")
Back to Journals » Cancer Management and Research » Volume 11
Atractylon induces apoptosis and suppresses metastasis in hepatic cancer cells and inhibits growth in vivo
Authors Cheng Y, Chen T, Yang X, Xue J, Chen J
Received 15 November 2018
Accepted for publication 12 May 2019
Published 28 June 2019 Volume 2019:11 Pages 5883—5894
DOI https://doi.org/10.2147/CMAR.S194795
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lu-Zhe Sun
Yang Cheng,1,2 Tianyang Chen,2 Xueli Yang,2 Jianhua Xue,1 Jianjie Chen1,2
1Department of Liver Disease, Hospital for Infectious Diseases of Pudong District, Shanghai 201299, People’s Republic of China; 2Institute of Liver Disease, Shuguang Hospital Affiliated to Shanghai University of Traditional Chinese Medicine, Shanghai 201203, People’s Republic of China
Background: Hepatic cancer is the most common primary liver malignancy, with high incidence and mortality worldwide. Atractylon is an active constituent isolated from Atractylodes lancea (Thunb.) DC. and Atractylodes chinensis (DC.) Koidz., which proved to have multiple activities.
Methods: In this study, we evaluated the antihepatic cancer (HCC) effect of atractylon in vitro and in vivo and investigated its underlying mechanism. Cell proliferation, colony formation, cell apoptosis, migration and invaison and was identified by MTT, crystal violet staining, flow cytometry analysis, and Transwell assay. The ∆Ψm of HepG2 and MHCC97H cells were detected by Rhodamine 123. The ROS level was determined by 2,7-Dichlorodi-hydrofluorescein diacetate (DCFH-DA) method. Protein expression was identified by Western blot analysis. The anti-HCC effect of atractylon in vivo was evaluated by a subcutaneous tumor model.
Results: The results suggested that atractylon significantly inhibits the proliferation and promotes apoptosis of hepatic cancer cell lines, including HepG2, SMCC7721, and MHCC97H. Moreover, the results showed that atractylon reduces the mitochondrial membrane potential (∆Ψm), increases ROS level, inhibits the expression of Bcl-2, and promotes the expression of Bax and cleaved caspase-3, indicating that atractylon induces HCC apoptosis through the mitochondrial apoptotic pathway. Our results also demonstrated that atractylon inhibits migration and invasion of hepatic cancer cells by inhibiting the epithelial-mesenchymal transition (EMT) process and downregulating MMP-2 and MMP-9 expression. In addition, atractylon inhibited the growth of hepatic cancer and showed an inhibition effect on EMT process in vivo.
Conclusion: In all, this study suggested that atractylon showed a promising anti-HCC effect with inhibiting proliferation, inducing apoptosis, and blocking invasion in vitro and inhibiting growth in vivo.
Keywords: hepatic cancer, atractylon, migration, apoptosis, EMT
Introduction
Hepatic cancer is the most common primary liver malignancy, with a prevalence of 5th in males and 7th in females, and is the 3rd cause of cancer-related death worldwide.1,2 In 2013, there were about 792,000 new cases of liver cancer in the world and 818,000 cases of liver cancer-related deaths, of which 86% were in developing countries. Among the developing countries, liver cancer is the 5th most common cause of morbidity and the 2nd most common cause of mortality.3 The occurrence and development of liver cancer are related to many factors. In the developed countries, such as Europe and United States, obesity, adult diabetes and internal environmental disorders are the main stimulating factors of liver cancer, while in the developing countries, hepatitis viruses (HBV and HCV), long-term intake of ethanol, and aflatoxin-contaminated food play an important role in the formation of liver cancer.4,5 Surgical resection is the preferred option for early-stage liver cancer patients and confers 5-year survival rates of 70%. However, for patients with intermediate or advanced stage hepatic cancer, only chemoembolization and chemotherapy were optional.6 The current therapies of hepatic cancer are still not ideal, commonly associated with recurrence, metastasis, severe side effect, and drug resistance.7 Hence, the safer and efficacious novel agents are urgently needed for the treatment of hepatic cancer.
Hepatic cancer is a malignant tumor with high metastatic potential, and its recurrence and metastasis are the main causes of treatment failure and death in most hepatic cancer patients.8,9 Metastasis is a complex and dynamic process, which includes the tumor cell exfoliation, vascularized metastasis, new site tumor formation, and other biological behaviors.10 Studies have shown that epithelial-mesenchymal transition (EMT) plays an important role in metastasis of malignant tumors, making the tumor cells more flexible, invasive and migratory, thus promoting tumor metastasis and spread.11 Therefore, inhibition of tumor cell EMT has a positive significance for the treatment of tumors.
In recent decades, natural original products or their derivatives have become an important source of discovering and developing new anticancer drugs.12 Atractylon is a sesquiterpenoid isolated from Atractylodes lancea (Thunb.) DC. and Atractylodes chinensis (DC.) Koidz., which used to treatment of several diseases such as rheumatic diseases, digestive disorders, hepatic protection and influenza for a long history in China.13 In the early 1980s, atractylon was found to show significant hepatoprotective activity, which could prevent liver damage induced by carbon tetrachloride (CCl4) and inhibit hepatocyte DNA damage induced by tert-butyl hydroperoxide.14–16 In recent years, the studies have reported that atractylon also has anti-inflammatory, antinociceptive, antiviral, anti-ulcer and Na+/K+-ATPase inhibition activity.17–19 However, only a few reports on antitumor activity except Yu et al reported that atractylon could inhibit tumor promotion in mouse skin two-stage carcinogenesis induced by 12-O-tetradecanoylphorbol-13-acetate following 7,12-dimethylbenz[a]anthracene initiation in mice.20 In this study, we investigated the effect of atractylon on apoptosis, invasion and migration of hepatic cancer.
Materials and methods
Cell lines and animals
The hepatic cancer cell lines HepG2, SMCC7721, and MHCC97H were obtained from ATCC (MD, USA). These cells were cultured in DMEM supplement with 10% FBS and 1% antibiotics at 5% CO2 at 37 °C. NOD/SCID mice (18~22 g, 6~8 weeks) were obtained from Beijing Huafukang bioscience Co. INC. (Beijing, China). The mice were housed in a standard laboratory condition (temperature 22±2 °C and humidity 50~60%). All animal experiments were performed in accordance with the guidelines of the Experimental Research Institute of Shuguang Hospital affiliated to Shanghai University of Traditional Chinese Medicine. The animal experiment was approved by the medical ethics committee of the Experimental Research Institute of Shuguang Hospital affiliated to Shanghai University of Traditional Chinese Medicine.
MTT assay
The cell viability was detected by MTT method. Briefly, the HepG2, SMCC7721, and MHCC97H cell suspensions were planted in 96-well culture plate at 1×104 cells/well respectively and grown for 12 h. Varying concentrations of atractylon (0, 5, 10, 20 μM) were added and incubated for 24, 48, and 72 h, respectively. Then 20 μl MTT (5 mg/ml) was added and incubated at 37 °C for an additional 4 h. The supernatants were removed and the formazan dissolved in 150 μl DMSO for 10 min. The absorbance was recorded at 570 nM by a micro-plate reader (Bio-Rad).
Colony formation analysis
The HepG2, SMCC7721, and MHCC97H cell suspensions were planted in a 6-well culture plate at 1×103 cells/well respectively and grown for 12 h. The supernatants were removed and the fresh media containing atractylon (5, 10, and 20 μM) were added. The media that contained 0.1% DMSO were added as a control. The cells were cultured for 2 weeks at 5% CO2 37 °C, and the media were replaced every three days. In the end, the supernatants were removed and the cells washed with PBS twice. Then the methanol was added to fix the colonies for 15 min. The colonies were stained with crystal violet solution (0.5%) for 30 min. Finally, the colonies with >10 cells were recorded under a microscope.
Flow cytometry analysis
The apoptosis of HepG2 and MHCC97H cells were detected by flow cytometry. Briefly, the HepG2 and MHCC97H cell suspensions were inoculated into the cell culture flask at 1×106 cells/flask respectively and grown for 12 h. The supernatants were removed, the fresh media containing atractylon (5, 10, and 20 μM) were added. The media that contained 0.1% DMSO were added as a control. The cells were cultured for 24 h. Then the cells were collected and washed with PBS twice and filtered through a 300 mesh nylon mesh to prepare a single cell suspension. Then the Annexin V-FITC apoptosis detection kit was used to determine the apoptosis rates following to manufacturer’s instructions by flow cytometry.
Mitochondrial membrane potential (∆Ψm) analysis
The ∆Ψm of HepG2 and MHCC97H cells were detected by Rhodamine 123. Briefly, the HepG2 and MHCC97H cell suspensions were inoculated into the cell culture flask at 1×106 cells/flask respectively and grown for 12 h. The supernatants were removed and the fresh media containing atractylon (5, 10, and 20 μM) were added. The media that contained 0.1% DMSO were added as a control. The cells were cultured for 36 h. Then the cells were collected and washed with cold PBS and filtered through a 300 mesh nylon mesh to prepare a single cell suspension. Then 1 ml Rhodamine (5 μg/ml) solution was added and incubated for 30 min protection from light. The cells washed with PBS three times and resuspended in fresh media, then subject to ∆Ψm detection by FCM.
Mitochondrial isolation
Mitochondria were isolated from HepG2 and MHCC97H cells by using a Qproteome Mitochondria Isolation Kit (QIAGEN) according to the manufacturer’s instructions. The isolated mitochondria were further purified using Percoll density gradient centrifugation. The supernatant that contains the cytosolic fraction was used for alternative experiments. Mitochondrial isolation was verified by staining for COX IV or ATP5A.
Migration and invasion analysis
The migration and invasion ability of HepG2 and MHCC97H cells were detected by a Transwell chamber that contains polycarbonate filters with 8-μm pores. For the migration assay, 200 μl HepG2 or MHCC97H cell suspensions in serum-free media were added in the upper chamber at 5×104 cells/well, 500 μl media that supplemented 10% FBS were added in the lower chamber. Different concentrations of atractylon were added in the upper and lower chambers to a final concentration of 5, 10, and 20 μM, respectively. The DMSO was added to 0.1% as a control. The cells were incubated at 37 °C for 24 h, then the unmigrated cells in the upper chamber were discarded. The migrated cells on the lower surface were fixed with ice-cold 95% ethanol and stained with 0.1% crystal violet. Then the chamber was inverted and the polycarbonate base film was cut and placed on a glass slide and mounted. The migrated cells in 5 randomly selected fields were quantitated under light microscopy. For the invasion assay, the 5 mg/ml Matrigel was diluted to a 1 mg/ml Matrigel with serum-free DMEM at 1:4. Then the upper chamber was coated with 50 μl of Matrigel (1 mg/ml). The following procedures were the same as the migration assay. The cells were incubated in 37 °C for 24 h, then the cells that did not penetrate through the membrane were removed with a cotton swab. The invaded cells on the underside were fixed with ice-cold 95% ethanol and stained with 0.1% crystal violet. Then the chamber was inverted and the polycarbonate base film was cut and placed on a glass slide and mounted. The cells in 5 randomly selected fields were quantitated under light microscopy.
Reactive oxygen species (ROS) analysis
The ROS level of HepG2 and MHCC97H cells was determined by DCFH-DA method. Briefly, the HepG2 and MHCC97H cell suspensions were planted in 6-well plates at 1×106 cells/well respectively and grown for 12 h. The supernatants were removed, the fresh media containing atractylon (5, 10, and 20 μM) were added. The media that contained 0.1% DMSO was added as a control. The cells were cultured for 48 h. Then 10 μl DCFH-DA (5 μM) was added and incubated for 10 min in dark at 37 °C. After washing with PBS, the cells were observed under Zeiss fluorescence microscopy (Germany).
Western blot
The expression of apoptosis- and migration-related proteins was determined by Western blot. Briefly, the HepG2 and MHCC97H cell suspensions were planted in 6-well plates at 1×106 cells/well respectively and grown for 12 h. The supernatants were removed, the fresh media containing atractylon (5, 10, and 20 μM) were added. The medium that contained 0.1% DMSO was added as a control. The cells were cultured for 48 h. The cells were washed with PBS and harvested. The cell harvests or tissue samples were lysed with RIPA lysis buffer containing 1 mmol/l PMSF. The protein concentration was determined by BCA method. Nonspecific binding was blocked with 5% skimmed milk for 1.5 h, and the membranes were incubated with primary antibodies (Bcl-2, Bax, cleaved caspase-3, α-SMA, Vimentin, N-cadherin, E-cadherin, MMP-2, MMP-9, TIMP2, and GAPDH) overnight at 4 °C. The secondary antibody was incubated for 1 h at room temperature. The protein bands were visualized by chemiluminescent reagents and quantified under an Amersham prime ECL Plus detection system (Pittsburgh, PA).
Subcutaneous tumor model
The NOD/SCID mice were implanted subcutaneously with 100 μl HepG2 cell suspensions (1×107 cells/mouse) into the left dorsal. When the visible tumors at the injection sites grown up to 50–100 mm3, the mice were randomly divided into 3 groups (n=6). The atractylon was dissolved in corn oil and given to the mice by intragastric administration at 5 and 10 mg/kg, which dosed at 0.1 ml/10 g of body weight for 15 d. The equal corn oil was given as a control. The tumor volume was measured every three days as follows the formula: Tumor volume (mm3) =0.5×L×W2, the L is the length and W is the width. In the end, the mice were sacrificed by cervical dislocation, the tumors, hearts, liver, lung, and kidney were excised. The tumors were photographed and weighed immediately. Then the tumors and organs were fixed in formaldehyde subject to HE and IHC analysis.
Immunohistochemistry analysis
The fixed tumor tissues embedded in paraffin. The paraffin blocks were cut into 5 μm thick sections and mounted on glass slides. Then the sections were deparaffinized and rehydrated through xylene and graded alcohols and then immersed in the target retrieval solution in a water bath for 30 min. The endogenous peroxidase was blocked with 3% H2O2 for 15 min, and the nonspecific bindings were blocked with goat serum for 50 min. Then, the slides stained with Ki-67 primary antibodies and secondary antibody polymer HRP successively. Then the slices stained with DAB and counterstained with methyl green. Images were taken with a Leica microscope.
Histopathology
The fixed hearts, liver, lung, and kidney were embedded in paraffin. The paraffin blocks were cut into 5 μm thick sections and mounted on glass slides. The sections were deparaffinized and rehydrated through xylene and graded alcohols. Then the sections were stained with hematoxylin and eosin (H&E) for morphological evaluation under a Leica microscope.
Statistical analysis
All data were presented as means ± SD. Statistical analysis was conducted by one-way ANOVA with SPSS 13.0. Statistical significance was accepted at P-values <0.05.
Results
Atractylon inhibits proliferation of hepatic cancer cells
In this study, we conducted the MTT method and colony formation assay to evaluate the proliferation inhibition effect of atractylon on the hepatic cancer cell lines, including HepG2, SMCC7721, and MHCC97H. As shown in Figure 1A, atractylon decreased the cell viability of HepG2, SMCC7721, and MHCC97H cells in a concentration-dependent manner. As shown in Figure 1B, the inhibition rate of atractylon on HepG2, SMCC7721, and MHCC97H cells was increased in a concentration-dependent manner. The IC50 values of atractylon on HepG2, SMCC7721, and MHCC97H cells were 26.19, 22.32, and 34.14 μM, respectively (Figure 1C). Furthermore, atractylon also significantly inhibited the colony formation of HepG2, SMCC7721, and MHCC97H cells at 10 and 20 μM (Figure 1D).
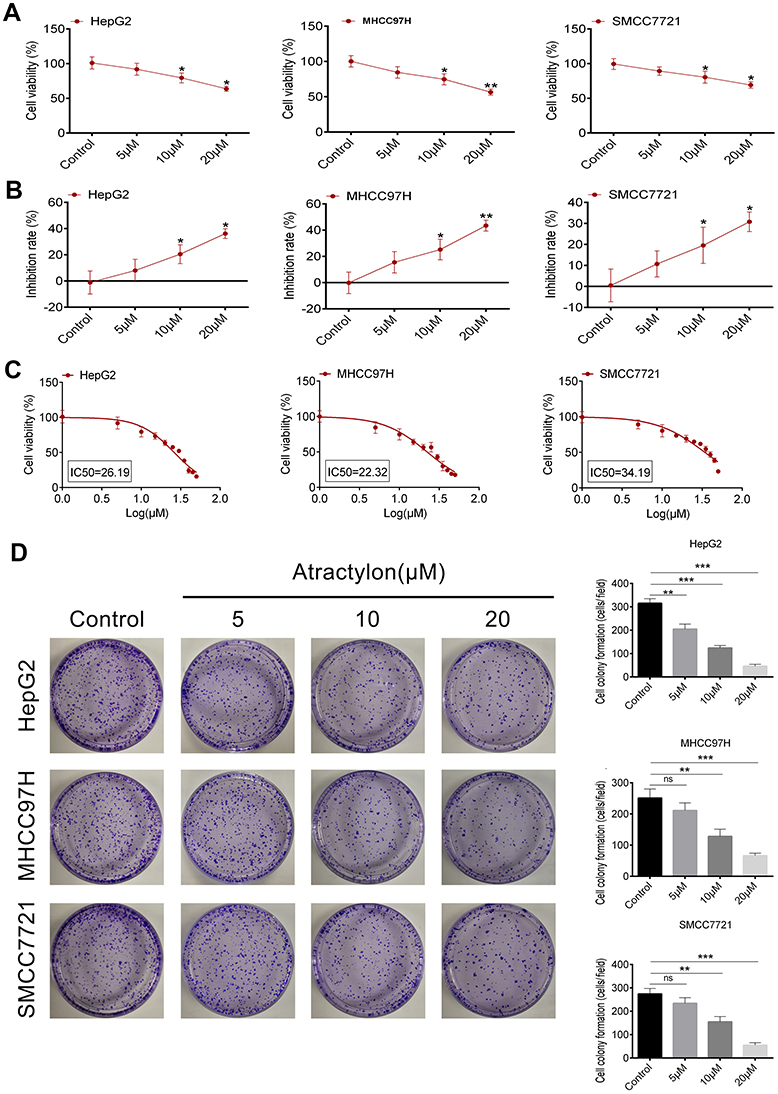 |
Figure 1 The effect of atractylon on hepatic cancer cell proliferation. (A) The cell viability of atractylon on HepG2, LM3, and MHCC97H cells at 0, 5, 10, and 20 μM. (B) The inhibition rate of atractylon on HepG2, LM3, and MHCC97H cells at 0, 5, 10, and 20 μM. (C) The IC50 value of atractylon on HepG2, LM3, and MHCC97H cells. (D) The colony formation inhibition of atractylon (0, 5, 10, 20 μM) on HepG2, LM3, and MHCC97H cells for 2 weeks. The surviving colonies >10 cells were counted. *P<0.05, **P<0.01, ***P<0.001.Abbreviation: ns, not significant. |
Atractylon promotes apoptosis of hepatic cancer cells
We have demonstrated that atractylon showed a significant inhibition effect on proliferation of hepatic cancer cell lines, so we further explored the effect of atractylon on apoptosis by flow cytometry. As shown in Figure 2A, the normal cells that not stained showed in the lower left part, and the early apoptotic cells that stained only by Annexin V-FITC showed in the lower right part, the necrotic cells and late apoptotic cells that stained by Annexin V-FITC and PI showed in the upper right part. For the HepG cells, the early apoptosis rate of the control group and atractylon (5, 10, and 20 μM) were 0.22%, 14.4%, 29.3%, and 49.9% and the late apoptosis rate were 0.087%, 0.016%, 0.55%, and 0.52%, respectively. Atractylon significantly increased the early apoptosis level of HepG2 cells. For the MHCC97H cells, the early apoptosis rate of the control group and atractylon (5, 10, and 20 μM) were 8.58%, 8.94%, 9.98%, and 14.2% and the late apoptosis rate were 2.67%, 5.24%, 15.6%, and 22.6%, respectively. Atractylon significantly increased the level of late apoptosis level in MHCC97H cells. The results suggested that although atractylon significantly increased the total apoptosis rate of HepG2 and MHCC97H cells, it mainly increased early apoptosis in HepG2 cells and late apoptosis in MHCC97H cells.
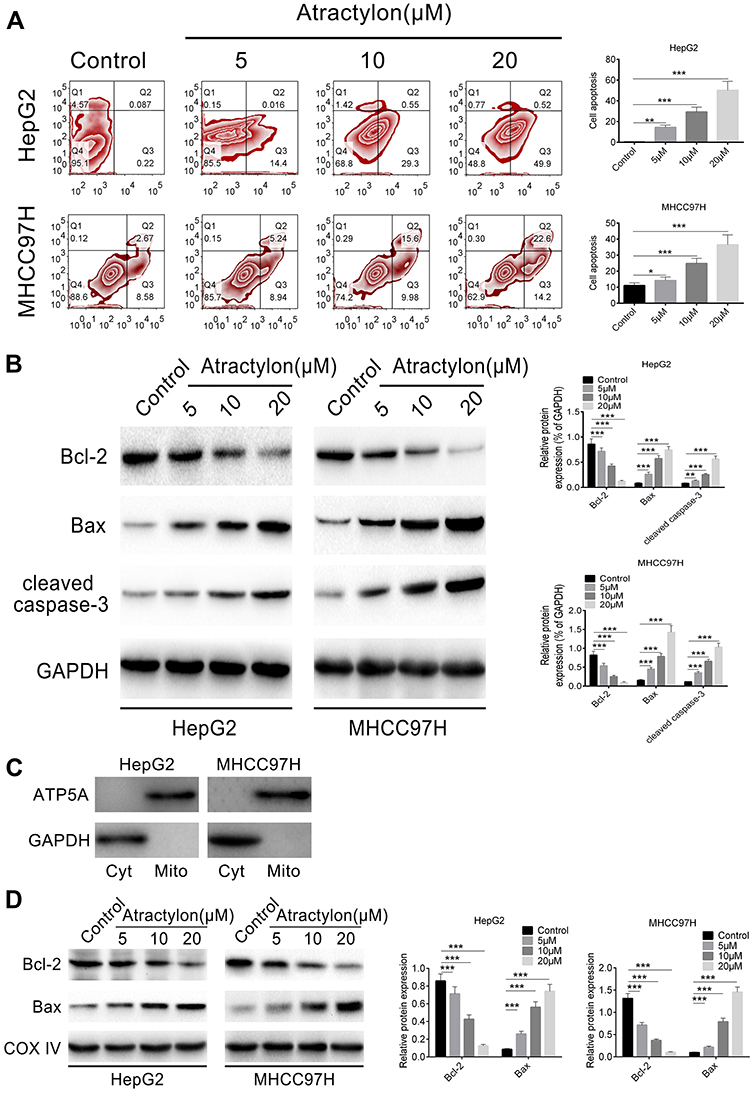 |
Figure 2 The effect of atractylon on hepatic cancer cell apoptosis and apoptosis-associated protein expression. (A) The apoptosis rate of HepG2 and MHCC97H cells that treatment with atractylon (0, 5, 10, 20 μM) for 24 h was determined by flow cytometry. (B) The expression of Bcl-2, Bax, and cleaved caspase-3 in HepG2 and MHCC97H cells that treatment with atractylon (0, 5, 10, 20 μM) for 24 h was detected by Western blot. (C) The expression of ATP5A and GAPDH in the mitochondrial (Mito) and cytosolic (Cyt) fractions that isolated from the cells was determined by Western blot. (D) The expression of Bcl-2 and Bax in the mitochondrial (Mito) was determined by Western blot. *P<0.05, **P<0.01, ***P<0.001. |
Moreover, the effect of atractylon on the expression of apoptosis-associated proteins such as Bcl-2, Bax and cleaved caspase-3 in the HepG2 and MHCC97H cells were also evaluated. As shown in Figure 2B, atractylon (0, 5, 10, 20 μM) significantly inhibited the expression of the anti-apoptotic factor Bcl-2 and promoted the expression of the pro-apoptotic factor Bax and cleaved caspase-3 in both hepatic cancer cell lines (P<0.001). Normally, Bax is localized to the cytosol, and it would translocate to the mitochondria after apoptotic stimulation. To provide convincing evidence to support our hypothesis, the mitochondrial and cytosolic fractions were isolated from the cells, and the contents of Bax and Bcl-2 were detected. As shown in Figure 2C, the ATP synthase-α (ATP5A), an indicator of mitochondrial, was enriched in the mitochondrial fractions, while GAPDH was only enriched in the cytosolic, suggesting the mitochondrial and cytosolic fraction was separated successfully. As shown in Figure 2D, the expression of Bax and Bcl-2 in the mitochondrial was consistent with the level in the cells, suggesting that Bax was translocated to the mitochondria. These results indicated that atractylon promotes apoptosis of hepatic cancer cells may via the mitochondrial pathway.
Atractylon promotes apoptosis by reducing mitochondrial membrane potential (∆Ψm) and increasing ROS
Our current study demonstrated that atractylon inhibits proliferation and promotes apoptosis of hepatic cancer cells. Many studies have shown that mitochondrial apoptotic pathway is closely related to tumor cells apoptosis, so we investigated the effect of atractylon on the disruption of mitochondrial transmembrane potential (∆Ψm), an early event in the apoptotic process, and intracellular ROS for the hepatic cancer cell lines.21 As shown in Figure 3A, 10.5%, 16.5%, and 19.2% of cells lost mitochondrial membrane potential after treatment with atractylon at 5, 10, and 20 μM as compared with control group (5.65%) in HepG2 cells and 10.8%, 13.3%, and 17.4% of cells lost mitochondrial membrane potential in MHCC97H cells, respectively. Moreover, ROS was considered to be able to trigger apoptosis in the mitochondria.22 So the effect of atractylon on the intracellular ROS levels was detected. As shown in Figure 3B, there was showed a low fluorescence intensity in the control group of both HepG2 and MHCC97H cells, however, the fluorescence intensities were increased significantly after treatment with atractylon, indicating an enhanced ROS level.
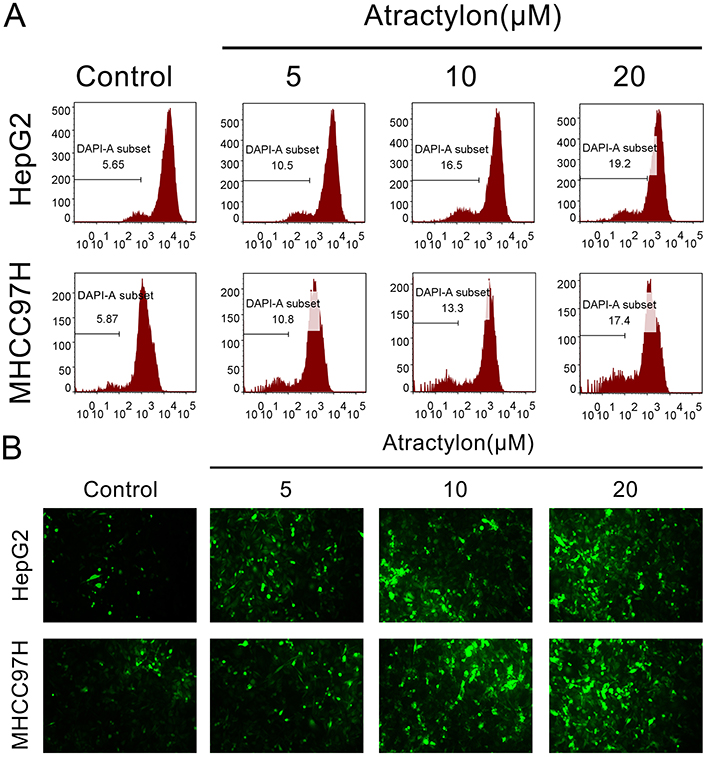 |
Figure 3 Atractylon induces losing of mitochondrial membrane potential (∆Ψm) and increasing ROS. (A) The mitochondrial membrane potential (∆Ψm) of HepG2 and MHCC97H cells that were treated with atractylon (0, 5, 10, 20 μM) was determined by flow cytometry. (B) The intracellular ROS level of HepG2 and MHCC97H cells after treatment with atractylon (0, 5, 10, 20 μM) was determined by 2,7-Dichlorodi-hydrofluorescein diacetate (DCFH-DA) method. *P<0.05, **P<0.01, ***P<0.001. |
Atractylon inhibits the migration and invasion of hepatic cancer cells via EMT process
The migration and invasion ability of tumor cells plays an important role in various tumor metastasis.23 Therefore, the effect of atractylon on the migration and invasion ability of HepG2 and MHCC97H cells were further determined. As shown in Figure 4A, a large number of HepG2 and MHCC97H cells were migrated from the upper chambers to the lower chambers in the control group. However, the migrated cells were sharply decreased when treated with different concentration of atractylon (5, 10, 20 μM). Similar results were also observed in the invasion assay (Figure 4B), atractylon significantly decreased the invasion ability of HepG2 and MHCC97H cells (P<0.001).
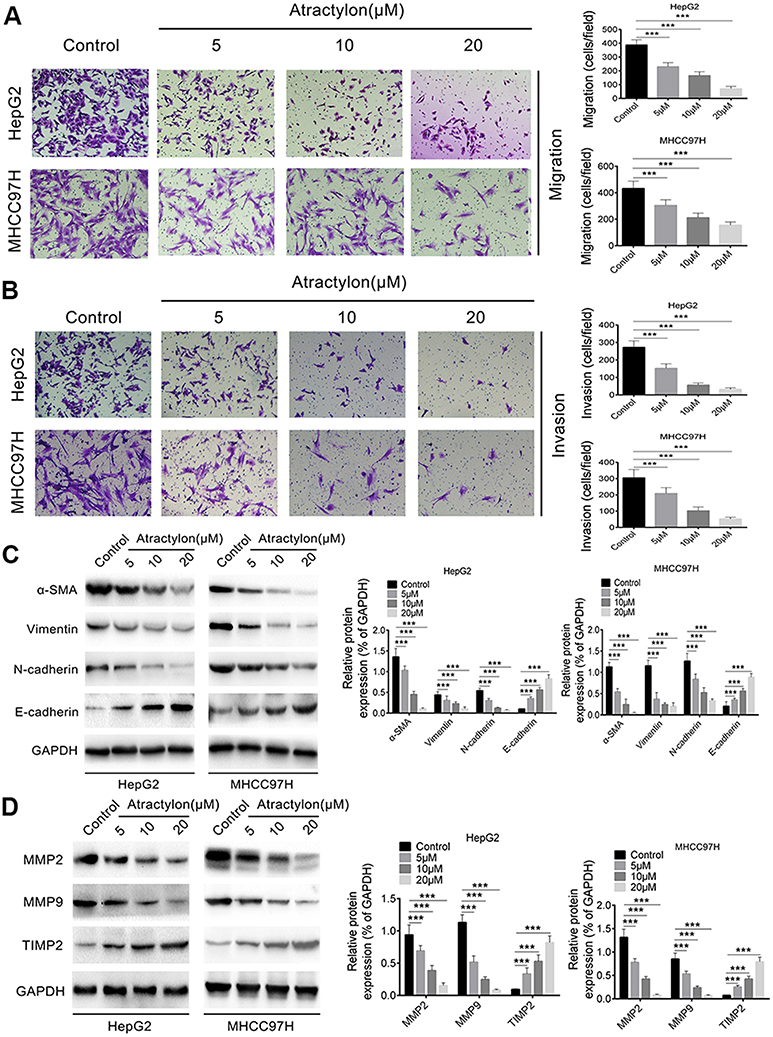 |
Figure 4 Atractylon inhibits HepG2 and MHCC97H cell migration and invasion. (A) The migration ability of HepG2 and MHCC97H cells that were treated with atractylon (0, 5, 10, 20 μM) was determined by the Transwell chamber. (B) The invasion ability of HepG2 and MHCC97H cells that were treated with atractylon (0, 5, 10, 20 μM) was determined by the Transwell chamber. (C) The expression of MMP-2, MMP-9, and TIMP2 in HepG2 and MHCC97H cells that were treated with atractylon (0, 5, 10, 20 μM) was detected by Western blot. (D) The expression of EMT markers (E-cadherin, N-cadherin, Vimentin, and α-SMA) in HepG2 and MHCC97H cells that were treated with atractylon (0, 5, 10, 20 μM) was detected by Western blot. *P<0.05, **P<0.01, ***P<0.001. |
Epithelial-mesenchymal transition (EMT) is considered to be the initial step in the spread of tumor cells, leading to invasion and metastasis of malignant tumors.24 So the expression of EMT markers, such as E-cadherin, N-cadherin, Vimentin, and α-SMA, in the hepatic cancer cell lines was determined. As shown in the Figure 4C, the control group showed a low level of E-cadherin and high levels of α-SMA, Vimentin, and N-cadherin, indicating that EMT occurred in HepG2 and MHCC97H cells and the epithelial phenotype was lost. However, the expression level of E-cadherin was increased and the expression levels of α-SMA, Vimentin, and N-cadherin were decreased significantly after treatment with atractylon (P<0.001). These results indicated that the atractylon inhibited the EMT process of HepG2 and MHCC97H cells. Furthermore, previous studies have shown that the MMPs such as MMP-2 and MMP-9 promotes the EMT process of degrading the basement membrane and promoting invasion.11,25 Therefore, the expression of MMP-2 and MMP-9, and TIMP2 in the hepatic cancer cell lines was also determined. As shown in Figure 4D, the control group showed a high level of MMP-2 and MMP-9 and a low level of TIMP2 in both HepG2 and MHCC97H cells. Atractylon decreased the expression of MMP-2 and MMP-9 and increased the expression of TIMP2, an inhibitor of MMP, significantly (P<0.001). Altogether, these results suggested that atractylon inhibited the migration and invasion of hepatic cancer cells via EMT process.
Atractylon inhibits hepatic cancer growth in vivo
Our present data have demonstrated that atractylon showed significant antitumor activity in vitro, in order to extend the in vitro effect of atractylon to the in vivo situation, a subcutaneous hepatic cancer tumor mouse model was established. The tumor-bearing mice were received intragastric administration of atractylon at the dose of 5 and 10 mg/kg for 15 d. The tumor volume and weight were recorded. As shown in Figure 5A, the tumors from the mice that treatment with atractylon was obviously smaller than the control group visually, the data of volume and weight were consistent with the pictures. The Ki-67, an indicator closely related to tumor cell proliferation, was determined by IHC. As shown in Figure 5B, the control group showed a large number of positive staining nuclear Ki-67, indicating that the subcutaneously injected HepG2 cells proliferated actively. Atractylon inhibited the proliferating cells that stained positive for nuclear Ki-67 significantly. Furthermore, the effect of atractylon on the expression of EMT markers (E-cadherin, N-cadherin, Vimentin, and α-SMA), EMT promoters (MMP-2 and MMP-9, and TIMP2), and apoptosis-associated factors (Bcl-2, Bax, and cleaved caspase-3) in vivo was also investigated. As shown in Figure 5C, the decrease of epithelial marker E-cadherin and increase of mesenchymal markers α-SMA, N-cadherin, and Vimentin were observed in the xenograft tumor tissues. Consistent with the data of in vitro tests, atractylon significantly increased the expression of E-cadherin and decreased the expression of α-SMA, Vimentin, and N-cadherin. Similarly, atractylon significantly increased the expression of TIMP2 and decreased the expression of MMP-2 and MMP-9. In addition, atractylon also significantly decreased the expression of Bcl-2 and increased the expression of Bax and cleaved caspase 3 in vivo (Figure 5D). These results indicated that atractylon inhibited the growth of hepatic cancer and showed an inhibition effect on EMT process.
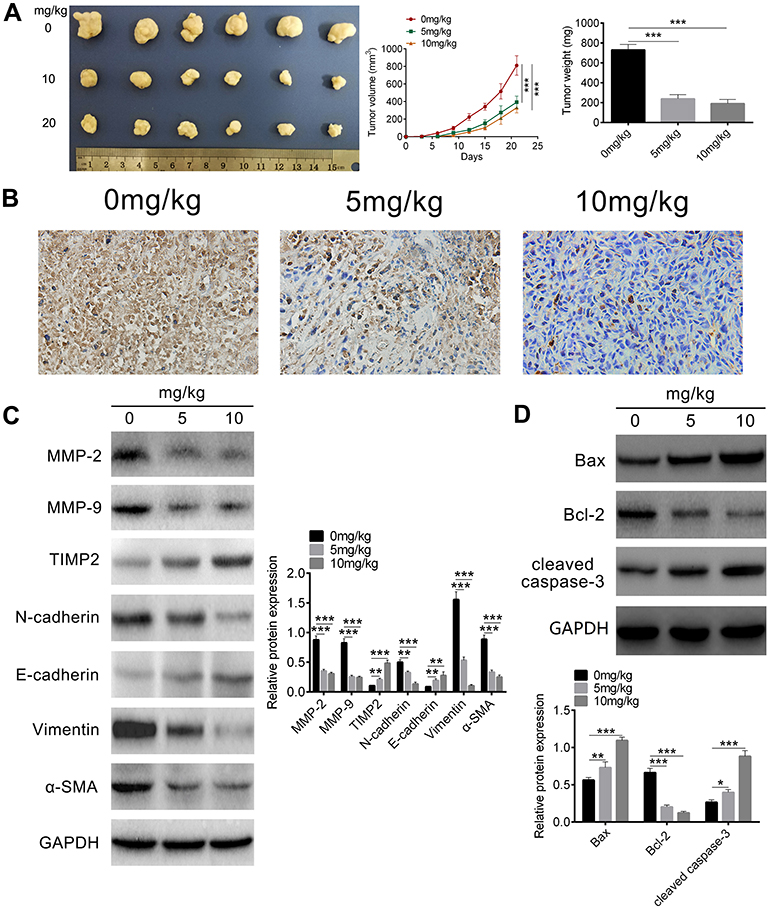 |
Figure 5 The antitumor effect of atractylon in vivo. (A) A in vivo hepatic cancer tumor mouse model was established by implanting subcutaneously with 100 μl HepG2 cell suspensions (1×107 cells/mouse) into the left dorsal. The tumor volume was measured every 3 days, the tumor tissues were excised, photographed, and weighed at the end. (B) The expression of Ki-67 in the tumor sections was detected by IHC staining (20×). (C) The expression of MMP-2, MMP-9, TIMP2, E-cadherin, N-cadherin, Vimentin, and α-SMA in xenograft tumor tissues was detected by Western blot. (D) The expression of Bcl-2, Bax, and cleaved caspase 3 in xenograft tumor tissues was detected by Western blot. *P<0.05, **P<0.01, ***P<0.001. |
Discussions
Hepatic cancer is one of the most common malignant worldwide with considerable metastatic potential. In the early-stage of hepatic cancer, the clinical symptoms were not obvious, most patients have developed in advanced stages when diagnosed. Distant metastasis, which promoted by the invasion and migration of tumor cells, is one of the major causes of high recurrence and mortality among hepatic cancer patients.9 Due to the complicated molecular mechanism and genetic differences, the present chemotherapy agents are limited and not effective for all the hepatic cancer patients. Therefore, the safer and more efficacious novel chemotherapy agents are urgently needed development for hepatic cancer.
Chinese medicine herb has been used to treat various diseases under the guidance of the traditional Chinese medicine theory for thousands of years in China. In recent decades, the natural original products from herbs or their derivatives have become an important source of discovering and developing new anticancer drugs.12 Atractylon is an active constituent isolated from Atractylodes lancea (Thunb.) DC. and Atractylodes chinensis (DC.) Koidz., which proved to have multiple activities, such as anti-inflammatory, antinociceptive, antiviral, anti-ulcer, antitumor, and Na+/K+-ATPase inhibition activity.17–20 Our current study expanded the potential antitumor activity of atractylon in hepatic cancer. The results showed that atractylon not only inhibited proliferation and induced apoptosis of hepatic cancer cells in vitro, and also showed a significant antitumor effect in vivo. Therefore, its underlying mechanism was further investigated.
Numerous evidence has demonstrated that mitochondria play an important role in apoptosis, tumorigenesis, and drug resistance. The mitochondrial signal transduction pathway has a special position in cellular energy metabolism and apoptosis initiation.26 Mitochondrial damage is characterized by losing mitochondrial membrane potential (Δψm), increasing membrane permeability, releasing of apoptogenic factors from mitochondria into the cytosol, and losing of oxidative phosphorylation, it would induce irreversible apoptosis ultimately.27 Our data showed that atractylon significantly promoted the apoptosis and induced the loss of membrane potential of hepatic cancer cells. In addition, atractylon also increased the level of ROS, which can directly result in activation of the mitochondrial membrane permeability and losing of mitochondrial transmembrane potential (∆Ψm).28 Studies have shown that Bcl-2 family proteins play a major regulatory role in mitochondria-dependent apoptosis. Under normal conditions, Bax is localized to the cytosol and combines with Bcl-2 to form a heterodimer, which stabilizes the cytoplasmic localization of pro-apoptotic proteins and inhibits apoptosis. However, Bcl-2/Bax balance can be broken by a variety of apoptosis-inducing signals, resulting in Bax to dissociate from the Bcl-2/Bax heterodimer and translocates to the mitochondrial membrane to initiate apoptotic pathway.29 Our studies suggested that atractylon significantly increased the pro-apoptotic proteins such as Bax and caspase-3 and decreased anti-apoptotic protein Bcl-2 levels in a dose-dependent manner. We also investigated the contents of Bax and Bcl-2 in the mitochondrial and cytosolic. The expression of Bax and Bcl-2 in the mitochondrial was consistent with the level in the cells, suggesting that Bax was translocated to the mitochondria. In addition, atractylon also decreased the expression of Bcl-2 and increased the expression of Bax and cleaved caspase 3 in vivo. These results indicated that atractylon promotes apoptosis of hepatic cancer cells may via the mitochondrial apoptotic pathway.
Numerous evidence has shown that the epithelial cell malignancies can acquire migration and invasion ability through epithelial-mesenchymal transition (EMT), which promotes tumor spread and metastasis. The main feature of EMT is the loss of epithelial markers such as E-cadherin and ZO-1 and accompanied by an increase of mesenchymal markers, such as α-SMA, N-cadherin, and Vimentin.30 Our present study revealed that atractylon inhibited the migration and invasion of hepatic cancer cells and increased the level of E-cadherin and decreased the level of α-SMA, N-cadherin, and Vimentin. Cancer metastasis is a multistep process that involves a series of complex interactions.31 Degradation of the extracellular matrix and basement membrane is a key step to promote tumor cell migration and invasion. Matrix metalloproteinases (MMPs) family is an important class of proteolytic enzymes that degrade the extracellular matrix. It plays an important role in the degradation of extracellular matrix, and high expression of MMPs can promote the invasion and migration of tumor cells.32 MMP-2 and MMP-9 are two important MMPs that highly expressed in a variety of tumors and play an important role in the degradation of extracellular matrix basic skeleton IV collagen.33 In this experiment, we found that atractylon can effectively inhibit the expression of MMP-2 and MMP-9 and increase the expression of TIMP2, an inhibitor of the MMPs. Furthermore, the effect of atractylon on the expression of EMT markers and EMT promoters in vivo was also investigated. Consistent with the in vitro data, atractylon significantly increased the expression of E-cadherin and decreased the expression of α-SMA, Vimentin, and N-cadherin. Similarly, atractylon significantly increased the expression of TIMP2 and decreased the expression of MMP-2 and MMP-9. Taken together, these results indicated that atractylon inhibits migration and invasion of hepatic cancer cells by inhibiting EMT and down-regulating MMP-2 and MMP-9 expression.
In conclusion, we evaluated the antitumor effect of atractylon in vitro and in vivo and investigated its underlying mechanism of invasion inhibition. The results suggested atractylon inhibited the proliferation and induced apoptosis of hepatic cancer cells via the mitochondrial apoptotic pathway. Furthermore, these results also suggested that atractylon inhibits migration and invasion of hepatic cancer cells by inhibiting EMT and down-regulating MMP-2 and MMP-9 expression. In addition, atractylon significantly inhibited hepatic cancer growth in vivo. In all, this study suggested that atractylon showed a promising antihepatic cancer effect with inhibiting proliferation, inducing apoptosis, and blocking invasion in vitro and inhibiting growth in vivo.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
This study was supported by grants from the “TCM Hepatology” Key Discipline Construction Project of Pudong Health Bureau of Shanghai (PWZxk2017-30) and the Shanghai Pudong New Area Science and Technology and Economic Committee (PKJ2017-Y55).
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no competing or conflicting interests in regard to this work.
References
1. Ferlay J, Shin HR, Bray F, et al. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. doi:10.1002/ijc.25516
2. El-Serag HB. Hepatocellular carcinoma. Hepat Mon. 2011;11(2):69–70.
3. Fitzmaurice C, Dicker D, Pain A, et al. The global burden of cancer 2013. JAMA Oncol. 2015;1(4):505. doi:10.1001/jamaoncol.2015.0735
4. Trojan J, Zangos S, Schnitzbauer AA. Diagnostics and treatment of hepatocellular carcinoma in 2016: standards and developments. Viszeralmedizin. 2016;32(2):116–120.
5. Yang C, Jian P, Chen J. Identification of potential gene network associated with HCV-related hepatocellular carcinoma using microarray analysis. Pathol Oncol Res. 2017;24(3):1–8.
6. Llovet JM, Zucman-Rossi J, Pikarsky E, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. doi:10.1038/nrdp.2016.18
7. Cervello M, Mccubrey JA, Cusimano A, et al. Targeted therapy for hepatocellular carcinoma: novel agents on the horizon. Oncotarget. 2012;3(3):236–260. doi:10.18632/oncotarget.466
8. Morise Z, Kawabe N, Tomishige H, et al. Recent advances in liver resection for hepatocellular carcinoma. Front Surg. 2014;1:21. doi:10.3389/fsurg.2014.00021
9. Yu Y, Shen H, Yu H, et al. Systematic proteomic analysis of human hepotacellular carcinoma cells reveals molecular pathways and networks involved in metastasis. Mol Biosyst. 2011;7(6):1908–1916. doi:10.1039/c0mb00265h
10. Perlikos F, Harrington KJ, Syrigos KN. Key molecular mechanisms in lung cancer invasion and metastasis: a comprehensive review. Crit Rev Oncol Hematol. 2013;87(1):1–11. doi:10.1016/j.critrevonc.2012.12.007
11. Orlichenko LS, Radisky DC. Matrix metalloproteinases stimulate epithelial-mesenchymal transition during tumor development. Clin Exp Metastasis. 2008;25(6):593–600. doi:10.1007/s10585-008-9143-9
12. Twilley D, Lall N. Chapter 7 – the role of natural products from plants in the development of anticancer agents. Nat Rev Drug Discovery. 2018;1:139–178.
13. Koonrungsesomboon N, Na-Bangchang K, Karbwang J. Therapeutic potential and pharmacological activities of Atractylodes lancea (Thunb.) DC. Asian Pac J Trop Med. 2014;7(6):421–428. doi:10.1016/S1995-7645(14)60069-9
14. Kiso Y, Tohkin M, Hikino H. Antihepatotoxic principles of atractylodes rhizomes. J Nat Prod. 1983;46(5):651. doi:10.1021/np50029a010
15. Kiso Y, Tohkin M, Hikino H. Mechanism of antihepatotoxic activity of atractylon, I: effect on free radical generation and lipid peroxidation. Planta Med. 1985;51(2):97–100. doi:10.1055/s-2007-969416
16. Hwang JM, Tseng TH, Hsieh YS, et al. Inhibitory effect of atractylon on tert-butyl hydroperoxide induced DNA damage and hepatic toxicity in rat hepatocytes. Arch Toxicol. 1996;70(10):640–644.
17. Satoh K, Nagai F, Ushiyama K, Kano I. Specific inhibition of Na+,K(+)-ATPase activity by atractylon, a major component of byaku-jutsu, by interaction with enzyme in the E2 state. Biochem Pharmacol. 1996;51(3):339. doi:10.1016/0006-2952(95)02188-4
18. Yang C, Mai JY, Hou TL, et al. Antiviral activities of atractylon from Atractylodis Rhizoma. Mol Med Rep. 2016;14(4):3704–3710. doi:10.3892/mmr.2016.5713
19. Chen LG, Jan Y, Tsai P, et al. Anti-inflammatory and antinociceptive constituents of Atractylodes japonica Koidzumi. J Agric Food Chem. 2016;64:11.
20. Yu S, Yasukawa K, Takido M. Atractylodis Rhizoma extract and its component, atractylon, inhibit tumor promotion in mouse skin two-stage carcinogenesis. Phytomedicine. 1994;1(1):55. doi:10.1016/S0944-7113(11)80023-1
21. Xiong Y, Ye T, Wang M, et al. A novel cinnamide YLT26 induces breast cancer cells Apoptosis via ROS-mitochondrial apoptotic pathway in vitro and inhibits lung metastasis in vivo. Cell Physiol Biochem. 2014;34(6):1863–1876. doi:10.1159/000366385
22. Finkel T. Signal transduction by reactive oxygen species in non-phagocytic cells. J Leukoc Biol. 1999;65(3):337–340. doi:10.1002/jlb.65.3.337
23. Zhang T, Li J, Dong Y, et al. Cucurbitacin E inhibits breast tumor metastasis by suppressing cell migration and invasion. Breast Cancer Res Treat. 2012;135(2):445–458. doi:10.1007/s10549-012-2175-5
24. Thiery JP, Acloque H, Huang RYJ, et al. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. doi:10.1016/j.cell.2009.11.007
25. Bhuvarahamurthy V, Kristiansen GO, Johannsen M, et al. In situ gene expression and localization of metalloproteinases MMP1, MMP2, MMP3, MMP9, and their inhibitors TIMP1 and TIMP2 in human renal cell carcinoma. Oncol Rep. 2006;15(5):1379.
26. Indran IR, Tufo G, Pervaiz S, et al. Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochim Biophys Acta. 2011;1807(6):735–745. doi:10.1016/j.bbabio.2011.03.010
27. Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (Δψm) in apoptosis; an update. Apoptosis. 2003;8(2):115–128.
28. Chung YM, Bae YS, Lee SY. Molecular ordering of ROS production, mitochondrial changes, and caspase activation during sodium salicylate-induced apoptosis. Free Radic Biol Med. 2003;34(4):434–442. doi:10.1016/S0891-5849(02)01301-1
29. Dewson G, Kluck RM. Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci. 2009;122(16):2801–2808.
30. Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013;27(20):2192–2206. doi:10.1101/gad.225334.113
31. Jin K, Gao W, Lu Y, et al. Mechanisms regulating colorectal cancer cell metastasis into liver (Review). Oncol Lett. 2012;3(1):11–15. doi:10.3892/ol.2011.432
32. Deryugina EI, Quigley JP. Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biol. 2015;44–46:94–112. doi:10.1016/j.matbio.2015.04.004
33. Kai K, Wang CY, Werb Z. Matrix metalloproteinases in stem cell regulation and cancer. Matrix Biol. 2015;44–46(46):184–190. doi:10.1016/j.matbio.2015.01.022
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.