (IvIg) is recommended for the treatment of warm antibody AIHA after CBT.
Keywords: autoimmune hemolytic anemia, cord blood transplantation, combination therapy, prognosis
Introduction
Cord blood (CB) is an important stem cell source for patients with hematologic disorders and non-hematologic diseases.1,2 One of the major complications after allogeneic hematopoietic stem cell transplantation (HSCT) is autoimmune cytopenia (AC), which includes autoimmune neutropenia (AIN), autoimmune hemolytic anemia (AIHA), immune thrombocytopenia (ITP), Evans syndrome (AIHA with ITP), and trilineage autoimmune cytopenia (AIN and AIHA with ITP).3 AIHA is the most common AC after HSCT, but accurate reporting of ITP is challenging because there are many causes of post-transplantation thrombocytopenia.4,5 These disorders have been recently described among pediatric and adult patients undergoing CB transplantation (CBT), in several case reports or a cohort of patients with malignant and non-malignant diseases.5–7
Studies of AIHA occurring after CBT are uncommon and based on case reports or small series, probably associated with graft-versus-host disease (GvHD).8 These hematologic autoimmune disorders may be related to the use of unrelated donors and development of GvHD, possibly reflecting a dysregulation of the immune system. CB-derived lymphocytes are very naive unlike adult lymphocytes, resulting in decreased stringency of HLA match and lower incidence of GvHD.8,9 The application of double-unit CBT to overcome the dose limitation of single unit in adults might modify the early immune reconstitution.10 These genetic and immunologic properties may affect the development of AIHA post-CBT.
This retrospective research was undertaken to characterize incidence, possible risk factors, and treatment options for AIHA developing after CBT in a small cohort of patients in China.
Patients and Methods
Patients
A total of 36 patients with hematologic disorders and non-hematologic diseases undergoing CBT from unrelated donors or matched related donors at Peking University First Hospital between January 2004 and July 2022 were included in this study. This study was conducted in accordance with the declaration of Helsinki. The institutional review board approved the protocol and written informed consent was obtained from all patients or their guardians. The treatment plans including graft selection, conditioning regimen, immune suppression and supportive care have been reported in detail previously.11–13
Conditioning Regimen
The pre-transplantation conditioning regimens varied according to the patient’s diagnosis, previous treatment, and disease status. Twenty-two patients were treated with modified busulfan/cyclophosphamide (Bu/CY) regimen. Eleven of them received antithymocyte globulin (ATG) at a total dose of 10 mg/kg. Two patients received a CY/total body irradiation (TBI) regimen. Twelve patients received non-myeloablative regimen. These conditioning protocols were described in detail previously.11–13
Prophylaxis and Treatment of GvHD
Twenty-nine patients received the combination of mycophenolate mofetil (MMF), cyclosporine A (CsA), and a short course of methotrexate (MTX) as prophylaxis of GvHD. Only seven patients received CsA and MMF as GvHD prophylaxis.11–13
Serologic Tests
ABO group typing and antibody screening tests were performed on donor and recipient samples before transplantation and whenever patients required blood components transfusion. The direct antiglobulin test (DAT) was performed as part of the routine pre-transfusion compatibility testing. If positive, further testing with specific anti-IgG and anti-C3d reagents was carried out.
Definitions
AIHA was diagnosed in patients fulfilling all of the following criteria: positive DAT, positive indirect antiglobulin test with broad reactivity to RBC in serum and eluate, clinical and laboratory evidence of hemolysis (increased lactate dehydrogenase and bilirubin levels, decreased Hb and haptoglobin levels and increased transfusion requirements) and exclusion of other causes of hemolysis.14 ITP was a diagnosis of exclusion, defined as isolated thrombocytopenia in the absence of other causes that may be associated with a low platelet count.15 The response to therapy was assessed according to previous criteria.14,16
Hematopoietic Recovery and Engraftment
Hematopoietic recovery was defined as time to ANC ≥ 0.5×109/L (first of the 3 consecutive days) and platelet count ≥ 20×109/L (first of the 7 days without transfusion).
Hematopoiesis by donor cells was ascertained by testing for cells with the donor’s ABO type, HLA antigen, sex chromosome, or a combination, in the recipient’s PB or BM. Donor chimerism was determined serially on BM and/or PB at days 30, 60, 100, 180, and 360 after transplantation, with additional time points as needed.
Results
Patient and Graft Characteristics
Thirty-six patients with leukemia, malignant lymphoma, aplastic anemia, metachromatic leukodystrophy (MLD), pyruvate kinase deficiency (PKD) and inflammatory bowel disease (IBD) underwent CBT. Patients and graft characteristics are summarized in Table 1. Twenty-eight patients received double-unit CBT, while eight patients received single-unit CBT. Median age was 27.5 years (range, 1.6–52). Majority of patients had a baseline diagnosis of acute myeloid leukemia (58.3%). Median number of total nucleated cells and CD34+ cells infused was 7.9×107/kg (range, 3.1–25.4) and 3.2×105/kg (range, 0.7–11.7), respectively. Most patients (80.6%) received a donor-recipient gender mismatched graft. All patients had a negative DAT at time of transplantation.
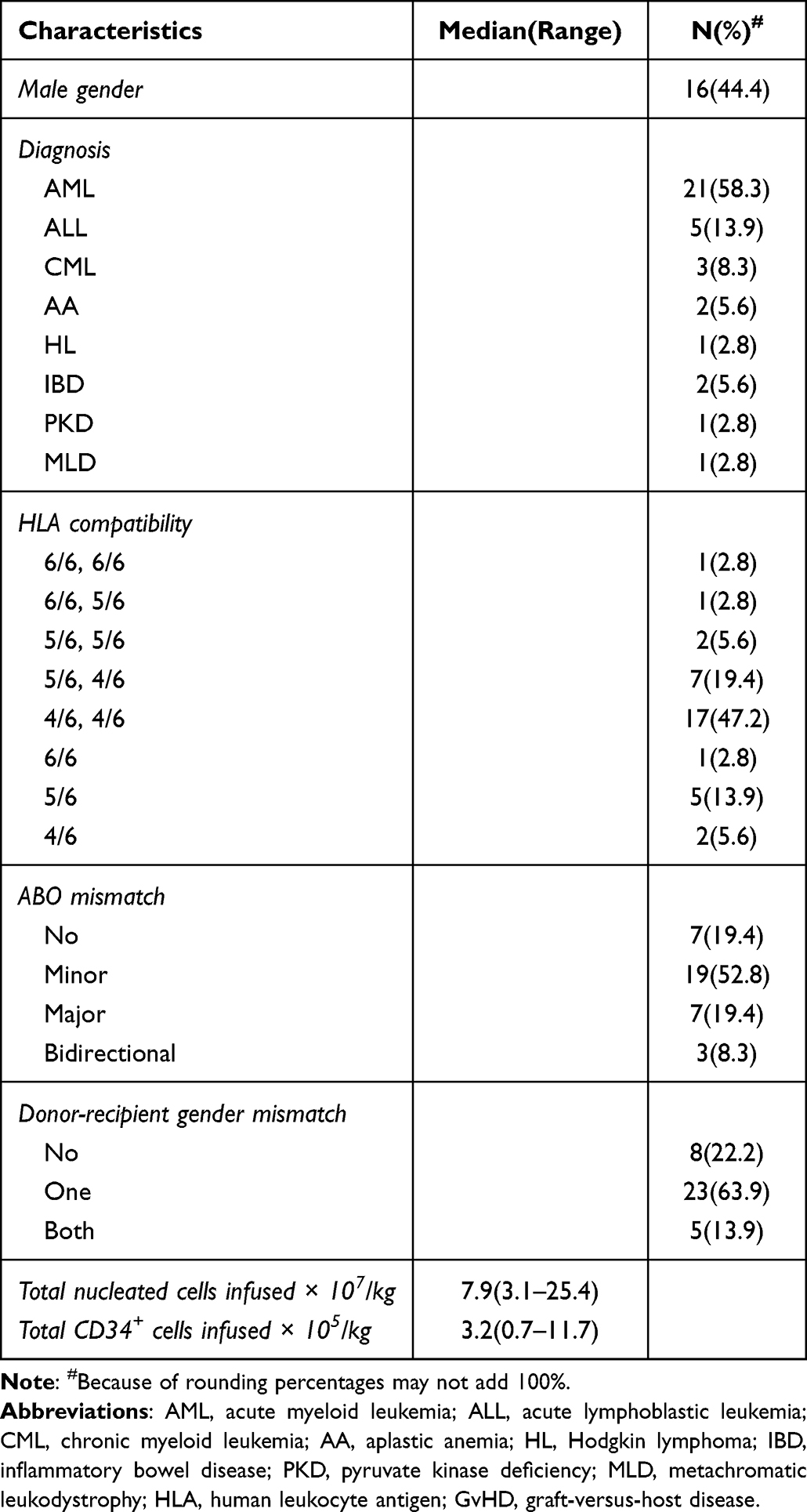 |
Table 1 Patients and Graft Characteristics |
Hematopoietic Recovery
Fifteen patients died at the end of follow-up (3/6 of the patients with AIHA, while 12/30 of the other patients). Ten patients failed to hematopoietic recovery after CBT. The other twenty-six evaluable patients had neutrophil engraftment at a median of 19 (range, 11–34) days, and platelet engraftment at a median of 36 (range, 16–209) days.
AIHA and Evans Syndrome
Incidence and Characteristics of AIHA and Evans Syndrome
AIHA was diagnosed in six patients who fulfilled the definition criteria, two of which with concomitant ITP were diagnosed Evans syndrome. Median time from transplant to AIHA or Evans syndrome was 168 (range, 122–264) days. At the onset of AIHA, median values of laboratory parameters were: Hb 5.1 g/dL (normal range, 2.8–9.2), reticulocyte count 161.5×109/L (normal range, 17.1–451.4), lactate dehydrogenase 482 U/L (normal range, 263–707), and indirect bilirubin 0.4 mg/dL (normal range, 0.1–0.6). Five patients required transfusion support. One of them died of acute severe hemolysis quickly and the other four received a median of 8 packs (2–19) RBCs. One patient received 3 packs platelets.
Characteristics of patients that developed AIHA or Evans syndrome are shown in Table 2. Five patients were in complete remission (CR) and had complete donor chimerism at time of diagnosis, while the other one relapsed. Three patients with early AIHA had acute GvHD (grade II in two patient and grade IV in the other one). Only one of these patients had chronic GvHD. Concomitant infection at the time of AIHA were present in all patients. Six patients had a cytomegalovirus (CMV) reactivation, including three cases of pulmonary polymicrobial infection (Pseudomonas aeruginosa, Klebsiella pneumoniae) and probable invasive fungal disease (IFD). One patient had pulmonary tuberculosis.
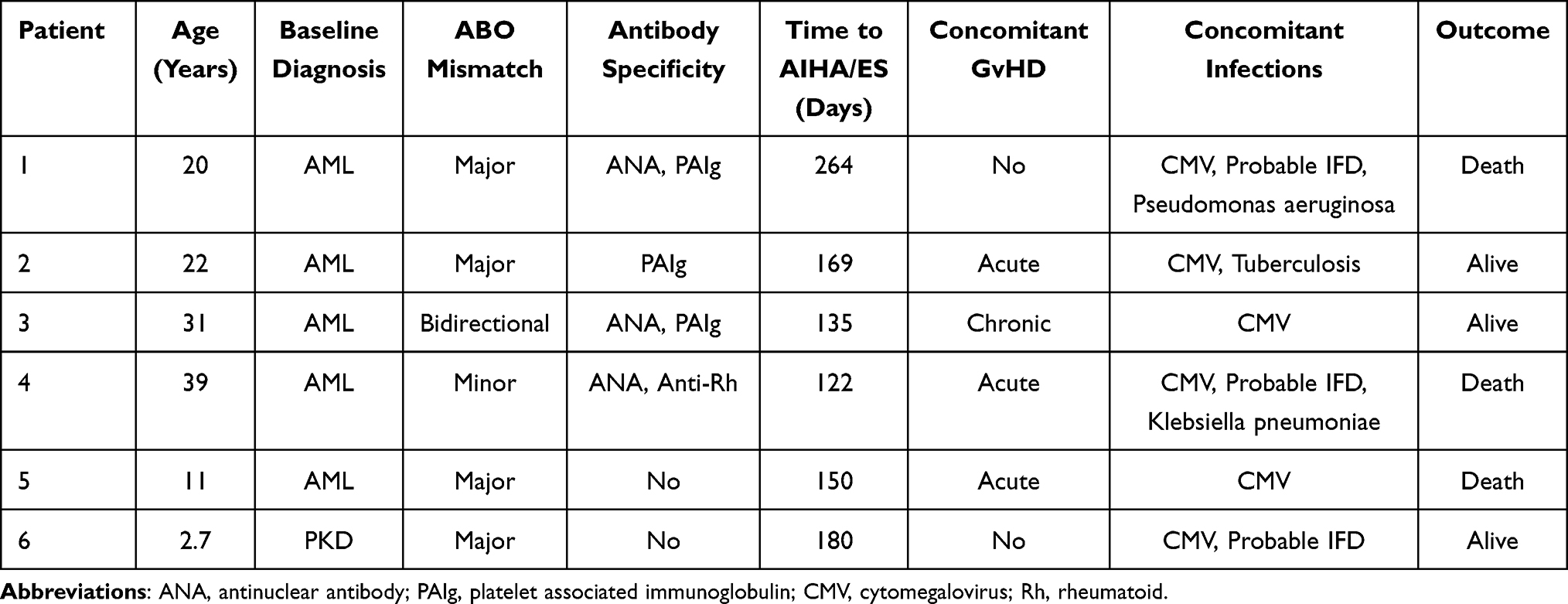 |
Table 2 Characteristics of Patients with AIHA and Evans Syndrome |
Serological Data
Major and bidirectional ABO mismatch between donor and recipient was present in five patients, whereas minor mismatch was present in one patient. All of the six patients in this study developed AIHA caused by warm antibodies (IgG/C3d). Three out of four patients had concomitant antinuclear antibody (ANA) and platelet associated immunoglobulin (PAIg), while one patient had multiple anti-rheumatoid (Rh) antibodies (including ANA, anti-nucleosome antibody, and anti-histone antibody). No antibodies against the ABO system were found in these patients.
Treatment and Outcome
All patients with AIHA and Evans syndrome were treated except for one patient who subsequently relapsed and died of acute severe hemolysis in two days. Intravenous immunoglobulin (IvIg, 0.4 g/kg/day for 5 days) was the first treatment administered in the remaining three patients. At the same time, prednisone or methylprednisolone (at doses ranging from 1–2 mg/kg/day) were administered. Only three of them achieved partial remission (PR), and the AIHA did not relapse at the end of follow-up.
Discussion
AIHA is a relatively common complication and may occur after any type of allogeneic HSCT, especially after CBT. Few cases on AIHA after CBT have been reported in China. The present research indicated its incidence was high to 16.7%. Meanwhile, the 3-year cumulative incidence density was 7.1%. AIHA after CBT occurred in approximately 5% of patients according to previous reports.7,8 The reasons for higher incidence may be as follows. Our study included 25% of pediatric patients, predominantly HLA mismatch donors, with a CMV reactivation rate up to 70%. González-Vicent et al found that patients less than 15-year-old, and patients using CB or an HLA mismatch donor were more likely to develop AIHA.17 The development of AC was strongly associated with the presence of chronic GvHD.8 The chronic GvHD was more frequently extensive after CBT, which led to the higher incidence. Our research mainly focused on adults that were transplanted for hematologic malignant diseases. In this regard, a retrospective single-center study may more reliably reflect the true incidence of the complications in China.
AIHA is also closely related to the presence of various infections. Concomitant infections were frequently observed at the time of diagnosis of AIHA. The CMV reactivation seems to be extremely common in this study. An intriguing possibility is that some of these infections could have triggered an abnormal immune response. In this aspect, the danger model of autoimmunity suggests that signals of damaged cells after exposure to infectious agents can bind to antigen-presenting cells (APCs) and activate a systemic immune response.
Most of patients with AIHA were ABO-mismatch between donor and recipient, which indicated blood group incompatibility was associated with hemolysis. Regarding serological data, all patients had IgG mediated warm antibodies directed against antigens of the rhesus system. This is similar with what was previously reported.18,19 No association between AIHA diagnosis and whether autoantibodies positive was noticed.
AIHA is a complication of allogeneic HSCT associated with poor prognosis. However, an optimal therapeutic approach is lacking. This study included a small group of pediatrics and adults with hematological and non-hematological disorders that received CBT at a single institution using a relatively homogeneous conditioning strategy, GvHD prophylaxis and supportive care, as well as monitoring for autoimmune complications. Response to therapy was disappointing and overall mortality was high. One patient died of concomitant infection, massive uncontrolled hemolysis was the cause of death in two patients who did not respond to first-line of treatment. Despite aggressive therapy, a similar clinical behavior was recently reported.20 Earlier identification and diagnosis of AIHA is key to improving efficacy and survival. In cases of sudden drop in Hb or an increase in transfusion requirements, early diagnosis of AIHA should be considered. All cases of warm antibodies AIHA may impact the choice of therapy. In particular, it is advisable to define the best choice, sequence and combination of drugs during different phases of disease. Corticosteroids combined with IvIg are preferred for early treatment. Our findings could help to increase awareness toward AIHA after CBT and guide therapy in these autoimmune complications. Most patients with AIHA failed to respond to corticosteroids or IvIg and needed further treatment. Rituximab and sirolimus are effective options, especially for patients with cold agglutinin disease.21
In conclusion, AIHA is a clinically significant common complication in recipients post-CBT. CMV reactivation, GvHD and HLA mismatch seem to increase the risk of developing AIHA. Earlier identification and diagnosis of AIHA is critical to improving efficacy and survival. Its prognosis was poor and mainly associated with concomitant infections. Corticosteroids combined with IvIg is recommended for the treatment of warm antibody AIHA after CBT. If ineffective, adjustment of immunosuppressant therapy should be initiated early.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ballen KK, Gluckman E, Broxmeyer HE. Umbilical cord blood transplantation: the first 25 years and beyond. Blood. 2013;122(4):491–498. doi:10.1182/blood-2013-02-453175
2. Doan PL, Chao NJ. Umbilical cord blood: biology and transplantation. Expert Rev Hematol. 2009;2(2):197–208. doi:10.1586/ehm.09.9
3. Faraci M, Dell’Orso G, Giardino S, Pierri F. Autoimmune diseases after allogeneic stem cell transplantation: a clinician’s guide and future outlook. Expert Rev Clin Immunol. 2022;12:1–14.
4. Michniacki TF, Ebens CL, Choi SW. Immune-mediated cytopenias after hematopoietic cell transplantation: pathophysiology, clinical manifestations, diagnosis, and treatment strategies. Curr Oncol Rep. 2019;21(10):87. doi:10.1007/s11912-019-0838-7
5. Page KM, Mendizabal AM, Prasad VK, et al. Posttransplant autoimmune hemolytic anemia and other autoimmune cytopenias are increased in very young infants undergoing unrelated donor umbilical cord blood transplantation. Biol Blood Marrow Transplant. 2008;14(10):1108–1117. doi:10.1016/j.bbmt.2008.07.006
6. Chen RL, Wu PL, Hsu YH, Kuo PL. Evans syndrome after unrelated cord blood transplantation for disseminated Langerhans cell histiocytosis in a child. J Pediatr Hematol Oncol. 2007;29(5):348–350. doi:10.1097/MPH.0b013e3180556467
7. Daikeler T, Labopin M, Ruggeri A, et al. New autoimmune diseases after cord blood transplantation: a retrospective study of EUROCORD and the Autoimmune Disease Working Party of the European Group for Blood and Marrow Transplantation. Blood. 2013;121(6):1059–1064. doi:10.1182/blood-2012-07-445965
8. Sanz J, Arango M, Carpio N, et al. Autoimmune cytopenias after umbilical cord blood transplantation in adults with hematological malignancies: a single-center experience. Bone Marrow Transplant. 2014;49(8):1084–1088. doi:10.1038/bmt.2014.107
9. Jacobson CA, Turki AT, McDonough SM, et al. Immune reconstitution after double umbilical cord blood stem cell transplantation: comparison with unrelated peripheral blood stem cell transplantation. Biology of Blood and Marrow Transplantation. 2012;18:565–574. doi:10.1016/j.bbmt.2011.08.018
10. Gluckman E. Milestones in umbilical cord blood transplantation. Blood Rev. 2011;25(6):255–259. doi:10.1016/j.blre.2011.06.003
11. Wang Q, Ren H, Liang Z, et al. Comparable outcomes in acquired severe aplastic anemia patients with haploidentical donor or matched related donor transplantation: a retrospective single-center experience. Front Med. 2022;8:807527. doi:10.3389/fmed.2021.807527
12. Zhang XH, Chen J, Han MZ, et al. The consensus from The Chinese Society of Hematology on indications, conditioning regimens and donor selection for allogeneic hematopoietic stem cell transplantation: 2021 update. J Hematol Oncol. 2021;14(1):145. doi:10.1186/s13045-021-01159-2
13. Yin Y, Ren HY, Cen XA, et al. Long-term outcomes in adults with leukemia treated with transplantation of two unrelated umbilical cord blood units. Chin Med J. 2011;124(16):2411–2416.
14. Jäger U, Barcellini W, Broome CM, et al. Diagnosis and treatment of autoimmune hemolytic anemia in adults: recommendations from the First International Consensus Meeting. Blood Rev. 2020;41:100648. doi:10.1016/j.blre.2019.100648
15. Neunert C, Terrell DR, Arnold DM, et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019;3(23):3829–3866. doi:10.1182/bloodadvances.2019000966
16. Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113(11):2386–2393. doi:10.1182/blood-2008-07-162503
17. González-Vicent M, Sanz J, Fuster JL, et al. Autoimmune hemolytic anemia (AIHA) following allogeneic hematopoietic stem cell transplantation (HSCT): a retrospective analysis and a proposal of treatment on behalf of the Grupo Español De Trasplante de Medula Osea en Niños (GETMON) and the Grupo Español de Trasplante Hematopoyetico (GETH). Transfus Med Rev. 2018;S0887-7963(17)30164–5. doi:10.1016/j.tmrv.2018.02.005
18. O’Brien TA, Eastlund T, Peters C, et al. Autoimmune haemolytic anaemia complicating haematopoietic cell transplantation in paediatric patients: high incidence and significant mortality in unrelated donor transplants for non-malignant diseases. Br J Haematol. 2004;127(1):67–75. doi:10.1111/j.1365-2141.2004.05138.x
19. Godder K, Pati AR, Abhyankar SH, Lamb LS, Armstrong W, Henslee-Downey PJ. De novo chronic graft-versus-host disease presenting as hemolytic anemia following partially mismatched related donor bone marrow transplant. Bone Marrow Transplant. 1997;19(8):813–817. doi:10.1038/sj.bmt.1700746
20. Yang Z, Wu B, Zhou Y, et al. Clinical and serological characterization of autoimmune hemolytic anemia after allogeneic hematopoietic stem cell transplantation. Chin Med J. 2014;127(7):1235–1238.
21. Park JA, Lee HH, Kwon HS, Baik CR, Song SA, Lee JN. Sirolimus for refractory autoimmune hemolytic anemia after allogeneic hematopoietic stem cell transplantation: a case report and literature review of the treatment of post-transplant autoimmune hemolytic anemia. Transfus Med Rev. 2016;30(1):6–14. doi:10.1016/j.tmrv.2015.09.001
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.