")
Back to Journals » OncoTargets and Therapy » Volume 13
ATF3 Demethylation Promotes the Transcription of ARL4C, Which Acts as a Tumor Suppressor in Human Breast Cancer
Authors Li L, Sun RM, Jiang GQ
Received 24 December 2019
Accepted for publication 14 April 2020
Published 23 April 2020 Volume 2020:13 Pages 3467—3476
DOI https://doi.org/10.2147/OTT.S243632
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Takuya Aoki
Liqi Li, 1, 2,* Rong-Mao Sun, 1,* Guo-Qin Jiang 1
1Department of Surgery, The Second Affiliated Hospital of Soochow University, Suzhou, Jiangsu 215004, People’s Republic of China; 2Department of Thyroid Breast Surgery, Affiliated Hospital of Jiangnan University, Wuxi, Jiangsu 214062, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Guo-Qin Jiang
Suzhou 215004, People’s Republic of China
Tel/Fax +86-512-67784797
Email [email protected]
Introduction: Breast cancer is a common malignancy in females worldwide. In this study, we investigated the role of activating transcription factor 3 (ATF3) and ADP-ribosylation factor like-4 (ARL4) in human breast cancer, and the associated mechanisms.
Materials and Methods: We measured ATF3 and ATL4C expressions in 15 paired breast cancer tissues using qRT-PCR, Western blotting and IHC. Cell growth, migration and invasion were tested in ATF3 or ARL4C overexpression breast cancer cells. TCGA database analysis was done to identify the correlation between ATF3 and ARL4C. We evaluated the binding of ATF3 to ARL4C promoter sequences and the effect of hypermethylation and demethylation of ATF3. A meta-analysis was done to investigate the relationship between the expression of ATF3 and/or ARL4C and the poor prognoses.
Results: Our results showed that ATF3 and ARL4C were decreased in breast cancer specimens at both mRNA and protein levels. Restoration of ATF3 or ARL4C reduced breast cancer tumorigenesis, evidenced by decreased cell growth, migration and invasion. The expression of ATF3 was positively correlated with ARL4C in breast cancer specimens, and ATF3 was shown to bind to the ARL4C promoter sequences. Furthermore, the expression of ATF3 was negatively regulated by hypermethylation, and demethylation of ATF3 stimulated ATF3 expression, which further promoted ARL4C transcription. Finally, a meta-analysis showed that patients with breast cancer with lower expression levels of ATF3 and/or ARL4C had worse prognoses.
Conclusion: Our results suggest that the ATF3/ARL4C axis may be a prospective biomarker for diagnosis and determination of prognosis, and a potential target for breast cancer treatment.
Keywords: ATF3, ARL4C, methylation, breast cancer, 5ʹ-Aza-dC
Introduction
Breast cancer is the leading cause of cancer-related death in women worldwide.1 Breast cancer subtypes include ER+, PR+, HER2+, and triple negative breast cancer (TNBC).2,3 Improvements have been made in early screening, diagnosis, and targeted therapy, but the detailed mechanisms of development of breast cancer are not well-characterized. Characterization of the molecular basis of breast cancer, and identification of reliable biomarkers, are critical to improvement of early diagnosis and treatment.
Activating transcription factor 3 (ATF3) is a member of the ATF/CREB transcription factor family, and contains a leucine zipper DNA binding domain that binds to a consensus sequence, TGACGTCA.4 Previous studies have shown that ATF3 can be stimulated by a variety of stress signals including anoxia,5 DNA damage, and hypoxia.6 Activating transcription factor 3 is considered an adaptive-response gene that participates in various biological processes,4,7 and it has been shown to act as a tumor suppressor or an oncogene in various cancers.8–10 Yin et al indicated that ATF3 enhanced TGFβ signaling and promoted breast cancer progression.11 In contrast Chen et al reported that ATF3 inhibited hepatocellular carcinoma carcinogenesis.12 However, the detailed molecular mechanisms of ATF3 in breast cancer tumorigenesis have not been characterized. In this study, we quantified the expression of ATF3 in breast cancer and investigated the effects of DNA methylation on ATF3 expression.
Adenosine diphosphate-ribosylation factor like-4 (ARL4C) is a member of the ARL4 family, and has been shown to play a role in several essential biological functions.13 Previous studies have shown that ARL4C plays a key role in cell proliferation, migration, and invasion of cancer cells, and may be a promising therapeutic target in several cancers.14,15 However, the role of ARL4C in breast cancer has not been characterized. In this study, we investigated role of ARL4C in development of breast cancer.
In the present study we showed that ATF3 expression was decreased in breast carcinoma. In addition, overexpression of ATF3 suppressed breast cancer tumorigenesis. Furthermore, ATF3 was shown to be a transcriptional regulator of ARL4C, which was also associated with breast cancer. We also showed that methylation of the ATF3 promoter led to ATF3 degradation and suppression of ARL4C expression. Downregulation of ARL4C was observed in breast carcinoma, and restoration of ARL4C expression inhibited tumor growth, migration, and invasion. Meta-analysis indicated that low expression of ATF3 or ARL4C was associated with poor prognosis.
Materials and Methods
Clinical Specimen
Fifteen paired human breast tissue samples were obtained from patients who underwent surgical procedures at Affiliated Hospital of Jiangnan University (Table 1). The study was approved by the ethical committee of the Affiliated Hospital of Jiangnan University. All study subjects provided informed written consent prior to initiation of the study.
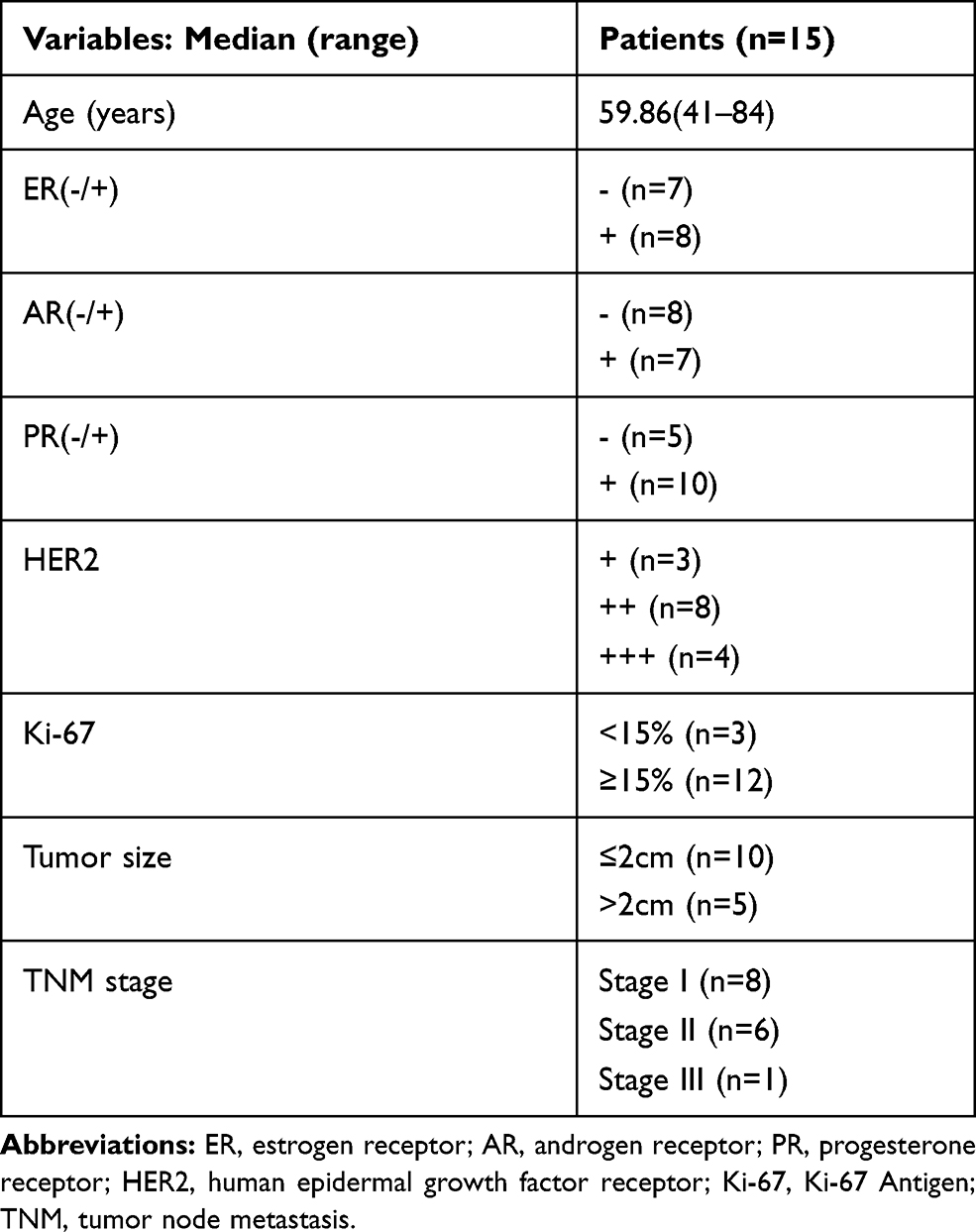 |
Table 1 Patient Clinical Information |
Cell Lines and Cell Culture
The breast cancer cell lines MCF7, CAMA1, BT474, BT20, MDA-MB-231, and MDA-MB-435 were purchased from the Cell Bank of Chinese Academy of Science (Shanghai, China). MCF7, CAMA1, BT474, and BT20 cells were cultured in DMEM medium containing 10% FBS, and MDA-MB-231 and MDA-MB-435 cells were cultured in RPMI-1640 medium with 10% FBS. The cells were incubated with 500 nM 5-aza-deoxycytidine (5ʹ-Aza-dC, Sigma Aldrich) for different lengths of time. All cells lines were cultured at 37 °C with 5% CO2.
Plasmid Construction
Plasmids were constructed using the lentiviral transferring plasmid, pLVX-CMV-MCS-Green (pLVX). Human ATF3 and ARL4C genes were amplified and inserted into pLVX to generate recombinant pLVX-ATF3 and pLVX-ARL4C for lentivirus production. The pGL3-ARL4C luciferase reporter plasmid was constructed by stitching the ARL4C promoter sequence and inserting it into pGL3 plasmid.
Production and Transfection of Lentivirus
The sequences of ATF3 and ARL4C were amplified and cloned into pLVX, then co-transfected into HEK293T cells with psPAX2 and pMD2.G. The viruses were harvested and filtered after 48 hours. Then, the breast cancer cell lines were infected using 8 μg/mL polybrene.16
Cell Proliferation Assay
Stable cell lines were constructed using the ATF3/ARL4C lentivirus. The cells were plated into 96-well plates at 1× 104 cells/well. Cell viability was measured at 0, 1, 3, and 5 days using CCK-8 (Cell Counting Kit-8) reagent (Sigma, 96992, USA) according to the manufacturer’s instructions. Optical density (OD) was measured at 450 nm using a Multi Mode plate reader (BioTek, USA).
Migration and Invasion Assay
Cell migration and invasion (using matrigel) were evaluated using the transwell assay (Corning, USA). Cells (6×105 cells/well) were seeded into the upper chamber without FBS, and media containing FBS was added into the lower chamber. After incubating for 24 hours, the cells were fixed and stained with crystal violet. Each treatment was performed in triplicate.
RNA Extraction and Quantitative RT-PCR
Total RNA was extracted using Trizol reagent (Invitrogen, USA) and reverse transcribed using the Reverse Transcription System (Vazyme, Nanjing, China). Quantitative RT-PCR was performed using Power SYBR Green Master Mix (ThermoFisher). Gene expression was normalized to GAPDH. The primer sequences used in this study were as follows:ARL4C (Forward: 5ʹ-ATGGGCAACATCTCCTCTAACAT-3ʹ, Reverse: 5ʹ-TCCACCGAGTCCACCACGTAGAT-3ʹ), ATF3 (Forward: 5ʹ- CTTCAACACCCAGGCCAGGTCTC-3ʹ, Reverse: 5ʹ-ATTTCTTTCTCGTCGCCTCTTTT-3ʹ), GAPDH (Forward: 5ʹ-AGAAGGCTGGGGCTCATTTG-3ʹ; Reverse: 5ʹ-AGGGGCCATCCACAGTCTTC-3ʹ).
Western Blot
Cells were harvested and extracted using RIPA lysis buffer (Thermo Fisher, USA). Thirty micrograms of protein from each sample was separated by 10% SDS-PAGE, then transferred to PVDF membranes (Millipore, USA). The membranes were blocked and incubated with primary antibodies (1:1000) against ARL4C (Invitrogen, PA5-56011, USA), ATF3 (Santa Cruz, sc-518032, USA), GAPDH (MultiSciences, MAB5465, China), and β-actin (Cell Signaling Technology 3700, USA). After washing with TBS-T, the membranes were treated with the secondary antibody (1:5000) at room temperature for 1 h. Blots were developed and visualized by enhanced chemiluminescence detection using a Tannon Western blotting detection system (Shanghai, China).
Dual-Luciferase Reporter Assay
For dual-luciferase assay, HEK293T cells (1×105 cells/well) were transfected with pLVX, pLVX-ATF3, pGL3-control, or pGL3-ARL4C-promoter. Cell lysates were collected after 48 h. Luciferase activity was analyzed using the dual-luciferase reporter assay kit according to the manufacturer’s instructions (Promega).
Immunohistochemical Analysis
After blocking endogenous peroxide and protein activity, 4-μm sections of clinical tissues were incubated at 4 °C with primary antibodies against ATF3 (1:200, Santa Cruz, sc-518032, USA) or ARL4C (1:500, Invitrogen, PA5-56011, USA). After washing with PBS, the sections were incubated with secondary antibody (1:2000) and stained using 3,3-diaminobenzidine as described previously.17
Chromatin Immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed using a ChIP KIT according to the manufacturer’s instructions (Cell signaling, 9002, USA). Cultured MDA-MB-231 and MDA-MB-435 cells were treated with 1% formaldehyde. Cell lysates were then sonicated and immunoprecipitated using an anti-ATF3 antibody (Santa Cruz, sc-518032, USA), or normal rabbit IgG (MultiSciences, GAR0072, China) as a control. The primer sequences used for ChIP analysis were as follows: forward primer (5ʹ-GGGCTGGTGTTGCCATTTCT-3ʹ), reverse primer (5ʹ-TTGGGGCAGGGTCGGACCTA-3ʹ). Amplified DNA was normalized to the input total DNA.
Analysis of Methylation
Total DNA was isolated using a DNA Tissue Kit (Generay Biotech, GK0221, China), then bisulfite modified using the EpiTect Bisulfite kit (QiaGen, Cat.no-59104, China). A 2000-bp sequence was selected from the promoter region of the ATF3 gene (Table S1). For methylation-specific polymerase chain reaction (MSP), PCR primers specific for unmethylated (U) or methylated (M) DNA were as follows: M-forward: 5ʹ-ATTATGTTGGTTAGGTTAGTTTCGA-3ʹ; M-reverse: 5ʹ-TTAACTAAATACGATAATTCACGCC-3ʹ. U-forward: 5ʹ-ATTATGTTGGTTAGGTTAGTTTTGA-3ʹ; M-reverse: 5ʹ-TTAACTAAATACAATAATTCACACC-3ʹ.
Statistical Analysis
All results are presented as means ± standard deviations. Two-tailed Student’s t-test or one-way ANOVA was used to determine statistical significance (Prizm, GraphPad Software Inc.). P < 0.05 was considered statistically significant. All experiments were performed at least in triplicate.
Results
ATF3 Was Downregulated in Human Breast Cancer Samples
We evaluated ATF3 mRNA levels in 15 breast carcinoma tissues and adjacent normal tissues using qRT-PCR. The results showed significantly lower ATF3 expression in 14 of the 15 human breast carcinoma samples compared to that in paired normal tissues (Figure 1A). We then randomly selected 5 tissues from each group for Western blot analysis. As shown in Figure 1B, protein expression of ATF3 in breast carcinoma tissues was significantly lower than that in paired normal tissues. Consistent with these observations, there were less ATF3-positive cells in the breast carcinoma lesions than in the normal breast tissues, as determined using immunohistochemistry staining (IHC) (Figure 1C). We then analyzed 1218 clinical samples (https://tcga.xenahubs.net/download/TCGA.BRCA.sampleMap/HiSeqV2.gz; full metadata). Consistent with the results obtained from our clinical specimens, ATF3 levels were decreased in breast tumors (Figure 1D). In addition, KM plotter was used to analyze prognostic results (n=1764) (http://kmplot.com/analysis/index.php?p=service). Patients with low ATF3 (Affy ID 1554420) levels had a poorer survival rate (Figure 1E). These results indicated that ATF3 was down-regulated in breast cancer, and may be a predictor of breast cancer prognosis.
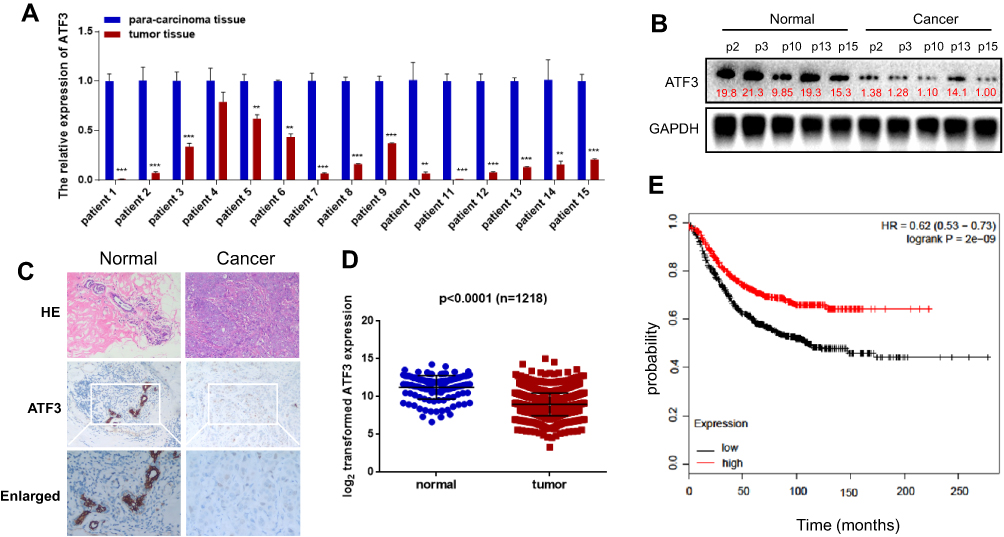 |
Figure 1 Expression of ATF3 in human breast cancer. (A) ATF3 expression levels were evaluated using qRT-PCR in 15 paired breast carcinoma and adjacent normal tissues. (B) The expression of ATF3 protein in breast carcinoma tissues and paired adjacent normal controls. (C) Hematoxylin and eosin (H&E) staining of normal (left panel) and breast cancer (right panel) tissue sections showed histologic features (original magnification, ×100, upper pannel), immunohistochemical staining (IHC) of ATF3 (original magnification, ×100, middle panel;×200, lower panel). (D) Analysis of TCGA data for 1218 breast cancer samples. (E) Kaplan-Meier plots were constructed for 1764 breast cancer patients stratified by the expression of ATF3 (low=884, high=880). **P < 0.01, ***P < 0.001 for t-test. Abbreviations: GAPHD, glyceraldehydes-3-phosphate dehydrogenase; HE, hematoxylin and eosin; HR, hazard ratio; TCGA, The Cancer Genome Atlas. |
ATF3 Regulated Growth, Migration, Invasion, and Apoptosis of Breast Cancer Cells
Several studies have shown that ATF3 promotes breast cancer tumorigenesis, but some studies have produced contradictory findings.18,19 To evaluate the role of ATF3 in breast cancer tumorigenesis, we overexpressed ATF3 in two breast cancer cells (Figure 2A). As shown in the results, restoration of ATF3 suppressed cell growth, induced G1/S cell cycle arrest (Figures 2B–D and S1A).
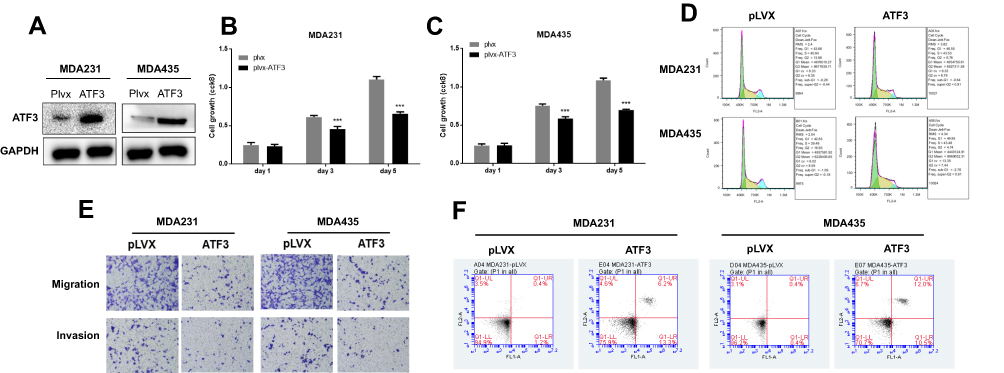 |
Figure 2 ATF3 suppressed tumorigenesis of breast carcinoma cell lines. (A) The expression of ATF3 proteins after transfection with lentivirus empty vector (pLVX) or lentivirus-ATF3 (ATF3), as detected using Western blot. (B and C) Cell growth was measured using CCK8 assay in MDA-MB-231 and MDA-MB-435 cells. (D) Representative experiments of cell cycle analysis performed in two cell lines. (E) Cell migration and invasion was analyzed in two cell lines using the transwell assay (n=3). (F) Apoptosis was measured using FACS (n = 3). ***P < 0.001 for t-test. Abbreviations: GAPHD, glyceraldehydes-3-phosphate dehydrogenase; FACS, fluorescence activated cell sorting; FL1A, FL1-A-FITC; FL2A, FL2-A-PI. |
In addition, we performed transwell and matrigel invasion assays on two cell lines stably transfected with ATF3. As seen in Figures 2E, S1B and C, overexpression of ATF3 suppressed cancer migration and invasion. Cell apoptosis has been shown to contribute to cell cycle suppression and reduced cell growth. We evaluated cell apoptosis following transfection with ATF3 using fluorescence-activated cell sorting. Transfection with ATF3 promote apoptosis in two breast cancer cell lines (Figures 2F and S1D). These results indicated that ATF3 regulated tumor metastasis by suppressing cell growth, inhibiting migration and invasion of cancer cells and promoting apoptosis.
ATF3 Modulated ARL4C Transcription
We investigated potential downstream targets of ATF3 using the TCGA database. As shown in Figure 3A, ATF3 and ARL4C were positively correlated in 1218 clinical samples. In addition, ATF3 and ARL4C were also positively correlated in human breast cancer cell lines (Figure 3B). We hypothesized that ARL4C expression was regulated by ATF3. As shown in Figures 2A, 3C–E, MDA-MB-231 and MDA-MB-435 cells transfected with lentivirus expressing ATF3 showed significantly increased ARL4C mRNA and protein levels. We then performed ChIP assay to further characterize the mechanisms by which ATF3 affected ARL4C expression. The results showed that overexpression of ATF3 enhanced the binding of ATF3 to the ARL4C promoter in two breast cancer cell lines (Figure 3F). Furthermore, we constructed a dual-luciferase report system by inserting the promoter sequence of ARL4 (Table S2) into the pGL3 plasmid (pGL3-control or pGL3-ARL4C promoter). Overexpression ofATF3 increased the luciferase activity of the ARL4 promoter (Figure 3G). In summary, these data suggested that ATF3 bound to the ARL4C promoter to induce ARL4C transcription.
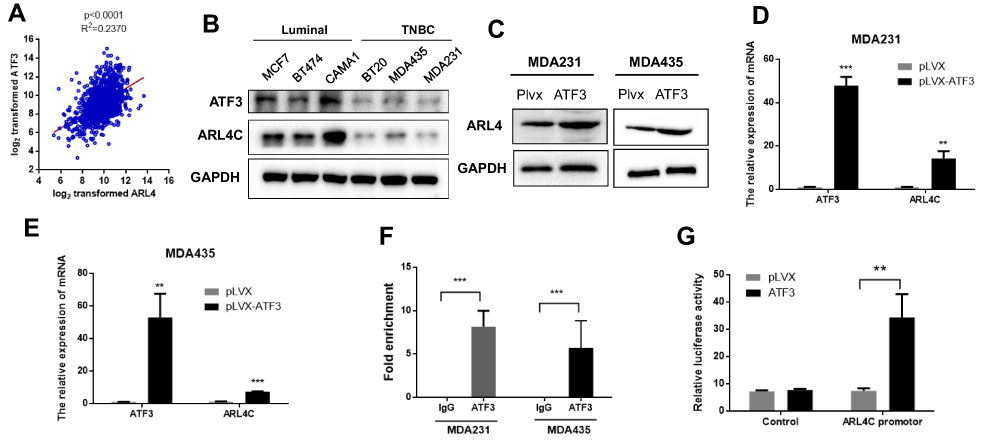 |
Figure 3 ATF3 modulated transcription of ATRL4C. (A) The expression of ATF3 was positively correlated with ATRL4C in the TCGA database. (B) The expression of ATF3 and ARL4C were reduced in TNBC cell lines than the luminal cells. (C) Overexpression of ATF3 promoted ARL4C protein expression. (D and E) ATF3 stimulated ARL4C mRNA expression in two cell lines.(F) Chromatin immunoprecipitation-qPCR of the promoter region of ARL4C locus in two cell lines. Antibody enrichment was quantified relative to the amount of input DNA. An antibody against IgG was used as a negative control. (G) Dual-luciferase activity was detected in HEK293T cells co-transfected with pLVX-ATF3 or empty vector (pLVX) with pGL3-Control or pGL3-ARL4C reporter. **P < 0.01, ***P < 0.001 for t-test, n ≥ 3. Abbreviations: GAPHD, glyceraldehydes-3-phosphate dehydrogenase; TNBC, triple negative breast cancer; pLVX, pLVX puro; IgG, immunoglobin G; TCGA, The Cancer Genome Atlas. |
Methylation of the ATF3 Promoter Affected ATF3 and ARL4C Expression
We evaluated methylation levels of the ATF3 promoter to further characterize the mechanisms of reduced ATF3 expression in breast carcinoma. As shown in Figure 4A, ATF3 promoter methylation levels were higher in tumor samples than those in adjacent control tissues, and methylation of the ATF3 promoter negatively correlated with ATF3 protein levels. We also evaluated methylation of the ATF3 promoter in different breast cancer cell lines, including luminal subtype (MCF7, CAMA1, and BT474), TNBC subtype (BT20, MDAMB231, and MDAMB435) cells. The ATF3 promoter was methylated to a greater extent in the TNBC subtype cell lines than in the luminal subtype cell lines (Figure 4B), and protein levels of ATF3 were higher in the luminal cell lines than those in the TNBC cell lines (Figure 3B). These results suggested that the low expression of ATF3 in breast cancer may result from high levels of ATF3 promoter methylation.
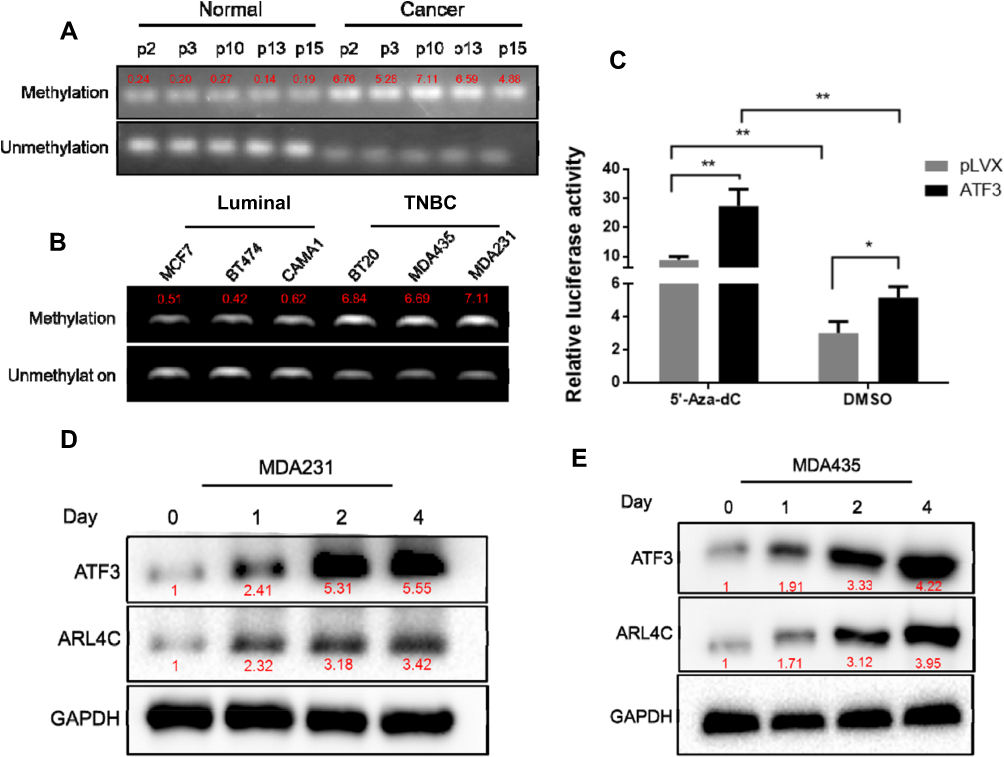 |
Figure 4 Methylation of ATF3 decreased the expression of ATF3 and ARL4C. (A) Methylation of the ATF3 promoter in breast carcinoma tissues and adjacent normal controls. The red font represents the relative methylation value of all CpG sites in each sample. (B) Methylation of the ATF3 promoter in human breast cancer cell lines. (C) Dual-luciferase assay in HEK293T cells treated with or without 5ʹ-Aza-dC. (D and E) Western blotting analysis of the expression of ATF3 and ARL4C in two human breast cancer cell lines treated with 5ʹ-Aza-dC. *P < 0.05, **P < 0.01 for t-test, n ≥ 3. Abbreviations: TNBC, triple negative breast cancer; GAPHD, glyceraldehydes-3-phosphate dehydrogenase; pLVX, pLVX puro; DMSO, dimethyl sulfoxide; CpG, cytosine-phosphoric acid-guanine. |
To investigate the role of ATF3 promoter methylation in regulation of ARL4C expression, we incubated breast cancer cells with 5-aza-deoxycytidine (5ʹ-Aza-dC), then performed dual-luciferase assay. As shown in Figure 4C, 5ʹ-Aza-dC treatment enhanced the luciferase activity in the ATF3 overexpression group compared to that in the control group. Incubation with 500 nM5ʹ-Aza-dC increased protein levels of ATF3 and ARL4C in a time-dependent manner (Figure 4D and E), which suggested that demethylation of ATF3 promoted ATF3 expression, and subsequently induced ARL4C transcription.
ARL4C Is a Potential Prognostic Biomarker of Breast Cancer
Breast carcinoma cells were analyzed for ARL4C using qRT-PCR, Western blot, and IHC. The results showed that ARL4C levels were lower in most of the human breast carcinoma samples compared to those in the paired control tissues (Figure 5A–C). Data from TCGA was analyzed to evaluate the expression of ARL4C in breast cancer (https://tcga.xenahubs.net/download/TCGA.BRCA.sampleMap/HiSeqV2.gz; Full metadata). As shown in Figure 5D, ARL4C expression was significantly lower in breast cancer tissues than that in adjacent control tissues. Kaplan-Meier plot analysis (n=3951) (http://kmplot.com/analysis/index.php?p=service) showed that ARL4C (Affy ID 213759) expression positively correlated with better outcomes (Figure 5E).
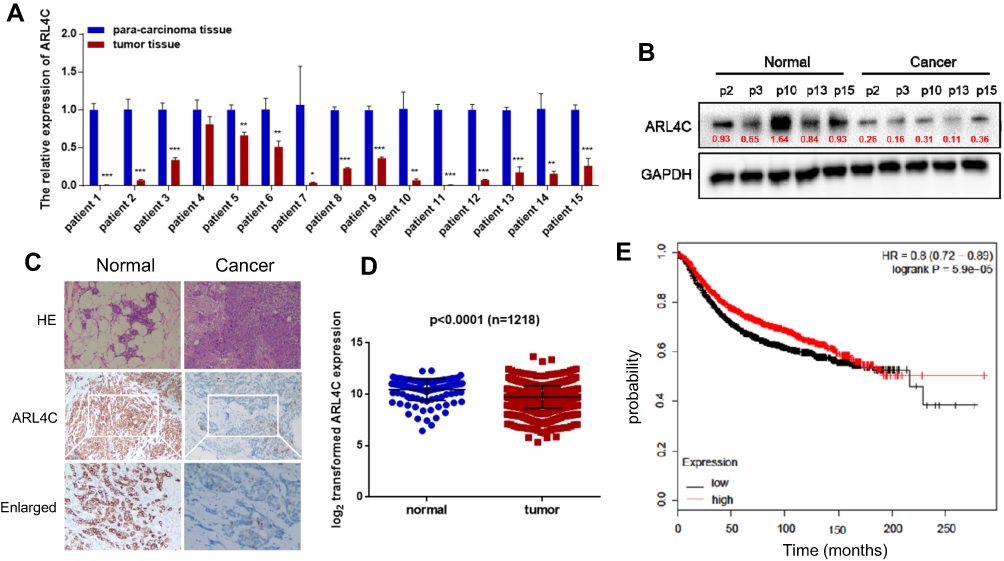 |
Figure 5 Expression of ARL4C in human breast cancer. (A) Expression levels of ARL4C were evaluated using qRT-PCR in breast carcinoma and adjacent normal tissues (n=15). (B) The protein level of ARL4C in breast carcinoma tissues and adjacent normal controls. (C) Hematoxylin and eosin staining, and IHC results for ARL4C. (D) Analysis of ARL4C expression in 1218 breast cancer samples. (E) Kaplan-Meier plots were constructed for 3951 breast cancer patients stratified by the expression of ATF3 (low = 1998, high = 1953). *P < 0.05, **P < 0.01, ***P < 0.001, n ≥ 3. Abbreviations: GAPHD, glyceraldehydes-3-phosphate dehydrogenase; HE, hematoxylin and eosin; HR, hazard ratio; IHC, immunohistochemical. |
Restoration of ARL4C Expression Suppressed Breast Carcinogenesis
To define the role of ARL4C in breast carcinogenesis, MDA-MB-231 and MDA-MB-435 cells were transfected with a lentivirus to form two stable cell lines that overexpressed ARL4C. Western blot analysis was used to confirm that ARL4C was overexpressed (Figure 6A). As shown in Figure 6B and C, overexpression of ARL4C significantly suppressed breast cancer cell growth. Moreover, flow cytometry analysis showed that ARL4C overexpression induced G1/S arrest and reduced the number of cells in G2 (Figure 6D, S2A).
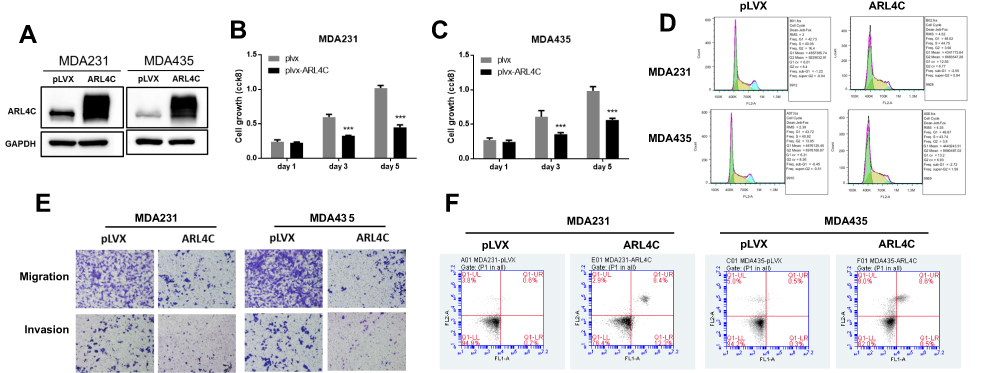 |
Figure 6 Restoration of ARL4C reduced cell proliferation, migration, and invasion, and promotes apoptosis in breast carcinoma cell lines. (A) Protein levels of ARL4C in two cell lines transfected with lentivirus empty vector (pLVX) or lentivirus-ARL4C (ARL4C). (B) and (C) Cell growth of MDA-MB-231 and MDA-MB-435 cells. (D) Representative cell cycle analysis performed in two cell lines stably transfected with ARL4C or empty vector (pLVX). (E) Cell migration and invasion in two cell lines were analyzed using transwell assay (n = 3). (F) Apoptosis was measured using FACS (n = 3). ***P < 0.001 for t-test, n ≥ 3. Abbreviations: GAPHD, glyceraldehydes-3-phosphate dehydrogenase; pLVX, pLVX puro; FACS, Fluorescence activated Cell Sorting; FL1A, FL1-A-FITC; FL2A, FL2-A-PI. |
We performed transwell assays on two cell lines stably transfected with ARL4C to evaluate the effects of ARL4C on cell migration and invasion. Overexpression of ARL4C reduced cell migration and invasion ability (Figures 6E, S2B and S2C). Furthermore, we evaluated the effects of ARL4C overexpression on breast cancer cell apoptosis. As shown in Figures 6F and S2D, ARL4C overexpression induced apoptosis in two breast cancer cell lines. These results suggested that ARL4C may suppress breast carcinogenesis.
Discussion
Activated transcription factor 3 is not expressed in most normal cell types.20–22 However, ATF3 is an “adaptive response gene,” and can be expressed in response to cell stress.23,24 Activated transcription factor 3 has been shown to act as an oncogene and as a tumor suppressor in several types of tumor, including in breast cancer.7,11 We found that ATF3 expression was markedly lower in breast cancer tissues compared to that in adjacent control tissues, and ATF3 overexpression reduced cell growth and invasion, which highlights its function as a tumor suppressor gene.
Methylation of DNA regulates a number of cellular processes, including cell growth and migration.25 A number of tumor suppressor genes are hypermethylated, and may be promising clinical target.26,27 We investigated whether downregulation of ATF3 was associated with methylation of the ATF promoter. The ATF promoter was methylated to a greater degree in tumor samples than in control samples, and hypermethylation was negatively correlated with ATF3 protein levels. We also showed that ARL4C expression was reduced in breast carcinoma tissue, and levels of ARL4C correlated with ATF3 levels. Furthermore, we showed that ATF3 could bind to the ARL4C promoter in MDA-MB-231 and MDA-MB-435 cells. In addition, ATF3 increased the activity of the ARL4C promoter, and methylation of ATF3 reduced ARL4C promoter activity. We also showed that treatment of MDA-MB-231 and MDA-MB-435 cells with 5ʹ-Aza-dC restored the expression of ATF3 and ARL4C in a time-dependent manner. These data suggested that ATF3 may regulate transcription of ARL4C, and methylation of ATF3 could induce degradation of ATF3, resulting in decreased transcriptional activity. In particular, ATF3 could regulates several genes, including TGF-beta1,28 BCL-X29 and ARL4C to suppress tumor growth in breast cancer.
Functional studies showed that restoration of ARL4C significantly reduced cell proliferation and promoted cell apoptosis. Moreover, ARL4C overexpression inhibited breast cancer cell metastasis, which indicated that ARL4C may play an anti-oncogenic role in breast carcinogenesis. However, the mechanisms by which ARL4C exerts anti-oncogenic effects has not been characterized.
In conclusion, our study showed that ATF3 and ARL4C may regulate biological processes in breast cancer, such as modulation of cell growth, cell cycle, apoptosis, and metastasis. We also showed that ATF3 was a transcriptional regulator of ARL4C expression. Furthermore, hypermethylation of ATF3 resulted in degradation of ATF3, which led to decreased ARL4C expression. These findings suggested that the ATF3/ARL4C axis may be a prognostic biomarker of breast carcinoma, and may also be a target for treatment of human breast cancer.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81873730), Jiangsu Women and Children Health Key Discipline Program (FXK201758), Suzhou Health Planning Commission’s Key Clinical Diagnosis and Treatment Program (LCZX201606).
Disclosure
The authors declare that they have no competing interests.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Blows FM, Driver KE, Schmidt MK, et al. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: a collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med. 2010;7(5):e1000279. doi:10.1371/journal.pmed.1000279
3. Wu Y, Meng Q, Yang Z, et al. Circulating HER-2 mRNA in the peripheral blood as a potential diagnostic and prognostic biomarker in females with breast cancer. Oncol Lett. 2018;16(3):3726–3734. doi:10.3892/ol.2018.9091
4. Hai T, Hartman MG. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene. 2001;273(1):1–11. doi:10.1016/S0378-1119(01)00551-0
5. Ameri K, Hammond EM, Culmsee C, et al. Induction of activating transcription factor 3 by anoxia is independent of p53 and the hypoxic HIF signalling pathway. Oncogene. 2007;26(2):284–289. doi:10.1038/sj.onc.1209781
6. Hai T, Wolfgang CD, Marsee DK, Allen AE, Sivaprasad U. ATF3 and stress responses. Gene Expr. 1999;7(4–6):321–335.
7. Yin X, Wolford CC, Chang YS, et al. ATF3, an adaptive-response gene, enhances TGF{beta} signaling and cancer-initiating cell features in breast cancer cells. J Cell Sci. 2010;123(Pt 20):3558–3565. doi:10.1242/jcs.064915
8. Ishiguro T, Nagawa H. ATF3 gene regulates cell form and migration potential of HT29 colon cancer cells. Oncol Res. 2001;12(8):343–346. doi:10.3727/096504001108747792
9. Bottone FG, Moon Y, Kim JS, Alston-Mills B, Ishibashi M, Eling TE. The anti-invasive activity of cyclooxygenase inhibitors is regulated by the transcription factor ATF3 (activating transcription factor 3). Mol Cancer Ther. 2005;4(5):693–703. doi:10.1158/1535-7163.MCT-04-0337
10. Lu D, Wolfgang CD, Hai T. Activating transcription factor 3, a stress-inducible gene, suppresses Ras-stimulated tumorigenesis. J Biol Chem. 2006;281(15):10473–10481. doi:10.1074/jbc.M509278200
11. Yin X, Dewille JW, Hai T. A potential dichotomous role of ATF3, an adaptive-response gene, in cancer development. Oncogene. 2008;27(15):2118–2127. doi:10.1038/sj.onc.1210861
12. Chen C, Ge C, Liu Z, et al. ATF3 inhibits the tumorigenesis and progression of hepatocellular carcinoma cells via upregulation of CYR61 expression. J Exp Clin Cancer Res. 2018;37(1):263. doi:10.1186/s13046-018-0919-8
13. Hofmann I, Thompson A, Sanderson CM, Munro S. The Arl4 family of small G proteins can recruit the cytohesin Arf6 exchange factors to the plasma membrane. Curr Biol. 2007;17(8):711–716. doi:10.1016/j.cub.2007.03.007
14. Fujii S, Matsumoto S, Nojima S, Morii E, Kikuchi A. Arl4c expression in colorectal and lung cancers promotes tumorigenesis and may represent a novel therapeutic target. Oncogene. 2015;34(37):4834–4844. doi:10.1038/onc.2014.402
15. Su D, Katsaros D, Xu S, et al. ADP-ribosylation factor-like 4C (ARL4C), a novel ovarian cancer metastasis suppressor, identified by integrated genomics. Am J Transl Res. 2015;7(2):242–256.
16. Xue X, Fei X, Hou W, Zhang Y, Liu L, Hu R. miR-342-3p suppresses cell proliferation and migration by targeting AGR2 in non-small cell lung cancer. Cancer Lett. 2018;412:170–178. doi:10.1016/j.canlet.2017.10.024
17. Huang H, Wang Y, Li Q, Fei X, Ma H, Hu R. miR-140-3p functions as a tumor suppressor in squamous cell lung cancer by regulating BRD9. Cancer Lett. 2019;446:81–89. doi:10.1016/j.canlet.2019.01.007
18. Chang YS, Jalgaonkar SP, Middleton JD, Hai T. Stress-inducible gene Atf3 in the noncancer host cells contributes to chemotherapy-exacerbated breast cancer metastasis. Proc Natl Acad Sci U S A. 2017;114(34):E7159–E7168. doi:10.1073/pnas.1700455114
19. Wolford CC, McConoughey SJ, Jalgaonkar SP, et al. Transcription factor ATF3 links host adaptive response to breast cancer metastasis. J Clin Invest. 2013;123(7):2893–2906. doi:10.1172/JCI64410
20. Hackl C, Lang SA, Moser C, et al. Activating transcription factor-3 (ATF3) functions as a tumor suppressor in colon cancer and is up-regulated upon heat-shock protein 90 (Hsp90) inhibition. BMC Cancer. 2010;10:668. doi:10.1186/1471-2407-10-668
21. Pan YX, Chen H, Thiaville MM, Kilberg MS. Activation of the ATF3 gene through a co-ordinated amino acid-sensing response programme that controls transcriptional regulation of responsive genes following amino acid limitation. Biochem J. 2007;401(1):299–307. doi:10.1042/BJ20061261
22. Chromik AM, Hahn SA, Daigeler A, et al. Gene expression analysis of cell death induction by taurolidine in different malignant cell lines. BMC Cancer. 2010;10:595. doi:10.1186/1471-2407-10-595
23. Kawai M, Jin M, Nishimura J, et al. Hepatocarcinogenic susceptibility of fenofibrate and its possible mechanism of carcinogenicity in a two-stage hepatocarcinogenesis model of rasH2 mice. Toxicol Pathol. 2008;36(7):950–957. doi:10.1177/0192623308327118
24. Kawai M, Saegusa Y, Jin M, et al. Mechanistic study on hepatocarcinogenesis of piperonyl butoxide in mice. Toxicol Pathol. 2009;37(6):761–769. doi:10.1177/0192623309344087
25. Muller HM, Widschwendter A, Fiegl H, et al. DNA methylation in serum of breast cancer patients: an independent prognostic marker. Cancer Res. 2003;63(22):7641–7645.
26. Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res. 1998;72:141–196.
27. Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet. 1999;21(2):163–167. doi:10.1038/5947
28. Rohini M, Arumugam B, Vairamani M, Selvamurugan N. Stimulation of ATF3 interaction with Smad4 via TGF-beta1 for matrix metalloproteinase 13 gene activation in human breast cancer cells. Int J Biol Macromol. 2019;134:954–961. doi:10.1016/j.ijbiomac.2019.05.062
29. Chueh AC, Tse JWT, Dickinson M, et al. ATF3 repression of BCL-XL determines apoptotic sensitivity to HDAC inhibitors across tumor types. Clin Cancer Res. 2017;23(18):5573–5584. doi:10.1158/1078-0432.CCR-17-0466
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.